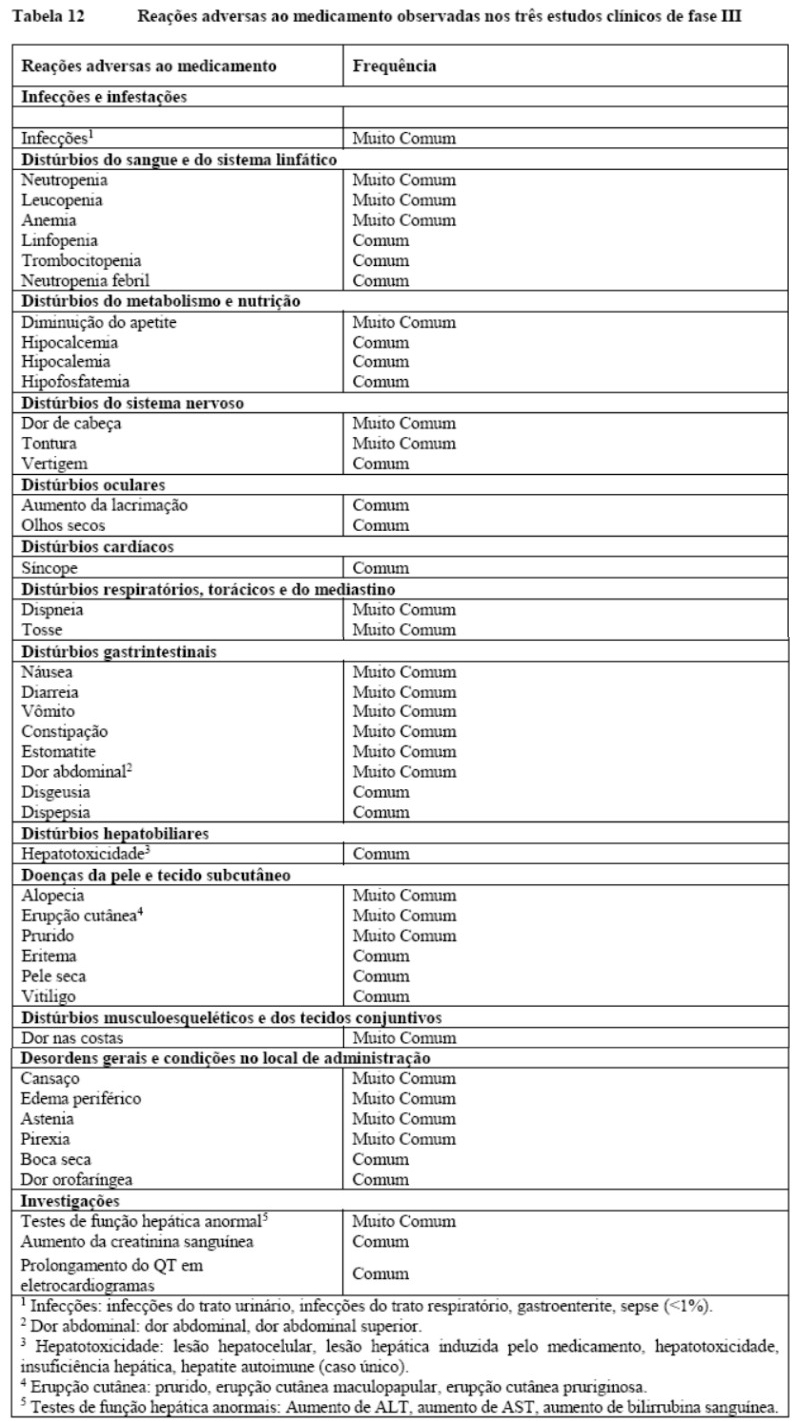
KISQALI
NOVARTIS
ribociclibe
Antineoplásico. Inibidor da proteína quinase.
Apresentações.
KisqaliTM 200 mg - embalagens contendo 21, 42 ou 63 comprimidos revestidos.
VIA ORAL
USO ADULTO
Composição.
Cada comprimido contém 254,40 mg de succinato de ribociclibe (equivalente a 200 mg de ribociclibe). Excipientes: Núcleo do comprimido: celulose microcristalina; hipromelose; povidona; dióxido de silício; estearato de magnésio. Composição do revestimento: álcool polivinílico; dióxido de titânio; óxido de ferro preto; óxido de ferro vermelho; talco: lecitina de soja; goma xantana.
Informações técnicas.
1. INDICAÇÕES
Kisqali (succinato de ribociclibe) é indicado para o tratamento de pacientes, com câncer de mama localmente avançado ou metastático, receptor hormonal (RH) positivo e receptor para o fator de crescimento epidérmico humano tipo 2 (HER2) negativo em combinação com um inibidor de aromatase ou fulvestranto.
Em mulheres na pré ou perimenopausa, a terapia endócrina deve ser combinada com um agonista do hormônio liberador do hormônio luteinizante (LHRH).
2. RESULTADOS DE EFICÁCIA
Segurança e eficácia clínica
Estudo CLEE011A2301 (MONALEESA-2)
Kisqali foi avaliado em um estudo clínico de fase III, randomizado, duplo-cego, controlado por placebo e multicêntrico, no tratamento de mulheres na pós-menopausa com câncer de mama avançado receptor hormonal (RH) positivoe HER2 negativo, que não haviam recebido tratamento anterior para a doença avançada, em combinação com letrozol versus letrozol isoladamente.
No total, 668 pacientes foram randomizados a uma razão de 1:1 para receber Kisqali 600 mg e letrozol (n = 334) ou placebo e letrozol (n = 334), estratificados de acordo com a presença de metástases hepáticas e/ou pulmonares (Sim [n = 292 (44%)]) vs. Não [n = 376 (56%)]). As características demográficas e basais foram equilibradas e comparáveis entre os braços do estudo. Kisqali foi administrado oralmente a uma dose de 600 mg por dia por 21 dias consecutivos, seguido de 7 dias sem tratamento, em combinação com letrozol 2,5 mg uma vez por dia por 28 dias. As pacientes não foram autorizadas a mudar de placebo para Kisqali durante o estudo ou após progressão da doença.
As pacientes recrutadas nesse estudo tinham, em mediana, 62 anos de idade (de 23 a 91). 44,2% das pacientes tinham mais de 65 anos, incluindo 69 pacientes com mais de 75 anos de idade. As pacientes incluídas eram caucasianas (82,2%), asiáticas (7,6%), e negras (2,5%). Todas as pacientes apresentaram um índice de desempenho na ECOG de 0 ou 1. No braço de Kisqali, 43,7% das pacientes haviam recebido quimioterapia em condição neoadjuvante ou adjuvante e 52,4% haviam recebido tratamento anti-hormonal em condição neoadjuvante ou adjuvante antes de entrar no estudo. 34,1% das pacientes eram pacientes "de novo", 20,7% das pacientes tinham apenas doença óssea e 59,0% das pacientes tinham doença visceral. Pacientes tratadas previamente com terapia (neo) adjuvante com anastrozol ou letrozol deveriam ter completado este tratamento pelo menos 12 meses antes de serem randomizadas para o estudo. O desfecho primário do estudo foi alcançado na análise interina planejada, realizada após observação de 80% dos eventos previstos de sobrevida livre de progressão (SLP) por meio do Critério de Avaliação da Resposta em Tumores Sólidos (RECIST v1.1), com base na avaliação da população total pelo investigador (todos os pacientes randomizados), e confirmado por uma avaliação radiológica central, independente e mascarada.
Os resultados de eficácia demonstraram um aumento estatisticamente significativo na sobrevida livre de progressão (SLP) em pacientes que receberam Kisqali mais letrozol em comparação com pacientes que receberam placebo mais letrozol no conjunto de análise completo (razão de risco de 0,556 IC de 95%: 0,429, 0,720, valor P de teste log-rank estratificado unilateral 0,00000329), com efeito de tratamento clinicamente significante.
Os dados de estado de saúde global / qualidade de vida não apresentaram diferenças relevantes entre o braço de Kisqali mais letrozol e o braço de placebo mais letrozol.
Uma atualização dos dados de eficácia (corte de 02 de janeiro de 2017) é fornecida nas Tabelas 1 e 2.
A SLP mediana foi de25,3 meses (IC de 95%: 23,0, 30,3) para pacientes que receberam ribociclibe mais letrozol e 16,0 meses (IC de 95%: 13,4, 18,2) para pacientes tratadas com placebo mais letrozol. 54,7% das pacientes que receberam ribociclibe mais letrozol estavam livres de progressão aos 24 meses em comparação com 35,9% no braço de placebo mais letrozol.
Não houve diferenças estatisticamente relevantes na sobrevida global (SG) entre o braço de Kisqali mais letrozol e o braço de placebo mais letrozol (RR 0,746 [IC de 95% 0,517, 1,078]). Os dados de SG permanecem imaturos.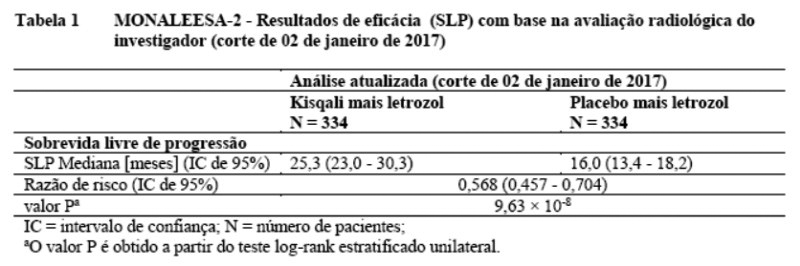
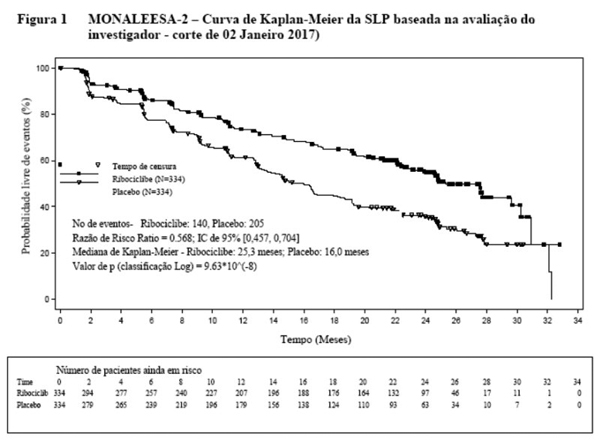
Uma série de análises de SLP de subgrupos pré-especificados foi realizada com base em fatores prognósticos e características basais para investigar a consistência interna do efeito do tratamento. Foi observada uma redução do risco de progressão da doença ou morte em favor do braço de Kisqali mais letrozol em todos os subgrupos individuais de pacientes em idade, etnia, quimioterapia adjuvante ou neoadjuvante prévia ou terapias hormonais, envolvimento hepático e/ou pulmonar e doença metastática exclusivamente óssea. Isto foi evidente em pacientes com metástase hepática e/ou pulmonar (RR de 0,561 [IC de 95%: 0,424, 0,743], com mediana de sobrevida livre de progressão [mSLP] de 24,8 meses para Kisqali mais letrozol versus 13,4 meses para letrozol em monoterapia) ou sem metástases hepáticas/pulmonares (RR de 0,597 [IC de 95%: 0,426, 0,837], com mSLP de 27,6 meses versus 18,2 meses).
Os resultados atualizados para a resposta global e as taxas de benefício clínico são apresentados na Tabela 2.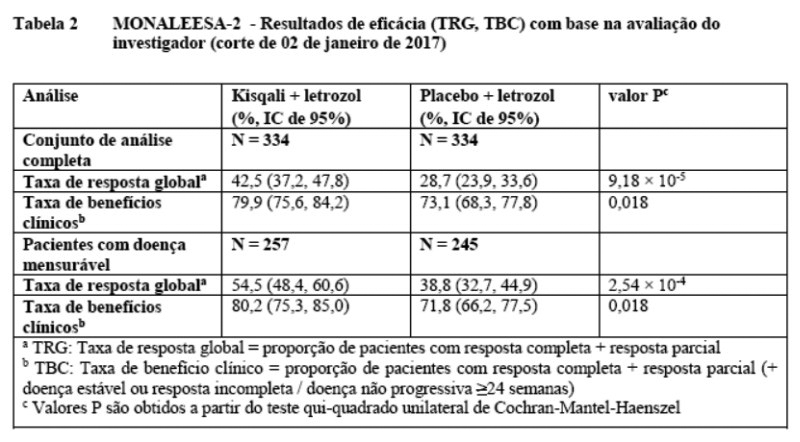
Estudo CLEE011E2301 (MONALEESA-7)
Kisqali foi avaliado em um estudo clínico fase III, ranzomizado, duplo-cego, controlado por placebo, multicêntrico no tratamento de mulheres na pré e perimenopausa com câncer de mama avançado, receptor hormonal positivo, HER2 negativo, em combinação com um IANE ou tamoxifeno mais gosserrelina versus placebo em combinação com um IANE ou tamoxifeno mais gosserrelina.
Um total de 672 pacientes foram randomizados em uma taxa de 1:1 para receber Kisqali 600 mg associado a IANE/tamoxifeno mais gosserrelina (n=335) ou placebo associado a IANE/tamoxifeno mais gosserrelina (n=337), estratificado de acordo com: a presença de metástase de fígado e/ou pulmão (Sim [n=344 (51,2%)] versus Não [n=328 (48,8%)]), antes de quimioterapia para doença avançada (Sim [n=120 (17,9%)] versus Não [n=552 (82,1%)]), e parceiro de combinação endócrina (IANE e gosserrelina [n=493 (73,4%)] versus tamoxifeno and gosserrelina [n=179 (26,6%)]). As características demográficas e basais foram balanceadas e comparadas entre os braços do estudo. Kisqali foi administrado oralmente em uma dose de 600 mg diariamente por 21 dias consecutivos seguidos por 7 dias sem tratamento em combinação com IANE (letrozol 2,5 mg ou anastrozol 1 mg) ou tamaxifeno (20 mg) oralmente uma vez ao dia por 28 dias, e gosserrelina (3,6 mg) subcutaneamente a cada 28 dias, até progressão da doença ou toxicidade inaceitável. A troca de pacientes do placebo para Kiqali não foi permitida durante o estudo ou após progressão da doença. A troca da combinação endócrina também não foi permitida.
Pacientes incluídos nesse estudo tinham idade mediana de 44 anos (intervalo de 25 a 58) e 27,7% de pacientes eram mais jovens do que 40 anos de idade. A maioria dos pacientes incluídos eram caucasianos (57,7%), asiáticos (29,5%) ou negros (2,8%) e quase todos os pacientes (99,0%) tinham um status de desempenho ECOG basal de 0 ou 1. Antes de entrar no estudo, dos 672 pacientes, 14% do pacientes receberam quimioterapia prévia para doença metastática, 32,6% dos pacientes receberam quimioterapia no contexto adjuvante e 18,0% no contexto neoadjuvante; 39,6% receberam terapia endócrina no contexto adjuvante e 0,7% no contexto neoadjuvante. No estudo E2301, 40,2% do pacientes tinham doença metastática de novo, 23,7% tinham doença óssea somente e 56,7% tinham doença visceral.
O estudo atingiu o desfecho primário na análise primária conduzida após 318 eventos de sobrevida livre de progressão (SLP) baseados na avaliação do investigador usando critários RECIST v 1.1 no grupo de análise completa (todos os pacientes randomizados). Os resultados de eficácia primária foram suportados pelos resultados de SLP com base na avaliação radiológica central independente cega. O tempo mediano de acompanhamento no momento da análise de SLP primária foi 19,2 meses.
Na população total do estudo, os resultados de eficácia demonstraram uma melhora estatisticamente significante na SLP em pacientes recebendo Kisqali mais IANE / tamoxifeno mais gosserrelina comparado a pacientes recebendo placebo mais IANE / tamoxifeno mais gosserrelina (razão de risco de 0,553, IC de 95%: 0,441, 0,694, teste de classificação de log estratificado unilateral, valor de p 9.83x10-8) com efeito de tratamento clinicamente significativo. A SLP mediana foi 23,8 meses (IC de 95%: 19,2, NE) para pacientes tratados com Kisqali mais IANE/tamoxifeno mais gosserrelina e 13,0 meses (IC de 95%: 11,0, 16,4) para pacientes recebendo placebo mais IANE/tamoxifeno mais gosserrelina.
A distribuição de SLP está resumida na curva de Kaplan Meier para SLP na Figura 2.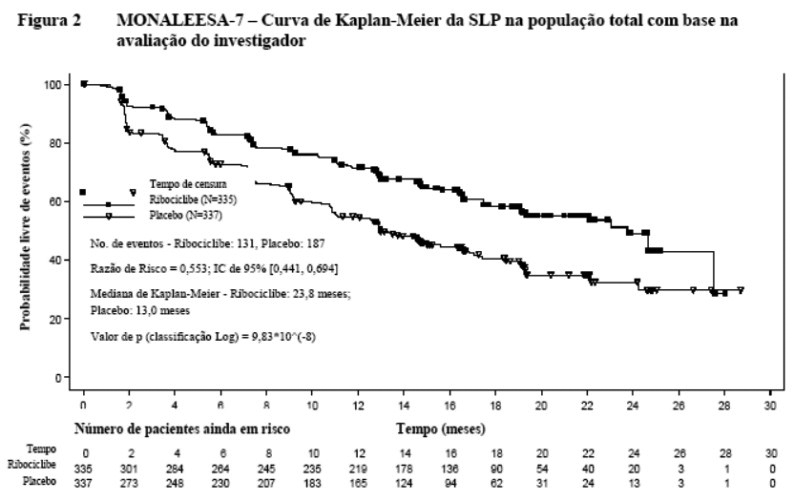
Os resultados para SLP baseados na avaliação radiológica central independente cega de um subgrupo selecionado randomicamente de aproximadamente 40% dos pacientes randomizados suportaram os resultados de eficácia primária com base na avaliação do investigador (razão de risco de 0,427; IC de 95%: 0,288, 0,633).
No momento da análise primária de SLP, os dados de sobrevida global não estavam maduros com 89 (35%) eventos (N=252, RR 0,916 [IC de 95%: 0,601, 1,396]).
A taxa de resposta global (TRG) pela avaliação do investigador baseada no RECIST v 1.1 foi maior no braço de Kisqali (40,9%; IC de 95%: 35,6, -46,2) comparado ao braço do placebo (29,7%; IC de 95%: 24,8 34,6, p=0,00098). A taxa de benefício clínico observada (TBC) foi maior no braço de Kisqali (79,1%; IC de 95%: 74,8:83,5) comparado ao braço do placebo (69,7%; IC de 95%: 64,8:74,6, p=0,002).
A principal medida de qualidade de vida (QoL) pré-especificada foi tempo para deterioração (TTD) no estado de saúde global. Deterioração definitiva de 10% foi definida como uma piora no escore (escore da escala de saúde global EORTC QLQ C30) por pelo menos 10% comparada ao basal, sem melhora posterior acima deste limite observado durante o perdíodo de tratamento, ou morte devido a qualquer causa.
A adição de Kisqali a IANE/tamoxifeno resultou em TTD tardio no escore da escala de saúde global EORTC QLQ C30 comparado com placebo mais tamoxifeno ou IANE (mediana não estimável versus 21,2 meses; RR de 0,699 [IC de 95%: 0,533,916]; p=0,004).
Na análise de subgrupo pré-espeficada de 495 pacientes que receberam Kisqali ou placebo em combinção com IANE mais gosserrelina, a SLP mediana foi 27,5 meses (IC de 95%: 19,1, NE) no subgrupo de Kisqali mais IANE e 13,8 meses (IC de 95%: 12,6, 17,4) no subgrupo de placebo mais IANE [RR: 0,569; IC de 95%: 0,436, 0,743]. Os resultados de eficácia estão resumidos na Tabela 3 e as curvas de Kaplan Meier para SLP são fornecidas na Figura 3.
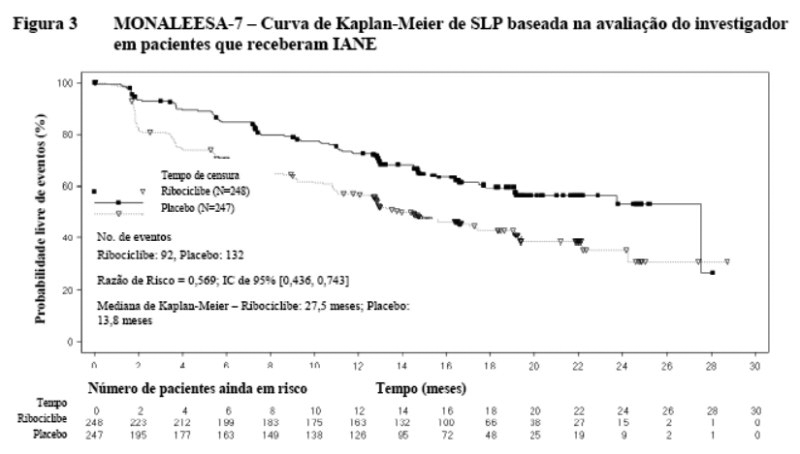
Os resultados de eficácia para a taxa de resposta global (TRG) e taxa de benefício clínico (TBC) na avaliação do investigador baseada no RECIST v1.1 estão fornecidos na Tabela 4.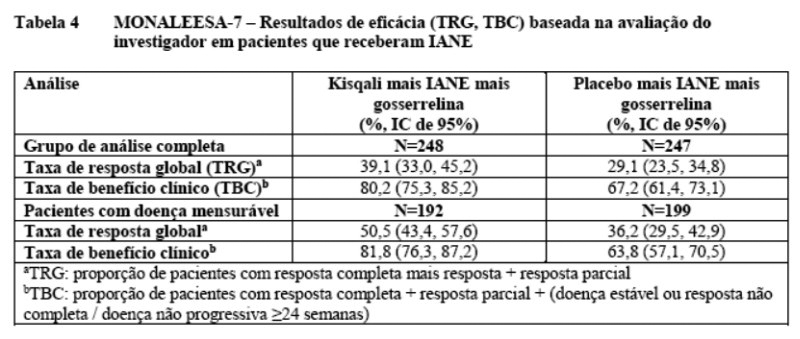
Os resultados no subgrupo de Kisqali mais IANE foram consistentes entre os subgrupos de idade, raça, qumioterapia adjuvante/neoadjuvante prévio ou terapias hormonais, envolvimento do fígado e/ou pulmão e doença óssea mestastática somente.
No subgrupo de IANE, o tempo mediano para resposta não foi atingido tanto no braço de Kisqali quanto no braço do placebo, e a probabilidade de resposta por 6 meses foi 34,7% (IC de 95%: 29,0, 41,1) no braço de Kisqali e 23,7% (IC de 95%: 18,8, 29,6) no braço placebo, indicando que uma maior proporção de pacientes obteve benefícios anteriores no braço de Kisqali.
No subgrupo de IANE, a duração mediana de resposta não foi atingida (IC de 95%: 18,3 meses, NE) no braço de Kisqali e foi de 17,5 meses (IC de 95% 12,0, NE) no braço do placebo. Entre os pacientes com resposta completa confirmada ou resposta parcial, a probabilidade de progressão subsequente foi 23,5% (IC de 95%: 15,6, 34,5) no braço de Kisqali e 36,4% (IC de 95%: 25,6, 49,8) no braço placebo aos 12 meses.
Estudo CLEE011F2301 (MONALEESA 3)
Kisqali foi avaliado em um estudo clínico fase III, ranzomizado, duplo-cego, controlado por placebo e multicêntrico no tratamento de homens e mulheres na pós-menopausa com câncer de mama avançado, receptor de hormônio positivo, HER2-negativo, que receberam nenhuma ou apenas uma linha de tratamento endócrino anterior, em combinação com fulvestranto versus fulvestranto sozinho.
Um total de 726 pacientes foram randomizados em uma taxa de 2:1 para receber tanto Kisqali 600 mg e fulvestranto (n= 484) ou placebo e fulvestranto (n= 242), estratificado de acordo com a presença de metástases de fígado e/ou pulmão (Sim [n= 351 (48,3%)] versus Não [n=375 (51,7%)]) e de acordo com a terapia endócrina prévia (A [n=354 (48,8%)] versus B [n=372 (51,2%)]). As características demográficas e basais da doença foram balanceadas e comparadas entre os braços do estudo. Kisqali 600 mg ou placebo foram administrados oralmente diariamente por 21 dias consecutivos seguidos de 7 dias sem tratamento em combinação com fulvestranto 500 mg intramuscular uma vez ao dia nos Dias 1 e 15 no Ciclo 1 e no Dia 1 de cada ciclo de 28 dias subsequente. Não foi permitido que os pacientes fizessem crossover do grupo placebo para o de Kisqali durante o estudo ou após progressão da doença.
Pacientes incluídos neste estudo tinham idade mediana de 63 anos (intervalo de 31 a 89). 46,7% dos pacientes tinham 65 anos de idade ou mais, incluindo 13,8% de pacientes com 75 anos ou mais. Os pacientes incluídos eram caucasianos (85,3%), asiáticos (8,7%) ou negros (0,7%) e quase todos os pacientes (99,7%) tinham um status de desempenho ECOG basal de 0 ou 1. Os pacientes de primeira e segunda linha foram incluídos nesse estudo (dos quais 19,1% tinham doença metastática de novo). Antes de entrar no estudo, receberam quimioterapia no contexto adjuvante e 13,1% no contexto neoadjuvante, enquanto 58,5% receberam terapia endócrina no contexto adjuvante e 1,4% no contexto neoadjuvante. No estudo F2301, 21,2% tinham somente doença óssea e 60,5% tinham doença visceral.
O estudo atingiu o desfecho primário na análise primária conduzida após 361 eventos de sobrevida livre de progressão (SLP) baseados na avaliação do investigador e usando critérios RECIST v 1.1 no grupo de análise completa (todos os pacientes randomizados). Os resultados de eficácia primária foram suportados pelos resultados de SLP com base na avaliação radiológica central independente cega. O tempo mediano de acompanhamento no momento da análise de SLP primária foi 20,4 meses.
Os resultados de eficácia primária demonstraram uma melhora estatisticamente significante em SLP nos pacientes recebendo Kisqali mais fulvestranto comparado a pacientes recebendo placebo mais fulvestranto em um grupo de análise completa (razão de risco de 0,593, IC de 95%: 0,480, 0,732, teste de classificação de log estratificado unilateral, valor de p 4.1x10-7), com uma redução estimada de 41% no risco relativo de progressão ou morte a favor do braço de Kisqali mais fulvestranto.
A SLP mediana foi 20,5 meses (IC de 95%: 18,5, 23,5) para pacientes tratados com Kisqali mais fulvestranto e 12,8 meses (IC de 95%: 10,9, 16,3) para pacientes recebendo placebo mais fulvestranto. 39,6% dos pacientes no braço de Kisqali foram estimados de estar livre de progressão aos 24 meses comparados com 17,6% no braço placebo.
A distribuição de SLP está resumida na Tabela 5 e na curva de Kaplan Meier para SLP na Figura 4.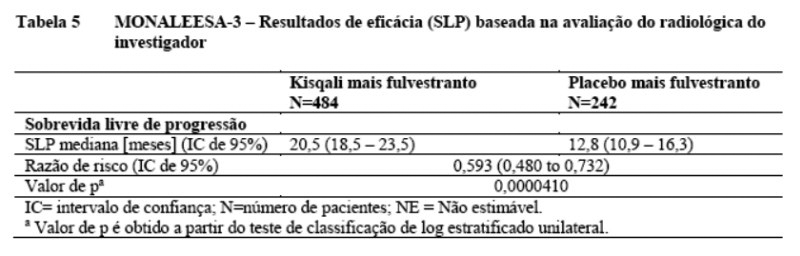
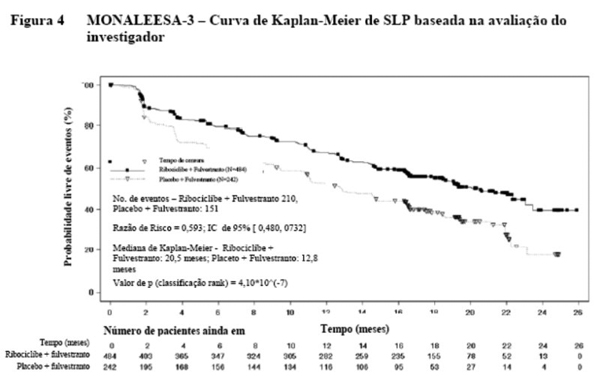
Os resultados para SLP baseados na avaliação radiológica central independente cega de um subgrupo randomicamente selecionado de aproximadamente 40% dos pacientes randomizados suportaram os resultados de eficácia primária com base na avaliação do investigador (razão de risco de 0,492; IC de 95%: 0,345, 0,703).
No momento da análise primária de SLP, os dados de sobrevida global não estavam maduros com 120 (34%) eventos (N=351, RR 0,670; [IC de 95%: 0,465, 0,964)].
A mediana de tempo para resposta não foi atingido tanto para o braço de Kisqali mais fulvestranto quanto para o braço de placebo mais fulvestranto, e a probabilidade de ter uma resposta por 6 meses foi 26,6% (IC de 95%: 22,7, 31,0) no braço de Kisqali mais fulvestranto e 16,2% (IC de 95%: 12,0, 21,6) no braço de placebo mais fulvestranto, indicando que uma maior proporção de pacientes obteve benefícios anteriores no braço de Kisqali mais fulvestranto.
A duração mediana de resposta não foi atingida (IC de 95%: 16,1, NE) tanto no braço de Kisqali mais fulvestranto quanto no braço de placebo mais fulvestranto (IC de 95%: 13,8, NE). Entre os pacientes com resposta completa confirmada ou resposta parcial, a probabilidade de progressão subsequente foi 21,7% (IC de 95%: 15,5, 30,0) no braço de Kisqali mais fulvestranto e 25,2% (IC de 95%: 15,1, 40,2) no braço de placebo mais fulvestranto aos 12 meses.
A principal medida de qualidade de vida (QoL) pré-especificada foi tempo para deterioração (TTD) no estudo global de saúde. Deterioração definitiva de 10% foi definida como uma piora no escore (escore da escala de saúde global EORTC QLQ C30) por pelo menos 10% comparada ao início, sem melhora posterior acima deste limite observado durante o perdíodo de tratamento, ou morte devido a qualquer causa.
A adição de Kisqali a fulvestranto resultou em TTD tardio no escore da escala de saúde global EORTC QLQ C30 comparado com placebo mais fulvestranto (mediana não estimável versus 19,4 meses; RR de 0,795 [IC de 95%: 0,602, 1,050); valor de p 0,051).
Os resultados de eficácia para a taxa de resposta global (TRG) e taxa de benefício clínico (TBC) pela avaliação do investigador baseadas no RECIST v 1.1 são fornecidas na Tabela 6.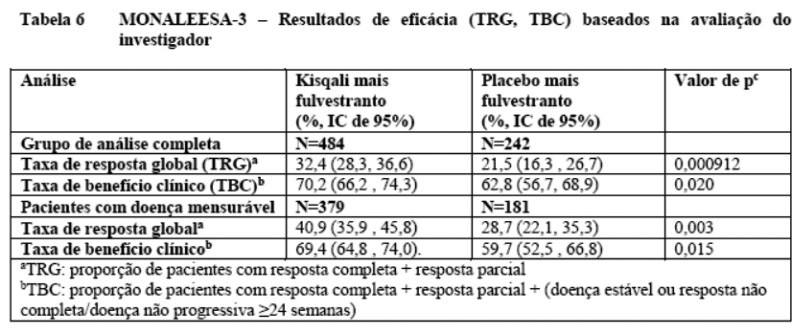
No subgrupo de pacientes sem tratamento prévio no contexto da doença avançada/metastática, a razão de risco foi de 0,577 (IC de 95%: 0,415, 0,802), com uma SLP mediana não alcançada no braço de Kisqali e 18,3 meses (IC de 95%: 14,8, 23,1) no braço do placebo.
No subgrupo de pacientes que receberam até uma linha de tratamento para doença avançada/metastática, a razão de risco foi de 0,565 (IC de 95%: 0,428, 0,744), com uma SLP mediana de 14,6 meses (IC de 95%: 12,5, 18,5) e 9,1 meses (IC de 95%: 6,1, 11,1) nos braços de Kisqali e placebo, respectivamente.
As razões de risco baseadas na análise de subgrupo pré-especificada de pacientes tratados com Kisqali mais fulvestranto mostraram benefícios consistentes entre os diferentes subgrupos incluindo idade, tratamento prévio (precoce ou avançado), quimioterapia adjuvante/neoadjuvante prévia ou terapias hormonais, envolvimento do fígado e/ou pulmão e doença metastática óssea somente.
Referências Bibliográficas
Hortobagyi GN et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med. 2016 Nov 3;375(18):1738-1748.
Resultados de eficácia do MONALEESA-2 (corte de 02 de janeiro de 2017) -Data on file
Updated results from MONALEESA-2, a phase 3 trial of first-line ribociclib + letrozole in hormone receptorpositive (HR+), HER2-negative (HER2-), advanced breast cancer (ABC). J Clin Oncol 35, 2017 (suppl; abstr 1038)
Tripathy D et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): a randomised phase 3 trial. Lancet Oncol. 2018 Jul;19(7):904-915.
Slamon DJ et al. Phase III Randomized Study of Ribocidib and Fulvestrant in Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: MONALEESA-3. J Clin Oncol. 2018 Aug 20;36(24):2465-2472.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: inibidores da proteína quinase. Código ATC: L01XE42
Farmacologia de segurança
Estudos de segurança cardíaca in vivo em cães demonstraram prolongamento do intervalo QTc relacionado com a dose e a concentração a uma exposição que seria esperada ser alcançada em pacientes que seguem a dose recomendada de 600 mg. Existe também potencial para induzir incidências de contrações ventriculares prematuras (CVP) em exposições elevadas (aproximadamente 5 vezes a Cmáx clínica antecipada).
Toxicidade de dose repetida
Estudos de toxicidade de dose repetida (esquema de 3 semanas em tratamento / 1 semana sem tratamento) de até 27 semanas de duração em ratos e até 39 semanas de duração em cães revelaram o sistema hepatobiliar (alterações proliferativas, colestase, cálculos da vesícula biliar semelhantes a areia e bile espessada) como o principal órgão-alvo primário da toxicidade do ribociclibe. Os órgãos alvo associados à ação farmacológica do ribociclibe em estudos de dose repetida incluem medula óssea (hipocelularidade), sistema linfoide (depleção linfoide), mucosa intestinal (atrofia), pele (atrofia), osso (diminuição da formação óssea), rim (degeneração e regeneração concomitante das células epiteliais tubulares) e testículos (atrofia). Além das alterações atróficas observadas nos testículos, que mostraram uma tendência à reversibilidade, todas as outras alterações foram totalmente reversíveis após um período de 4 semanas sem tratamento. A exposição ao ribociclibe em animais nos estudos de toxicidade foi geralmente menor ou igual à observada em pacientes que receberam doses múltiplas de 600 mg/dia (com base na ASC).
Propriedades farmacodinâmicas
Mecanismo de ação
O ribociclibe é um inibidor seletivo da quinase dependente de ciclina (CDK) 4 e 6, resultando em valores de inibição de 50% (IC50) de 0,01 (4,3ng/ml)e 0,039 mM (16,9ng/ml)emensaiosbioquímicos,respectivamente. Estas quinases são ativadas por ligação às D-ciclinas e desempenham um papel crucial nas vias de sinalização que conduzem à progressão do ciclo celular e à proliferação celular. O complexo ciclina D-CDK4/6 regula a progressão do ciclo celular através da fosforilação da proteína retinoblastoma (pRb).
In vitro, o ribociclibe diminuiu a fosforilação de pRb, levando à detenção na fase G1 do ciclo celular, e reduziu a proliferação celular em linhas celulares de câncer de mama. In vivo, o tratamento com o fármaco ribociclibe isoladamente levou a regressões tumorais que se correlacionaram com a inibição da fosforilação de pRb.
Estudos in vivo utilizando combinações do modelo de xenoenxerto de câncer de mama receptor positivo para estrogênio derivado do paciente de ribociclibe e antiestrogênios (ou seja, letrozol) resultaram em uma inibição de crescimento tumoral superior com regressão tumoral sustentada e novo crescimento do tumor depois de interromper a administração em comparação com cada substância isolada. Adicionalmente, atividade antitumoral in vivo de ribociclibe em combinação com fulvestranto foi avaliada em camundongos imunodeficientes portadores dos xenoenxertos de câncer de mama humano ZR751 ER e a combinação com fulvestranto resultou em inibição completa de crescimento tumoral.
Quando testado em um painel de linhas celulares de câncer de mama com estado de RE conhecido, o ribociclibe demonstrou ser mais eficaz em linhas celulares de câncer de mama RE+ do que nos RE-.
Eletrofisiologia cardíaca
Foram coletados ECG em série, triplicados, após uma única dose e em estado estacionário, para avaliar o efeito do ribociclibe no intervalo QTc em pacientes com câncer avançado. Uma análise farmacocinéticafarmacodinâmica incluiu um total de 997 pacientes tratados com ribociclibe em doses variando de 50 a 1200 mg. A análise sugeriu que o ribociclibe provoca aumentos dependentes da concentração no intervalo QTc. A variação mediana estimada do QTcF em relação à linha basal para Kisqali 600 mg em combinação com IANE ou fulvestranto foi de 22,0 msec (IC de 90%: 20,56, 23,44) e 23,7 msec (IC de 90%: 22,31, 25,08), respectivamente. Na Cmáx mediana geométrica em estado estacionário comparado a 34,7 msec (IC de 90%: 31,64, 37,78) em combinação com tamoxifeno (ver seção 5. Precauções e Advertências).
Propriedades farmacocinéticas
A farmacocinética do ribociclibe foi investigada em pacientes com câncer avançado após doses diárias de 50 mg a 1200 mg por via oral. Os indivíduos saudáveis receberam doses orais únicas variando de 400 mg a 600 mg ou doses diárias repetidas (8 dias) de 400 mg.
- Absorção
A biodisponibilidade absoluta do ribociclibe é desconhecida.
O tempo para atingir a Cmáx (Tmáx) após a administração oral de ribociclibe foi entre 1 e 4 horas. O ribociclibe apresentou aumentos ligeiramente excessivos na exposição (Cmáx e ASC) em todo o intervalo de dose testado (50 a 1.200 mg). Após administração repetida uma vez por dia, o estado estacionário foi geralmente alcançado após 8 dias e o ribociclibe acumulado com uma mediana geométrica de razão de acumulação de 2,51 (intervalo: 0,97 a 6,40).
- Efeitos alimentares
Em comparação com o estado de jejum, a administração oral de uma dose única de 600 mg de ribociclibe em comprimidos revestidos com uma refeição rica em gordura e calorias não teve qualquer efeito sobre a taxa e a extensão da absorção de ribociclibe.
A ligação do ribociclibe às proteínas plasmáticas humanas in vitro foi de aproximadamente 70% e foi independente da concentração (10 a 10000 ng/ml). O ribociclibe foi igualmente distribuído entre glóbulos vermelhos e plasma com uma relação mediana sangue-plasma in vivo de 1,04. O volume aparente de distribuição no estado estacionário (Vss/F) foi de 1.090 L com base na análise farmacocinética da população.
- Biotransformação
Estudos in vitro e in vivo indicaram que o ribociclibe sofre metabolismo hepático extensivo principalmente via CYP3A4 em seres humanos. Após a administração oral de uma única dose de 600 mg de [14C] ribociclibe a seres humanos, as principais vias metabólicas para o ribociclibe envolveram oxidação (desalquilação, C e/ou N-oxigenação, oxidação (-2H)) e suas combinações. Os conjugados de fase II de metabolitos de ribociclibe de fase 1 envolveram N-acetilação, sulfatação, conjugação de cisteína, glicosilação e glucuronidação. O ribociclibe foi a principal entidade derivada do medicamento circulante no plasma (43,5%). Os principais metabolitos circulantes incluíram o metabolito M13 (CCI284, N-hidroxilação), M4 (LEQ803, N-desmetilação) e M1 (glucuronido secundário). A atividade clínica (farmacológica e de segurança) do ribociclibe deveu-se principalmente ao medicamento original, com uma contribuição insignificante dos metabolitos circulantes.
O ribociclibe foi extensivamente metabolizado com o medicamento inalterado responsável por 17,3% e 12,1% da dose nas fezes e urina, respectivamente. O metabolito LEQ803 foi um metabolito significativo nas excreções e representou aproximadamente 13,9% e 3,74% da dose administrada nas fezes e urina, respectivamente.
Muitos outros metabolitos foram detectados em fezes e urina em pequenas quantidades (≤ 2,78% da dose administrada).
- Eliminação
A mediana geométrica da meia-vida plasmática efetiva (baseada na taxa de acumulação) foi de 32,0 horas (63% CV) e a mediana geométrica de eliminação oral aparente (CL/F) foi de 25,5 l/hr (66% CV) em estado estacionário a 600 mg em pacientes com câncer avançado. A mediana geométrica da meia-vida plasmática aparente (T1/2) do ribociclibe variou de 29,7 a 54,7 horas e a mediana geométrica CL/F do ribociclibe variou de 39,9 a 77,5 l/h a 600 mg entre os estudos em indivíduos saudáveis.
Ribociclibe e seus metabólitos são eliminados, principalmente, através de fezes, com uma pequena contribuição da via renal. Em 6 indivíduos saudáveis do sexo masculino, após uma única dose oral de [14C] ribociclibe, 91,7% da dose radioativa total administrada foi recuperada dentro de 22 dias; a principal via de excreção foi através das fezes (69,1%), com 22,6% da dose recuperada na urina.
- Linearidade/não linearidade
O ribociclibe apresentou aumentos ligeiramente excessivos na exposição (Cmáx e ASC) em todo o intervalo de 50 a 1200 mg, após dose única e doses repetidas. Esta análise é limitada pelos pequenos tamanhos de amostra para a maioria das coortes de dose, com a maioria dos dados provenientes da coorte de dose de 600 mg.
Populações especiais
Insuficiência renal
Com base em uma análise farmacocinética populacional que incluiu 77 pacientes com função renal normal
(eGFR ≥ 90ml/min/1,73m), 76 pacientes com insuficiência renal leve (eGFR 60 a < 90 ml/min/1,73 m2) e 35 pacientes com insuficiência renal moderada (eGFR 30 a < 60 ml/min/ 1,73 m2), insuficiência renal leve e moderada não tiveram qualquer efeito na exposição de ribociclibe. A farmacocinética de ribociclibe em pacientes com insuficiência renal grave não foi estudada.
Insuficiência hepática
Com base em um estudo farmacocinético em pacientes com insuficiência hepática, a insuficiência hepática leve não teve qualquer efeito na exposição de ribociclibe. A mediana de exposição ao ribociclibe foi aumentada menos de 2 vezes em pacientes com insuficiência hepática moderada (razão de mediana geométrica [GMR]: 1,50 para Cmáx, 1,32 para ASCinf) e grave (GMR: 1,34 para Cmáx e 1,29 para ASCinf). Com base em uma análise farmacocinética populacional que incluiu 160 pacientes com câncer de mama com função hepática normal e 47 pacientes com insuficiência hepática leve, a insuficiência hepática leve não teve qualquer efeito na exposição de ribociclibe, apoiando ainda mais os resultados do estudo de insuficiência hepática dedicada (vide seção 8. Posologia e modo de usar).
Efeito de idade, peso, gênero e etnia
A análise farmacocinética da população mostrou que não há efeitos clinicamente relevantes da idade, peso corporal ou sexo na exposição sistêmica de ribociclibe que necessite de um ajuste de dose. Os dados sobre as diferenças na farmacocinética devido à etnia são muito limitados para tirar conclusões.
Uso geriátrico
Dos 334 pacientes que receberam Kisqali no primeiro estudo fase III (MONALEESA 2, no braço de ribociclibe mais letrozol), 150 pacientes (44,9%) tinham ≥65 anos de idade e 35 pacientes (10,5%) tinham ≥75 anos de idade. Dos 484 pacientes que receberam Kisqali em outro estudo fase III (MONALEESA 3, no braço de ribiciclibe mais fulvestranto), 226 pacientes (46,7%) tinham ≥65 anos de idade e 65 pacientes (13,4%) tinham ≥75 anos de idade. Não foram observadas diferenças globais na segurança ou eficácia de Kisqali entre esses pacientes e os pacientes mais jovens (vide seção 8. Posologia e modo de usar).
Dados sobre interações em vitro Efeito do ribociclibe sobre enzimas do citocromo P450
In vitro, ribociclibe é um inibidor reversível da CYP1A2, CYP2E1 e CYP3A4/5 e um inibidor dependente do tempo da CYP3A4/5, em concentrações clinicamente relevantes. Avaliações in vitro indicaram que ribociclibe não tem potencial para inibir as atividades da CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, e CYP2D6 em concentrações clinicamente relevantes. Ribociclibe não tem potencial para inibição tempo-dependente da CYP1A2, CYP2C9 e CYP2D6.
Os dados in vitro indicam que ribociclibe não tem potencial para induzir enzimas UGT ou as enzimas CYP, CYP2C9, CYP2C19 e CYP3A4 via PXR. Assim, é pouco provável que Kisqali afete os substratos destas enzimas. Os dados in vitro não são suficientes para excluir o potencial de ribociclibe para induzir a CYP2B6 via CAR.
Efeito dos transportadores sobre ribociclibe
Ribociclibe é um substrato para P-gp in vitro, mas com base nos dados de equilíbrio de massa a inibição de Pgp ou BCRP é improvável que afete a extensão da absorção oral de ribociclibe em doses terapêuticas. Ribociclibe não é um substrato para os transportadores da absorção hepática OATP1B1, OATP1B3 ou OCT1 in vitro.
Efeito do ribociclibe sobre os transportadores
Avaliações in vitro indicaram que ribociclibe tem potencial para inibir as atividades dos transportadores P-gp, BCRP, OATP1B1/B3, OCT1 OCT2, MATE1 e BSEP. Ribociclibe não inibiu OAT1, OAT3 ou MRP2 em concentrações clinicamente relevantes in vitro.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa, ou a amendoim, soja ou a qualquer um dos excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Doença visceral crítica
A eficácia e segurança de ribociclibe não foram estudadas em pacientes com doença visceral crítica.
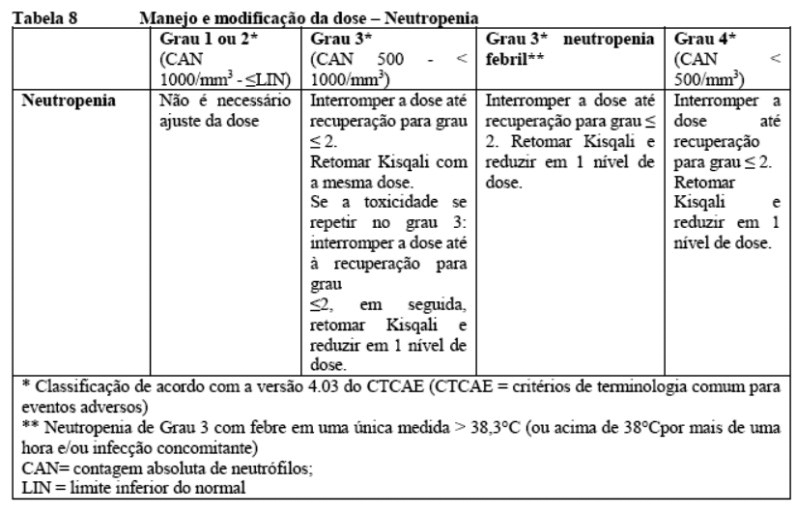
Neutropenia
Com base na gravidade da neutropenia, o tratamento com Kisqali pode precisar ser interrompido, reduzido ou descontinuado (como descrito na Tabela 2 da seção 8. Posologia e modo de usar e seção 9. Reações adversas).
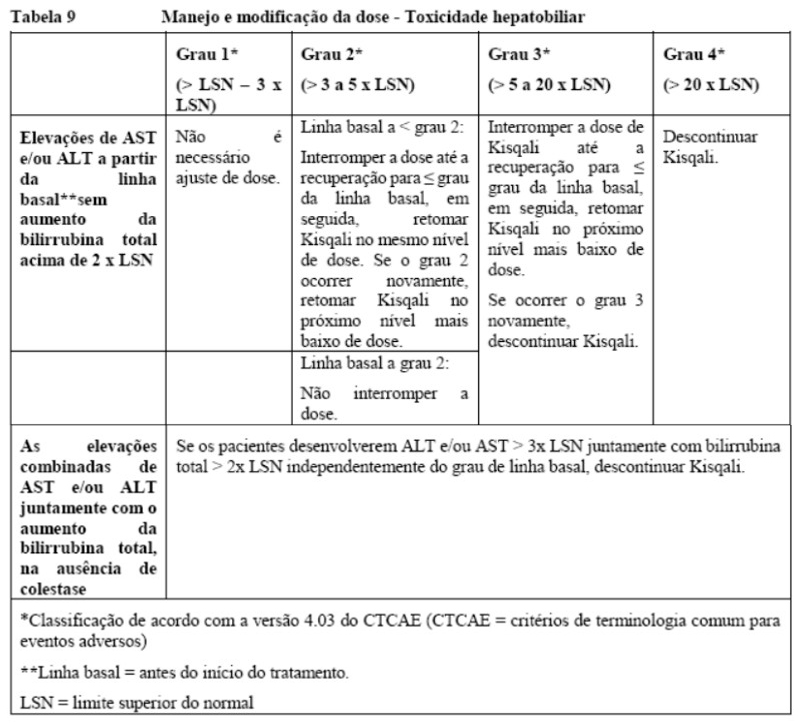
Toxicidade hepatobiliar
Devem ser realizados testes de função hepática antes de iniciar o tratamento com Kisqali. Após iniciar o tratamento com Kisqali, a função hepática deve ser monitorada (vide seção 8. Posologia e modo de usar e 9. Reações adversas).
Com base na gravidade da elevação das transaminases, o tratamento com Kisqali pode precisar ser interrompido, reduzido ou descontinuado (como descrito na Tabela 3 da seção 8. Posologia e modo de usar e seção 9. Reações adversas). Não foram estabelecidas recomendações para pacientes com níveis elevados de AST/ALT grau ≥ 3 no início do estudo.
Prolongamento do intervalo QT
O ECG deve ser avaliado antes de iniciar o tratamento. O tratamento com Kisqali deve ser iniciado apenas em pacientes com valores de QTcF inferiores a 450 mseg. O ECG deve ser repetido aproximadamente no dia 14 do primeiro ciclo e no início do segundo ciclo, e posteriormente conforme clinicamente indicado (ver seção 8. Posologia e modo de usar e 9. Reações adversas).
Deve ser realizado um monitoramento adequado dos eletrólitos séricos (incluindo potássio, cálcio, fósforo e magnésio) antes de iniciar o tratamento, no início dos primeiros 6 ciclos e, em seguida, conforme indicado clinicamente. Qualquer anormalidade deve ser corrigida antes de iniciar o tratamento com Kisqali e durante o tratamento com Kisqali.
O uso de Kisqali deve ser evitado em pacientes que já tenham ou que estejam em risco significativo de desenvolver prolongamento de QTc. Isso inclui pacientes:
· com síndrome de QT longo;
· com doença cardíaca não controlada ou significativa, incluindo infarto do miocárdio recente, insuficiência cardíaca congestiva, angina instável e bradiarritmias;
· com anormalidades nos eletrólitos
O uso de Kisqali com medicamentos conhecidos por prolongar o intervalo QTc e/ou inibidores potentes de CYP3A4 devem ser evitados, uma vez que podem levar a um prolongamento clinicamente significativo do intervalo QTcF (vide seção 8. Posologia e modo de usar, 6. Interações medicamentosas e Propriedades farmacodinâmicas). Se o tratamento com um inibidor potente de CYP3A4 não puder ser evitado, a dose deve ser reduzida para 400 mg uma vez por dia (vide seção 8. Posologia e modo de usar e 6. Interações medicamentosas).
Com base no prolongamento de QT observado durante o tratamento, o tratamento com Kisqali pode precisar ser interrompido, reduzido ou descontinuado como descrito na Tabela 4 (vide seção 8. Posologia e modo de usar, 9. Reações adversas e Propriedades farmacocinéticas).
No estudo E2301 (MONALEESA 7), o aumento médio observado de QTcF a partir da linha de base foi aproximadamente 10 msec maior no subgrupo de tamoxifeno mais placebo comparado com o subgrupo de inibidor de aromatase não esteroidal (IANE) mais placebo, sugerindo que o tamoxifeno sozinho teve um efeito de prolongamento QTcF que pode contribuir para os valores de QTcF observados no grupo Kisqali mais tamoxifeno. No braço do placebo, um aumento do intervalo QTcF > 60 msec da linha de base ocorreu em 6/90 (6,7%) pacientes recebendo tamoxifeno e em nenhum dos pacientes que receberam um IANE (vide seção 3. Características farmacológicas), Um aumento do intervalo QTcF > 60 ms a partir da linha de base foi observado em 14/87 (16,1%) do pacientes recebendo Kisqali mais tamoxifeno e em 18/245 (7,3%) pacientes recebendo Kisqali mais IANE. Kisqali não é recomendado para ser usado em combinação com tamoxifeno (vide seção 3. Características farmacológicas).
Substratos da CYP3A4
Ribociclibe é um inibidor potente da CYP3A4 na dose de 600 mg e um inibidor moderado da CYP3A4 na dose de 400 mg. Assim, o ribociclibe pode interagir com medicamentos que são metabolizados através da CYP3A4, o que pode levar a um aumento das concentrações séricas de substratos da CYP3A4. Recomenda-se precaução em caso de utilização concomitante com substratos da CYP3A4 sensíveis com uma janela terapêutica estreita e deve ser consultado a bula do outro produto em relação às recomendações relativas à coadministração com inibidores da CYP3A4.
Mulheres com potencial reprodutivo
Mulheres com potencial reprodutivo devem ser aconselhadas a usar um método eficaz de contracepção enquanto estiver tomando Kisqali e por pelo menos 21 dias após a última dose.
Lecitina de soja
Kisqali contém lecitina de soja. Os pacientes que possuem hipersensibilidade a amendoim ou soja não devem tomar Kisqali (vide seção 4. Contraindicações).
Fertilidade, gravidez e aleitamento Mulheres com potencial reprodutivo e contracepção
A possibilidade de gravidez deve ser verificada antes de iniciar o tratamento com Kisqali.
Mulheres com potencial reprodutivo recebendo Kisqali devem usar contracepção eficaz (métodos que resultam em taxas de gravidez < 1%) durante a terapia e por pelo menos 21 dias após parar o tratamento com Kisqali.
Gravidez
Não existem estudos adequados e bem controlados em mulheres grávidas. Com base nos achados em animais, o ribociclibe pode causar danos fetais quando administrado a mulheres grávidas (vide seção 3. Características farmacológicas). Kisqali não é recomendado durante a gravidez e em mulheres com potencial reprodutivo que não utilizam métodos contraceptivos.
Amamentação
Não se sabe se o ribociclibe está presente no leite materno. Não existem dados sobre os efeitos de ribociclibe em lactentes ou sobre os efeitos de ribociclibe na produção de leite. O ribociclibe e seus metabólitos passaram rapidamente para o leite em ratos lactantes. Devido ao potencial para reações adversas graves em lactentes, deve-se tomar uma decisão sobre descontinuar a amamentação ou descontinuar ribociclibe, levando em consideração a importância do tratamento para a mãe. É recomendado que mulheres usando Kisqali não devem amamentar por pelo menos 21 dias após a última dose.
Ferti