KOSELUGO
ASTRAZENECA
selumetinibe, sulfato
Tratamento da neurofibromatose tipo 1.
Apresentações.
Cápsulas duras de 10 mg em uma embalagem com 1 frasco de 60 cápsulas duras
Cápsulas duras de 25 mg em uma embalagem com 1 frasco de 60 cápsulas duras
VIA ORAL
USO PEDIÁTRICO A PARTIR DE 2 ANOS DE IDADE
Composição.
Cada cápsula dura de 10 mg de KOSELUGO contém 10 mg de selumetinibe (que equivalem a 12,10 mg de sulfato de selumetinibe);
Cada cápsula dura de 25 mg de KOSELUGO contém 25 mg de selumetinibe (que equivalem a 30,25 mg de sulfato de selumetinibe)
Excipientes das cápsulas duras de 10 mg: tocofersolana; hipromelose, carragenina, cloreto de potássio, dióxido de titânio, cera de carnaúba, goma laca, óxido de ferro preto, propilenoglicol, hidróxido de amônia 28%
Excipientes das cápsulas duras de 25 mg: tocofersolana; hipromelose, carragenina, cloreto de potássio, dióxido de titânio, azul de indigotina, óxido de ferro amarelo, cera de carnaúba e/ou amido de milho, óxido de ferro vermelho, azul de indigotina 132 laca de alumínio, goma laca, monoleato de glicerila
Informações técnicas.
1. INDICAÇÕES
KOSELUGO é indicado para o tratamento de pacientes pediátricos a partir de 2 anos de idade, com neurofibromatose tipo 1 (NF1) que apresentem neurofibromas plexiformes (NP) sintomáticos e inoperáveis.
2. RESULTADOS DE EFICÁCIA
Eficácia clínica
SPRINT
A eficácia de KOSELUGO foi avaliada em um estudo aberto, multicêntrico, de braço único [Estrato 1 do SPRINT Fase II (NCT01362803)] de 50 pacientes pediátricos com NF1 e NP inoperável que causava morbidade significativa. NP inoperável foi definido como um NP que não poderia ser completamente removido cirurgicamente sem risco de morbidade substancial devido ao revestimento, ou proximidade, de estruturas vitais, grau de invasão ou alta vascularização do NP. Pacientes receberam 25 mg/m2 (ASC) duas vezes ao dia, por 28 dias (1 ciclo de tratamento) em um esquema posológico contínuo. O tratamento foi descontinuado se o paciente não estivesse mais obtendo benefício clínico, apresentasse toxicidade inaceitável ou progressão do NP ou a critério do investigador.
O NP alvo, o NP que causou sintomas clínicos ou complicações relevantes (morbidades relacionadas com NP) foi avaliado para taxa de resposta usando análise de ressonância magnética nuclear (RMN) volumétrica de leitura centralizada de acordo com os critérios de Avaliação de Resposta em Neurofibromatose e Schwannomatose (REiNS). A resposta do tumor foi avaliada no basal e durante o tratamento a cada 4 ciclos por 2 anos e então a cada 6 ciclos.
Os pacientes tiveram avaliações de RMN volumétrica no NP alvo e avaliações de resultado clínico, que incluíram avaliações funcionais e resultados relatados pelos pacientes.
A mediana de idade dos pacientes foi de 10,2 anos (intervalo: 3,5 - 17,4 anos), 60% eram homens, 84% eram Caucasianos.
As características da doença no basal são fornecidas na Tabela 1.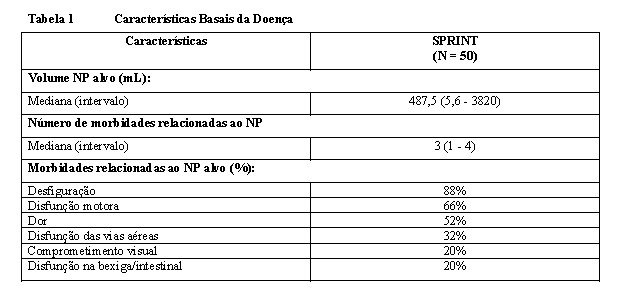
O desfecho primário de eficácia foi Taxa de Resposta Objetiva (TRO), definida como o percentual de pacientes com resposta completa (definida como desaparecimento do NP alvo) ou resposta parcial confirmada (definida como redução ≥20% no volume do NP, confirmada por uma avaliação tumoral subsequente dentro de 3-6 meses), com base na avaliação centralizada do NCI (National Cancer Insitute). A Duração da Resposta (DR) também foi avaliada.
O desfecho primário, TRO foi de 66% (IC 95%, 51,2 - 78,8). O tempo para início da resposta para a maioria dos pacientes (24/33 [72,7%]) ocorreu dentro de 8 ciclos (intervalo 4 - 20 ciclos).
A mediana da DR desde o início da resposta não foi atingida; no momento do corte de dados, a mediana do tempo de acompanhamento foi de 24 ciclos. Dos 33 pacientes que tiveram respostas parciais confirmadas, 29 (87,9%) permaneceram em resposta após 12 ciclos; 4 pacientes foram censurados não devido à progressão. A probabilidade de permanecer em resposta após 12 e 16 ciclos, estimada usando o método de Kaplan-Meier, foi de 100% (IC 95% não estimado) e 96,2% (IC 95% 75,7 - 99,4), respectivamente. A mediana do tempo do início do tratamento até progressão da doença durante o tratamento não foi atingida.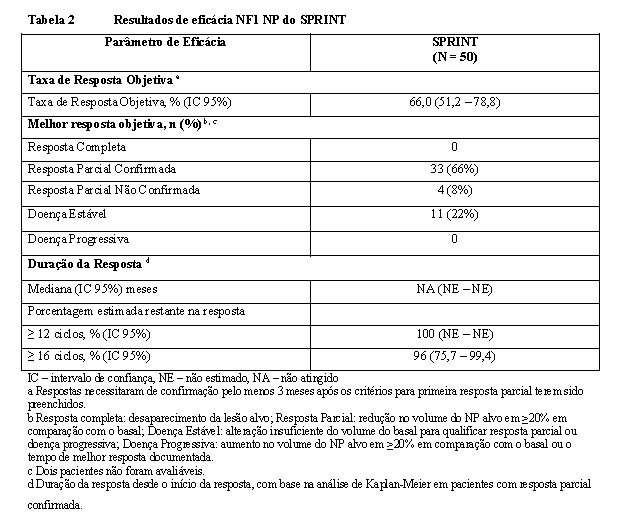
No momento do corte de dados, 28 (56%) pacientes permaneciam em resposta parcial confirmada, 2 (4%) apresentavam resposta parcial não confirmada, 15 (30%) tinham doença estável e 3 (6%) tinham doença progressiva.
A mediana da melhor alteração percentual no volume de NP do basal foi -27,85% (intervalo: 2,2% a -54,5%). A Figura 1 demonstra a melhor alteração percentual no volume de NP alvo para cada paciente.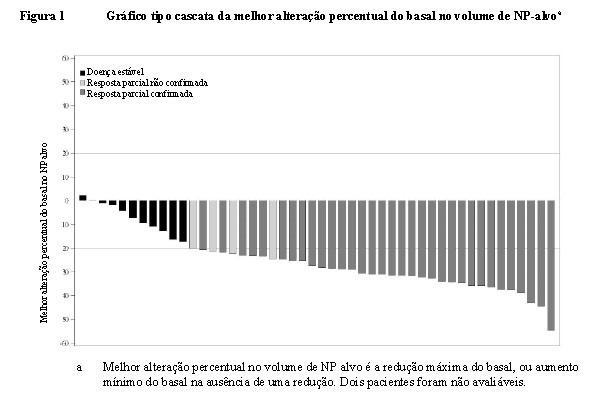
Avaliações de Resultado Clínico
A intensidade da dor do NP alvo foi autorrelatada pelos pacientes ≥8 anos de idade usando uma Escala de Classificação Numérica de 11 pontos (NRS-11). Uma redução clinicamente significativa na dor (definida como uma redução ≥2 pontos do basal) foi reportada por 50% dos pacientes (n=12) no pré-Ciclo 13; 12 pacientes (50%) não reportaram alteração (10 dos quais tinham um escore basal ≤1) e nenhum paciente mostrou deterioração.
A qualidade de vida relacionada à saúde (QVRS) reportada pelos pais (todos os pacientes) e reportada pelo paciente (≥8 anos de idade) foi avaliada usando o questionário Peds-QL. Com base em uma análise MMRM, uma melhora clinicamente significativa na QVRS (limiar clinicamente significativo 11,9) foi reportada pelos pais no pré-Ciclo 13 com uma alteração média do basal no escore total do Peds-QL de 12,7 (IC 95% 8,91 - 16,55). A melhora na QVRS também foi reportada pelos pacientes com uma alteração média do basal de 6,68 (IC 95% 1,34 - 12,02).
Referências Bibliográficas
Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550-2560.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Selumetinibe é um potente e seletivo inibidor das proteínas quinases ativadas por mitógenos, quinases 1 e 2 (MEK1/2), não competitivo com relação ao ATP e disponível por via oral. As proteínas MEK1/2 são componentes críticos da via RAF-MEK-ERK regulada por RAS, que é frequentemente ativada em diferentes tipos de tumores. Selumetinibe bloqueia a atividade da MEK e inibe o crescimento de linhagens celulares ativadas pela via RAF-MEK-ERK. Portanto, a inibição de MEK pode bloquear a proliferação e sobrevivência das células tumorais, nas quais a via RAF-MEK-ERK está ativada.
Propriedades Farmacodinâmicas
Em modelos de camundongos geneticamente modificados de NF1 que geram neurofibromas que recapitulam o genótipo e fenótipo de neurofibromas humanos do tipo 1, a administração oral de selumetinibe inibe a fosforilação de ERK, reduz o volume, proliferação, número e crescimento de neurofibroma.
Eletrofisiologia cardíaca
Em um estudo positivo (moxifloxacino) controlado por placebo, o efeito de selumetinibe no intervalo QTc, em 48 adultos sadios, após uma dose única oral de 75 mg, não demonstrou ser clinicamente relevante (alteração < 10 ms). Uma análise farmacocinética-farmacodinâmica previu uma alteração < 10 ms com a dose de 150 mg (3 vezes maior do que a dose máxima recomendada de 50 mg em pacientes pediátricos com NF1).
Propriedades Farmacocinéticas
Na posologia recomendada de 25 mg/m2 duas vezes ao dia em pacientes pediátricos (3 a ≤18 anos de idade), a média geométrica (coeficiente de variação [CV%]) da concentração plasmática máxima (Cmax) foi 731 (62%) ng/mL e a área sob a curva da concentração plasmática (AUC0-12) após a primeira dose foi 2009 (35%) ng·h/mL. Acúmulo mínimo de ~1,1 vezes foi observado no estado estacionário com a administração duas vezes ao dia.
Em pacientes pediátricos, com um nível de dose de 25 mg/m2, selumetinibe tem uma depuração oral aparente de 8,8 L/h, volume médio aparente de distribuição no estado de estacionário de 78 L e uma meia-vida média de eliminação de ~6,2 horas.
Absorção
Em indivíduos adultos sadios, a média da biodisponibilidade absoluta oral de selumetinibe foi 62%. Após a administração oral, selumetinibe é rapidamente absorvido, produzindo concentrações plasmáticas máximas no estado de estacionário (Tmax) entre 1-1,5 horas após a dose.
Efeito do alimento
Em estudos clínicos separados, em indivíduos adultos sadios e em pacientes adultos com malignidades sólidas avançadas, a administração concomitante de 75 mg de selumetinibe com uma refeição com alto teor de gordura resultou em uma redução média na Cmax de 50% e 62%, respectivamente em comparação com a administração em jejum. A média da AUC de selumetinibe foi reduzida em 16% e 19%, respectivamente, e o tempo para atingir a concentração máxima (Tmax) foi retardado em aproximadamente 1,5 a 3 horas (consulte seção 8. POSOLOGIA E MODO DE USAR).
Em indivíduos adultos sadios, a administração concomitante de 50 mg de selumetinibe com uma refeição com baixo teor de gordura resultou em Cmax 60% menor em comparação com a administração em jejum. A AUC de selumetinibe foi reduzida em 38% e o tempo para atingir a concentração máxima (Tmax) foi retardado em aproximadamente 0,9 horas (consulte seção 8. POSOLOGIA E MODO DE USAR).
Efeito de agentes redutores de ácido gástrico no selumetinibe
As cápsulas de selumetinibe não apresentam dissolução dependente de pH. KOSELUGO pode ser usado concomitantemente com agentes modificadores do pH gástrico (ou seja, antagonistas do receptor H2 e inibidores da bomba de prótons) sem restrições.
Distribuição
O volume médio aparente de distribuição de selumetinibe entre 20 a 30 mg/m2 no estado estacionário variou de 78 a 171 L em pacientes pediátricos, indicando distribuição moderada no tecido.
A ligação in vitro à proteína plasmática é de 98,4% em humanos. Selumetinibe se liga principalmente à albumina sérica (96,1%) do que à a-1 glicoproteína ácida ( < 35%).
Biotransformação / Metabolismo
In vitro, selumetinibe passa por reações metabólicas de Fase 1 incluindo oxidação da cadeia lateral, N-desmetilação e perda da cadeia lateral para formar metabólitos amido e ácido.
CYP3A4 é a isoforma predominante responsável pelo metabolismo oxidativo de selumetinibe com CYP2C19, CYP1A2, CYP2C9, CYP2E1 e CYP3A5 envolvidas em menor extensão. Os estudos in vitro indicam que selumetinibe também passa por reações metabólicas diretas de Fase 2 para formar conjugados glicuronídeos envolvendo principalmente as enzimas UGT1A1 e UGT1A3. Glicuronidação é uma via significativa de eliminação para os metabólitos de Fase 1 de selumetinibe envolvendo diversas isoformas de UGT.
Após a administração oral de 14C-selumetinibe por indivíduos saudáveis do sexo masculino, o selumetinibe inalterado (~40% da radioatividade) e outros metabólitos, incluindo glicuronídeo do metabólito imidazoindazol (M2; 22%), glicuronídeo de selumetinibe (M4; 7%), N-desmetil selumetinibe (M8; 3%) e ácido N-desmetil carboxílico (M11; 4%), foram responsáveis pela maior parte da radioatividade circulante no plasma humano. N-desmetil selumetinibe representa menos que 10% dos níveis de selumetinibe no plasma humano, mas é aproximadamente 3 a 5 vezes mais potente que o composto original, contribuindo para cerca de 21% a 35% da atividade farmacológica geral.
Eliminação
Em voluntários adultos sadios, após uma dose única oral de 75 mg de selumetinibe radiomarcado, 59% foi recuperado nas fezes (19% inalterado) enquanto que 33% da dose administrada ( < 1% como original) foi encontrada na urina 9 dias após a coleta da amostra.
Interações
In vitro, o selumetinibe não é um inibidor de CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4 e CYP2E1. In vitro, o selumetinibe não é um indutor de CYP3A4, CYP1A2 e CYP2B6. In vitro, o selumetinibe inibe UGT1A3, UGT1A4, UGT1A6 and UGT1A9, mas não se espera que esses efeitos sejam clinicamente relevantes.
Interações com proteínas transportadoras
Com base nos estudos in vitro, o selumetinibe é um substrato para transportadores PRCM e gp-P, mas é improvável que seja submetido a interações medicamentosas clinicamente significativas na dose pediátrica recomendada. Estudos in vitro sugerem que selumetinibe não inibe a proteína de resistência ao câncer de mama (PRCM), glicoproteína P (gp-P), OATP1B1, OATP1B3, OCT2, OAT1, MATE1 e MATE2K na dose pediátrica recomendada. Um efeito clinicamente relevante na farmacocinética de substratos de OAT3 administrados concomitantemente não pode ser excluído.
Populações especiais
Insuficiência renal
A exposição de 50 mg de selumetinibe oral foi investigada em indivíduos adultos com função renal normal (n=11) e indivíduos com doença renal em estágio terminal (DRET) (n=12). O grupo DRET demonstrou Cmax e AUC 16% e 28% menores, respectivamente, com a fração de selumetinibe não ligado sendo 35% maior em indivíduos DRET. Como resultado, as razões de Cmax e AUC não ligado foram 0,97 e 1,13 no grupo DRET quando comparadas com o grupo com função renal normal. Um pequeno aumento, aproximadamente 20% da AUC, na razão do metabólito N-desmetil/original foi detectado no grupo DRET quando comparado com o grupo normal. Como a exposição em indivíduos DRET foi similar àquela com função renal normal, investigações em indivíduos com comprometimento renal leve, moderado e grave não foram realizadas. Não é esperado que a insuficiência renal tenha influência significativa na exposição de selumetinibe (consulte seção 8. POSOLOGIA E MODO DE USAR).
Insuficiência hepática
Indivíduos adultos com função hepática normal (n=8) e insuficiência hepática leve (Child-Pugh A, n=8) receberam uma dose de 50 mg de selumetinibe, indivíduos com insuficiência hepática moderada (Child-Pugh B, n=8) uma dose de 50 ou 25 mg e indivíduos com insuficiência renal grave (Child-Pugh C, n=8) uma dose de 20 mg. A AUC da dose total normalizada de selumetinibe e AUC da dose não ligada foi 86% e 69% respectivamente, nos pacientes com insuficiência hepática leve em comparação com os valores de AUC dos indivíduos com função hepática normal. A exposição ao selumetinibe (AUC) foi maior em pacientes com insuficiência hepática moderada (Child-Pugh B) e grave (Child-Pugh C); os valores de AUC total e AUC não ligada dos indivíduos com função hepática normal foram, respectivamente, 159% e 141% (Child-Pugh B) e 157% e 317% (Child-Pugh C), (consulte seção 8. POSOLOGIA E MODO DE USAR).
Etnicidade
Após dose única, a exposição ao selumetinibe parece ser maior em voluntários adultos sadios japoneses, asiáticos não japoneses e indianos em comparação com voluntários adultos ocidentais. No entanto, há sobreposição considerável com indivíduos ocidentais quando corrigido para peso corporal ou ASC (consulte seção 8. POSOLOGIA E MODO DE USAR).
Pacientes adultos ( > 18 anos de idade)
Os parâmetros farmacocinéticos em voluntários sadios adultos e pacientes adultos com malignidades sólidas avançadas, são similares àqueles em pacientes pediátricos (3 a ≤18 anos de idade) com NF1.
Em pacientes adultos com malignidades sólidas, a uma dose de 75 mg duas vezes ao dia, Cmax e média geométrica da AUC (%CV) foram 1307 (76%) ng/mL e 4736 (37%) ng·h/mL, respectivamente. As concentrações plasmáticas máximas de selumetinibe foram atingidas 1,5 horas após a dose com uma meia-vida média de eliminação de 7,8 horas. Cmax e AUC aumentaram proporcionalmente ao longo de um intervalo de dose de 25 mg a 100 mg e a administração de 75 mg de selumetinibe duas vezes ao dia resultou em acúmulo mínimo de ~1,2 vezes.
Dados pré-clínicos de segurança
Mutagenicidade
Selumetinibe não demonstrou potencial mutagênico ou clastogênico in vitro, mas produziu um aumento nos eritrócitos imaturos micronucleados (aberrações cromossômicas) em estudos de micronúcleos em camundongo, predominantemente via um modo de ação aneugênico. A exposição média livre (Cmax) no Nível Sem Eventos Adversos Observados (NOEL) foi aproximadamente 27 vezes maior do que a exposição clínica livre na dose máxima recomendada para humanos (DMRH) de 25 mg/m2.
Carcinogênese
Selumetinibe não foi carcinogênico em um estudo de 6 meses em camundongos transgênicos rasH2 em exposições livres 24 vezes (fêmeas) e 16 vezes (machos) a AUC clínica livre no DMRH e em um estudo de carcinogenicidade de 2 anos em ratos em exposições livres 2,9 vezes (fêmeas) e 3,7 vezes (machos) a AUC clínica livre no DMRH.
Toxicidade de dose repetida
Nos estudos de toxicidade de dose repetida em camundongos e ratos, os principais efeitos observados após a exposição de selumetinibe foram na pele, crostas de feridas associadas com erosões microscópicas e ulceração em ratos em uma exposição livre similar à exposição clínica (AUC livre) no DMRH; achados inflamatórios e ulcerativos no trato GI de camundongos associados com alterações secundárias no fígado e sistema linforeticular nas exposições livres aproximadamente 28 vezes a exposição clínica livre no DMRH; displasia de placa de crescimento (fiseal) em ratos machos em uma exposição livre 11 vezes a exposição livre clínica no DMRH. Os achados GI demonstraram evidência de reversibilidade após um período de recuperação. A reversibilidade para toxicidades cutâneas e displasia fiseal não foram avaliadas.
Toxicologia Reprodutiva
Fertilidade
Em um estudo de 6 meses com camundongos, selumetinibe não afetou o desempenho de acasalamento de machos em qualquer dose até 20 mg/kg duas vezes ao dia correspondente a aproximadamente 22 vezes a exposição clínica humana com base no AUC livre no DMRH. Em camundongos fêmeas expostas a selumetinibe a 12,5 mg/kg duas vezes ao dia, o desempenho de acasalamento e fertilidade não foram afetados, mas o número de fetos vivos foi levemente reduzido. Após o período de descontinuação de tratamento de três semanas, nenhum efeito foi aparente em qualquer parâmetro. O Nível Sem Evento Adverso Observável NOAEL) para a toxicidade materna e efeitos no desempenho reprodutivo foi 2,5 mg/kg duas vezes ao dia (aproximadamente, 3,5 vezes a exposição livre em humanos na DMRH).
Toxicidade embriofetal
Nos estudos de desenvolvimento embriofetal em camundongos, selumetinibe causou uma redução no número de fetos vivos devido a um aumento na perda pós-implantação, uma redução na média dos pesos fetais e da ninhada, ocorrência aumentada de olho aberto e fenda palatina em níveis de dose que não induziram toxicidade materna significativa. Estes efeitos foram observados a uma exposição > 3,5 vezes a exposição clínica no DMRH com base no AUC livre e indicam que selumetinibe pode ter potencial para causar defeitos no feto.
Desenvolvimento pré e pós-natal
A administração de selumetinibe em camundongos prenhes a partir do Dia 6 da gestação até o Dia 20 da lactação resultou em pesos corporais reduzidos dos filhotes e menos filhotes dentro dos critérios de constrição de pupila no Dia 21 após o parto. A incidência de malformações (olhos prematuramente abertos e fenda palatina) foi aumentada em todos os níveis de dose. As malformações ocorreram em concentração materna (Cmax) 0,4 vezes abaixo da média de concentração clínica livre na DMRH.
Selumetinibe e seu metabólito ativo foram excretados no leite de camundongos amamentando em concentrações aproximadamente iguais às do plasma.
4. CONTRAINDICAÇÕES
Não há contraindicações conhecidas para KOSELUGO.
5. ADVERTÊNCIAS E PRECAUÇÕES
Redução de FEVE
Reduções assintomáticas na fração de ejeção foram reportadas em 22% dos pacientes pediátricos no estudo clínico pivotal (9. REAÇÕES ADVERSAS). A mediana do tempo para início dos eventos foi de 226 dias.
Pacientes pediátricos com histórico de comprometimento da função ventricular esquerda ou uma FEVE basal abaixo do LIN não foram estudados. FEVE deve ser avaliado antes do início do tratamento para estabelecer valores basais. Antes do início do tratamento com selumetinibe, pacientes devem ter uma fração de ejeção acima do LIN institucional.
Avaliar FEVE em intervalos de aproximadamente 3 meses ou mais frequentemente conforme clinicamente indicado, durante o tratamento. A redução na FEVE pode ser controlada por meio de interrupção do tratamento, redução da dose ou descontinuação do tratamento (consulte seção 8. POSOLOGIA E MODO DE USAR).
Toxicidade ocular
Aconselhar pacientes a relatarem qualquer novo distúrbio visual. Os eventos adversos de visão turva foram reportados em pacientes pediátricos recebendo KOSELUGO. Casos isolados de descolamento do epitélio pigmentar da retina (DEPR), retinopatia serosa central (RSC) e oclusão da veia da retina (OVR) foram observados em pacientes adultos com múltiplos tipos de tumores, recebendo tratamento com KOSELUGO em monoterapia e em combinação com outros agentes oncológicos e em um único paciente pediátrico com astrocitoma pilocítico recebendo tratamento com KOSELUGO em monoterapia (consulte seção 9. REAÇÕES ADVERSAS).
Em linha com a prática clínica, recomenda-se uma avaliação oftalmológica antes do início do tratamento e a qualquer momento que um paciente relatar um novo distúrbio visual. Em pacientes diagnosticados com DEPR ou RSC sem acuidade visual reduzida, avaliação oftalmológica deve ser realizada a cada 3 semanas até a resolução. Se DEPR ou RSC for diagnosticado e a acuidade visual for afetada, a terapia com KOSELUGO deve ser interrompida e a dose reduzida quando o tratamento for retomado (consulte Tabela 4). Se OVR for diagnosticado, o tratamento com KOSELUGO deve ser permanentemente descontinuado (consulte seção 8. POSOLOGIA E MODO DE USAR).
Suplementação com Vitamina E
Aconselhar pacientes a não tomarem qualquer suplementação com vitamina E.
As cápsulas de 10 mg de KOSELUGO contêm 32 mg de vitamina E como excipiente, tocofersolana. As cápsulas de 25 mg de KOSELUGO contêm 36 mg de vitamina E como tocofersolana. Doses altas de vitamina
E podem aumentar o risco de sangramento em pacientes recebendo concomitantemente medicamentos anticoagulantes ou antiplaquetários (por exemplo, varfarina ou aspirina). As avaliações anticoagulantes, incluindo relação internacional normalizada ou tempo de protrombina, devem ser realizadas mais frequentemente para detectar quando ajustes de dose do medicamento anticoagulante ou antiplaquetário são necessários (consulte seção 6. INTERAÇÃO MEDICAMENTOSA).
Efeitos sobre a capacidade de dirigir veículos e operar máquinas:
Nenhum estudo sobre o efeito na capacidade de dirigir e operar máquinas foi realizado. KOSELUGO pode ter um efeito menor na capacidade de dirigir e operar máquinas. Fadiga, astenia e distúrbios visuais foram reportados durante o tratamento com selumetinibe e pacientes que apresentam estes sintomas devem ter cautela ao dirigir ou operar máquinas.
Uso durante a gravidez e lactação
Contracepção em homens e mulheres
Mulheres férteis devem ser aconselhadas a evitar a gravidez enquanto estiverem recebendo KOSELUGO. Pacientes homens e mulheres (com potencial reprodutivo) devem ser aconselhados a usar contracepção efetiva durante e por pelo menos 1 semana após a conclusão do tratamento com KOSELUGO.
KOSELUGO não é recomendado em mulheres férteis que não estão usando métodos contraceptivos.
Gravidez
Não há dados sobre o uso de selumetinibe em mulheres grávidas. Estudos em animais demonstraram toxicidade reprodutiva incluindo morte embriofetal, defeitos estruturais e pesos fetais reduzidos (consulte seção 3. CARACTERÍSTICAS FARMACOLÓGICAS). KOSELUGO não é recomendado durante a gravidez.
Recomenda-se que um teste de gravidez seja realizado em mulheres férteis antes do início do tratamento.
Mulheres férteis devem ser orientadas a evitar a gravidez enquanto estiverem recebendo selumetinibe. Se uma paciente ou a parceira de um paciente que estiver recebendo KOSELUGO engravidar, ela deve ser informada sobre os riscos potenciais para o feto.
Categoria de risco na gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
Selumetinibe e seu metabólito ativo são excretados no leite de camundongos amamentando (consulte seção 3. CARACTERÍSTICAS FARMACOLÓGICAS). Não se sabe se selumetinibe, ou seus metabólitos ativos, são excretados no leite humano. Um risco para o lactente não pode ser descartado, portanto as mães amamentando devem ser aconselhadas a não amamentarem durante o tratamento com KOSELUGO.
Fertilidade
Não há dados sobre o efeito de KOSELUGO na fertilidade humana.
Selumetinibe não tem impacto na fertilidade e desempenho de acasalamento em camundongos machos e fêmeas, apesar de uma redução na sobrevivência embriônica ter sido observada em camundongos fêmeas (consulte seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
6. INTERAÇÕES MEDICAMENTOSAS
Interações farmacocinéticas
Os estudos de interação foram realizados apenas em adultos sadios (idade ≥18 anos).
Princípios ativos que podem aumentar as concentrações plasmáticas de selumetinibe
A administração concomitante com um inibidor forte de CYP3A4 (200 mg de itraconazol duas vezes ao dia por 4 dias) aumentou a Cmax de selumetinibe em 19% (IC 90% 4, 35) e AUC em 49% (IC 90% 40, 59) em voluntários adultos sadios.
A administração concomitante com um inibidor forte de CYP2C19/moderado de CYP3A4 (200 mg de fluconazol uma vez ao dia por 4 dias) aumentou a Cmax de selumetinibe em 26% (IC 90% 10, 43) e AUC em 53% (IC 90% 44, 63) em voluntários adultos sadios, respectivamente.
Evite a administração concomitante de KOSELUGOTM com medicamentos que sejam inibidores fortes de CYP3A4 (por exemplo, claritromicina, suco de toranja (grapefruit), cetoconazol oral) e CYP2C19 (por exemplo, ticlopidina). Se a administração concomitante for inevitável, pacientes devem ser monitorados cuidadosamente com relação a eventos adversos (consulte seção 8. POSOLOGIA E MODO DE USAR).
Nenhum ajuste de dose é necessário para uso de KOSELUGOTM com inibidores moderados de CYP3A4 ou CYP2C19. Pacientes devem ser cuidadosamente monitorados para eventos adversos quando administrados concomitantemente com inibidores moderados de CYP3A4 ou CYP2C19.
Princípios ativos que podem reduzir as concentrações plasmáticas de selumetinibe
A administração concomitante com um indutor forte de CYP3A4 (600 mg de rifampicina diariamente por 8 dias) reduziu a Cmax de selumetinibe em -26% (IC 90% -17, -34) e AUC em -51% (IC 90% -47, -54).
Evite a administração concomitante de KOSELUGO com indutores fortes de CYP3A4 (por exemplo, fenitoína, rifampicina, carbamazepina, erva de São João) ou indutores moderados de CYP3A4.
Princípios ativos cujas concentrações plasmáticas podem ser alteradas pelo selumetinibe
In vitro, selumetinibe é um inibidor de OAT3, portanto um potencial efeito clinicamente relevante na farmacocinética de substratos de OAT3 administrados concomitantemente não pode ser excluído (consulte seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Vitamina E
As cápsulas de KOSELUGO contêm vitamina E como o excipiente tocofersolana. Portanto, pacientes devem evitar tomar suplementos de vitamina E e avaliações anticoagulantes devem ser realizadas mais frequentemente em pacientes recebendo concomitantemente medicamentos anticoagulantes ou antiplaquetários (consulte seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
KOSELUGO deve ser armazenado à temperatura ambiente (15° a 30°C). Armazene no frasco original para proteger da umidade. Mantenha o frasco bem fechado. Não remova o dessecante.
KOSELUGO tem validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
KOSELUGO é apresentado em frasco plástico HDPE com tampa resistente a crianças e sílica gel dessecante.
Após aberto, válido por 4 semanas.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
A terapia deve ser iniciada por um médico experiente no diagnóstico e tratamento de pacientes com tumores relacionados a NF1.
Posologia
A dose recomendada de KOSELUGO é 25 mg/m2 da área de superfície corporal (ASC), administrada por via oral duas veze ao dia (aproximadamente a cada 12 horas).
A dose é individualizada com base na ASC (mg/m2) e arredondada para a dose mais próxima que se consiga alcançar de 5 mg ou 10 mg (até uma dose única máxima de 50 mg). As diferentes concentrações de KOSELUGO podem ser combinadas para se obter a dose desejada (Tabela 3). KOSELUGOTM não é recomendado para pacientes com ASC < 0,55 m2.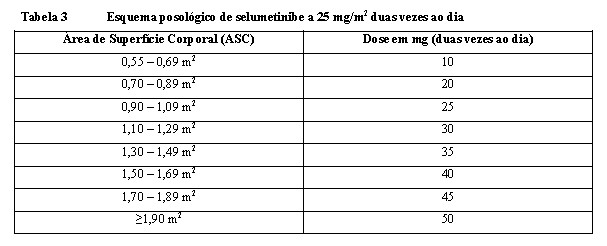
O tratamento com KOSELUGO deve continuar enquanto o benefício clínico for observado, ou até progressão do NP ou desenvolvimento de toxicidade inaceitável.
Modo de usar
KOSELUGO deve ser administrado com o estômago vazio sem alimento ou bebida, além de água.
Não consumir alimento 2 horas antes da administração e 1 hora após a administração (consulte seções 6. INTERAÇÕES MEDICAMENTOSAS e 3. CARACTERÍSTICAS FARMACOLÓGICAS).
As cápsulas de KOSELUGO devem ser engolidas por inteiro com água, e não devem ser mastigadas, dissolvidas ou abertas.
Dose esquecida
Se uma dose de selumetinibe for esquecida, ela deve ser administrada apenas se tiver mais de 6 horas até a próxima dose programada.
Vômitos
Não administrar uma dose adicional se ocorrer vômito após a administração de KOSELUGO, mas continuar com a próxima dose programada.
Ajustes de dose
Para eventos adversos
A interrupção e/ou redução da dose ou descontinuação permanente de selumetinibe pode ser necessária com base na segurança e tolerabilidade individual (consulte 5. ADVERTÊNCIAS E PRECAUÇÕES e 9. REAÇÕES ADVERSAS).
As reduções recomendadas da dose são apresentadas na Tabela 4 e podem necessitar que as doses diárias sejam divididas em duas administrações de concentrações diferentes ou que o tratamento seja administrado uma vez ao dia.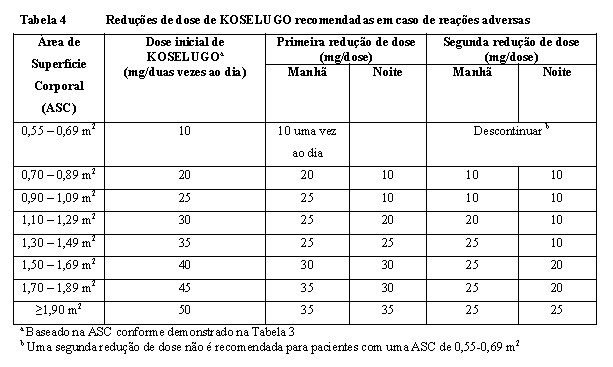

Recomendação de modificação de dose em caso de redução da fração de ejeção ventricular esquerda (FEVE)
Em casos de redução assintomática da FEVE de ≥ 10 pontos percentuais em relação ao basal e abaixo do limite inferior de normalidade (LIN) institucional, o tratamento com KOSELUGO deve ser interrompido até a resolução. Assim que resolvido, reduzir KOSELUGO em um nível de dose quando retomar a terapia (consulte Tabela 4).
Em pacientes que desenvolverem redução sintomática da FEVE ou uma redução da FEVE de grau 3 ou 4, KOSELUGO deve ser descontinuado e um encaminhamento imediato para o cardiologista deve ser realizado (consulte seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Recomendação de modificação de dose em caso de toxicidades oculares
O tratamento com KOSELUGO deve ser interrompido em pacientes diagnosticados com descolamento epitelial de pigmento da retina (DEPR) ou retinopatia serosa central (RSC) com acuidade visual reduzida, até a resolução; reduzir KOSELUGO em um nível de dose quando retomar a terapia (consulte Tabela 4). Em pacientes diagnosticados com DEPR ou RSC sem acuidade visual reduzida, avaliação oftalmológica deve ser realizada a cada 3 semanas até a resolução. Em pacientes diagnosticados com oclusão da veia da retina (OVR), o tratamento com KOSELUGO deve ser descontinuado permanentemente (consulte seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Populações especiais de pacientes
Insuficiência renal
Com base nos estudos clínicos, nenhum ajuste de dose é recomendado em pacientes com insuficiência renal leve, moderada, grave ou naqueles com doença renal em estágio terminal (DRET) (consulte seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência hepática
Com base nos estudos clínicos, nenhum ajuste de dose é recomendado em pacientes com insuficiência hepática leve. A dose inicial deve ser reduzida em pacientes com insuficiência hepática moderada para 20 mg/m2 ASC, duas vezes ao dia (vide tabela 6). KOSELUGO não é recomendado para uso em pacientes com insuficiência hepática grave (consulte seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Etnia
Exposição sistêmica aumentada foi observada em indivíduos adultos asiáticos, apesar de haver sobreposição considerável com indivíduos ocidentais quando corrigido para peso corporal. Nenhum ajuste específico da dose inicial é recomendado para pacientes pediátricos asiáticos, no entanto, estes pacientes devem ser monitorados de perto para eventos adversos (consulte seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
População pediátrica
A segurança e a eficácia de KOSELUGO em crianças menores de 2 anos de idade não foram estabelecidas. Não há dados atualmente disponíveis.
Este medicamento não deve ser partido, aberto ou mastigado.
Este medicamento não deve ser cortado.
9. REAÇÕES ADVERSAS
Resumo geral do perfil de segurança
A segurança de selumetinibe em monoterapia foi avaliada em uma população combinada de 74 pacientes pediátricos (20-30 mg/m2 duas vezes ao dia) com NF1 NP e 347 pacientes adultos (75-100 mg duas vezes ao dia) com múltiplos tipos de tumor.
A mediana de duração total do tratamento com selumetinibe em pacientes pediátricos com NF1 NP foi de 28 meses (intervalo: < 1 - 71 meses), 23% dos pacientes foram expostos ao tratamento com selumetinibe por > 48 meses.
No Estrato 1 do estudo de Fase II (SPRINT), 50 pacientes pediátricos com NF1 NP foram tratados com selumetinibe 25 mg/m2 duas vezes ao dia, consulte seção 3 CARACTERÍSTICAS FARMACOLÓGICAS. As reações adversas mais comuns de qualquer grau (incidência ≥45%) foram vômitos, erupção cutânea, creatina fosfoquinase aumentada no sangue, diarreia, náusea, pele seca, eventos astênicos, pirexia, erupção cutânea acneiforme, hipoalbuminemia, estomatite, aspartato aminotransferase aumentado e paroníquia. As interrupções e reduções de dose devido a eventos adversos foram reportadas em 80% e 24% dos pacientes, respectivamente. As reações adversas medicamentosas (RAMs) mais comumente reportadas que levaram à modificação de dose de selumetinibe foram vômitos (14 [24,0%]), paroníquia (7 [14,0%]), diarreia (6 [12,0%]) e náusea (5 [10,0%]). A descontinuação permanente devido a eventos adversos foi reportada em 12% dos pacientes.
Com base na análise da altura e do crescimento dos pacientes no estudo SPRINT, nenhum efeito do tratamento com selumetinibe no crescimento estaturo-ponderal foi observado. Não há dados do efeito do medicamento sobre o desenvolvimento neuro-psico-motor e maturação sexual dos pacientes.
Tabela das reações adversas
A Tabela 6 apresenta as reações adversas identificadas no Estrato 1 do estudo de Fase II SPRINT. As RAMs estão organizadas pela Classe de Sistema de Órgãos (CSO) do MedDRA. Dentro de cada CSO, os termos preferidos estão organizados por ordem decrescente de frequência e então por ordem decrescente de gravidade. As frequências das reações adversas são definidas como: muito comum (≥1/10); comum (≥1/100 a < 1/10); incomum (≥1/1000 a < 1/100); rara (≥1/10000 a < 1/1000); muito rara ( < 1/10000) e desconhecida (não pode ser estimada a partir dos dados disponíveis), incluindo relatos isolados.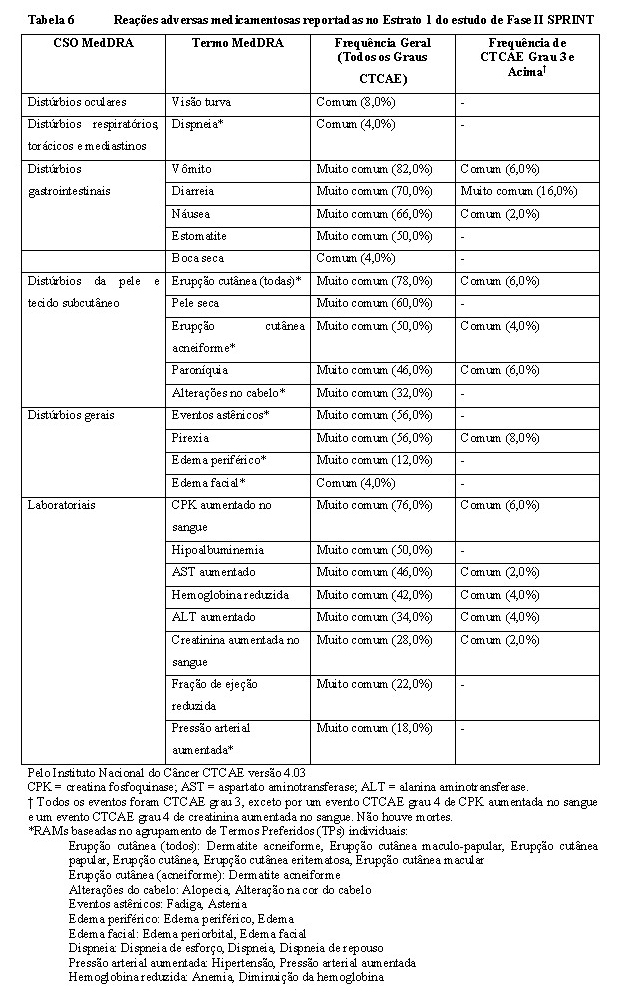
Reações Adversas Medicamentosas Identificadas em Outros Estudos Clínicos
A Tabela 7 apresenta as reações adversas medicamentosas identificadas em outros estudos clínicos em pacientes adultos (N=347), com múltiplos tipos de tumor, recebendo tratamento com selumetinibe (75 mg duas vezes ao dia):
Além disso, um único evento de DEPR foi reportado em um paciente pediátrico recebendo selumetinibe em monoterapia (25 mg/m2 duas vezes ao dia) para astrocitoma pilocítico envolvendo a via ótica em um estudo pediátrico com patrocínio externo, consulte seção 5. ADVERTÊNCIAS E PRECAUÇÕES e 8. POSOLOGIA E MODO DE USAR).
Descrição das reações adversas selecionadas
Redução da FEVE
No SPRINT, redução da FEVE (TP: fração de ejeção reduzida) foi reportada em 11 (22%) pacientes; todos os casos foram de grau 2, assintomáticos e não levaram à interrupção, redução ou descontinuação da dose. Dos 11 pacientes, 6 pacientes se recuperaram e 5 pacientes não tiveram o desfecho relatado. A mediana de tempo para a primeira ocorrência de redução de FEVE foi de 226 dias (mediana da duração de 78 dias). A maioria dos eventos adversos de redução de FEVE foi reportada como reduções do basal (redução ≥10%), mas foram consideradas como permanecendo no intervalo normal. Pacientes com FEVE menor que o LIN institucional basal não foram incluídos no estudo pivotal.
A redução na FEVE deve ser tratada por meio de interrupção do tratamento, redução da dose ou descontinuação do tratamento (consulte seções 5. ADVERTÊNCIAS E PRECAUÇÕES e 8. POSOLOGIA E MODO DE USAR).
Visão turva
No SPRINT, eventos de grau 1 e 2 de visão turva foram reportados em 4 (8%) pacientes. Dois pacientes necessitaram de interrupção de dose. Todos os eventos foram tratados sem redução de dose. Não foi observado envolvimento da retina nos exames oftalmológicos de pacientes pediátricos.
Se pacientes relatarem novos distúrbios visuais, recomenda-se uma avaliação oftalmológica completa. As toxicidades de retina podem ser controladas com interrupção do tratamento, redução da dose ou descontinuação do tratamento (consulte seções 5. ADVERTÊNCIAS E PRECAUÇÕES e 8. POSOLOGIA E MODO DE USAR).
Paroníquia
No SPRINT, paroníquia foi reportada em 23 (46%) pacientes, a mediana de tempo para o início do primeiro evento adverso de paroníquia de grau 4 foi 306 dias e a mediana da duração dos eventos foi de 96 dias. A maioria destes eventos foi de grau 1 ou 2 e foi tratada com terapia suporte ou sintomática e modificação de dose. Os eventos de grau ≥3 ocorreram em três (6%) pacientes. Sete pacientes tiveram interrupção da dose de KOSELUGO pelos eventos de paroníquia, dos quais 3 tiveram interrupção da dose seguida de redução da dose (2 pacientes necessitaram de uma segunda redução de dose). Em um paciente (2%) o evento levou à descontinuação.
Aumento da creatina fosfoquinase (CPK) no sangue
Os eventos adversos de elevação de CPK no sangue ocorreram em 76% dos pacientes no SPRINT. A mediana de tempo para o início do primeiro aumento de CPK de grau máximo foi de 106 dias e a mediana da duração dos eventos foi de 126 dias. A maioria dos eventos foi de grau 1 ou 2 e foi resolvido sem alteração na dose de KOSELUGO. Eventos grau ≥3 ocorreram em três (6%) pacientes. Um evento grau 4 levou à interrupção do tratamento seguida por redução de dose.
Toxicidades gastrointest