KIENDRA
NOVARTIS
siponimode
Tratamento da esclerose múltipla.
Apresentações.
Kiendra® 0,25 mg - embalagem contendo 12 ou 120 comprimidos revestidos de 0,25 mg. Kiendra® 2,0 mg - embalagem contendo 28 comprimidos revestidos de 2,0 mg
VIA ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém 0,25 mg ou 2,0 mg de siponimode equivalente a 0,278 mg ou 2,224 mg de ácido fumárico siponimode, respectivamente. Excipientes: lactose monoidratada, celulose microcristalina, crospovidona, dibeenato de glicerila e dióxido de silício. Excipientes do revestimento: álcool polivinílico, dióxido de titânio, óxido de ferro vermelho, óxido de ferro preto, óxido de ferro amarelo talco, lecitina (soja), goma xantana.
Informações técnicas.
1. INDICAÇÕES
Kiendra® é indicado no tratamento de pacientes com esclerose múltipla secundária progressiva (EMSP) com doença ativa evidenciada por recidivas ou características de imagem de atividade inflamatória.
2. RESULTADOS DE EFICÁCIA
A eficácia de Kiendra® foi demonstrada em um estudo de fase 3 que avaliou doses diárias de 2 mg de Kiendra® em pacientes com EMSP. Estudo A2304 (EXPAND) O estudo A2304 foi um estudo de fase 3, randomizado, duplo-cego, controlado por placebo, guiado por desfecho e duração do acompanhamento, em pacientes com EMSP que apresentavam evidências documentadas de progressão nos 2 anos anteriores na ausência de recidivas ou independente delas, nenhuma evidência de recidiva em 3 meses antes da inclusão no estudo e com pontuação na Escala Expandida no Estado de Incapacidade (EDSS) de 3,0 a 6,5 na entrada do estudo. O EDSS mediano foi de 6,0 no baseline. Pacientes acima de 61 anos de idade não foram incluídos. No que diz respeito à atividade da doença, as características de imagem da atividade inflamatória na EMSP podem ser relacionadas à recidiva ou imagem (ou seja, lesões T1 realçadas por gadolínio (Gd) ou lesões T2 ativas [novas ou aumentadas]).
Os pacientes foram randomizados na razão de 2:1 para receber Kiendra® 2 mg uma vez ao dia ou placebo. As avaliações foram realizadas na seleção e a cada 3 meses e no momento da recidiva. Avaliações por RM foram realizadas na seleção e a cada 12 meses.
O desfecho primário do estudo foi o tempo até a progressão da incapacidade confirmada (CDP) em 3 meses, determinado como um aumento de pelo menos 1 ponto em relação ao valor de baseline na EDSS (aumento de 0,5 ponto para pacientes com EDSS basal de 5,5 ou mais) mantido por 3 meses. Os principais desfechos secundários foram o tempo até o agravamento confirmado em 3 meses de pelo menos 20% em relação ao valor de baseline no teste de caminhada cronometrado de 25 pés (T25FW) e a mudança em relação ao valor de baseline no volume da lesão em T2. Os desfechos secundários adicionais incluíram tempo até CDP em 6 meses, alteração percentual do volume cerebral, medidas da atividade inflamatória da doença (taxa de recidiva anualizada, lesões por ressonância magnética). A alteração da pontuação oral de velocidade de processamento cognitivo do Symbol Digit Modality Test [Teste de Modalidades de Símbolos e Dígitos] foi um desfecho exploratório.
A duração do estudo foi variável para pacientes individuais (a duração mediana do estudo foi de 21 meses, intervalo de 1 dia a 37 meses).
O estudo randomizou 1.651 pacientes para Kiendra® 2 mg (N=1.105) ou placebo (N=546); 82% dos pacientes tratados com Kiendra® e 78% dos pacientes tratados com placebo concluíram o estudo. A idade mediana foi de 49,0 anos, a duração mediana da doença foi de 16,0 anos e a pontuação mediana na EDSS foi de 6,0 no baseline; 63,9% dos pacientes não apresentaram recidivas nos 2 anos anteriores à inclusão no estudo e 76% não tiveram lesões realçadas por gadolínio (Gd) na ressonância magnética de baseline; 78,3% dos pacientes haviam sido previamente tratados com uma terapia para EM.
O tempo até o início da progressão da incapacidade confirmada em 3 meses (desfecho primário) foi retardado de forma significativa para Kiendra® com uma redução de risco de 21,2% em comparação com
o placebo (razão de risco (HR) 0,79, p < 0,0134) e redução no risco de CDP de 6 meses em 26% em comparação com o placebo (HR 0,74, p = 0,0058). Os resultados deste estudo estão resumidos na Tabela 2-1 e na Figura 2-1.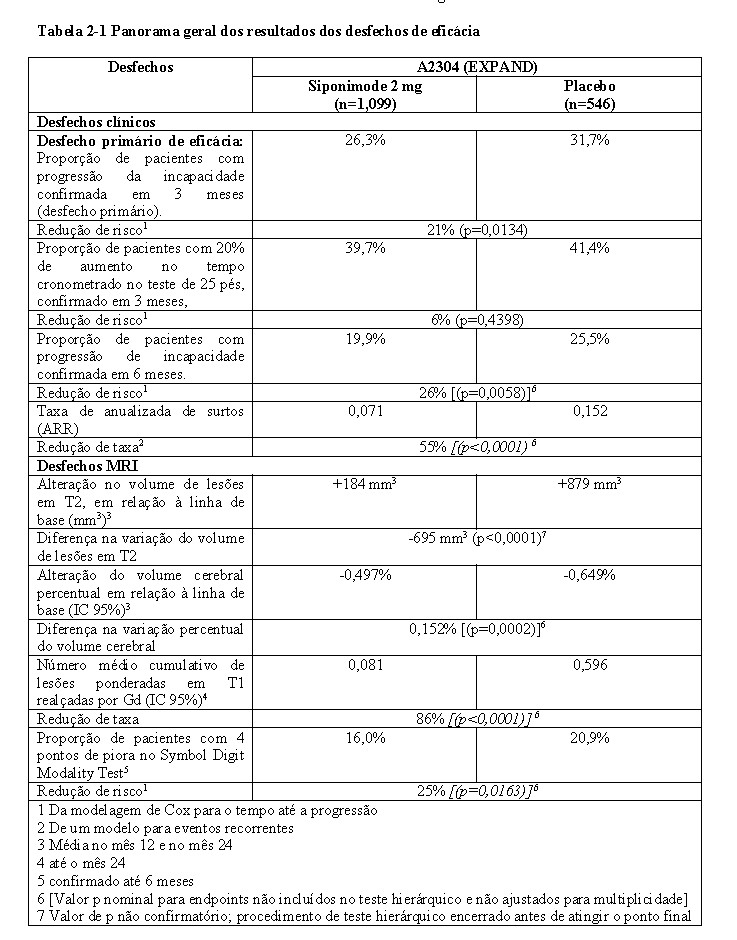
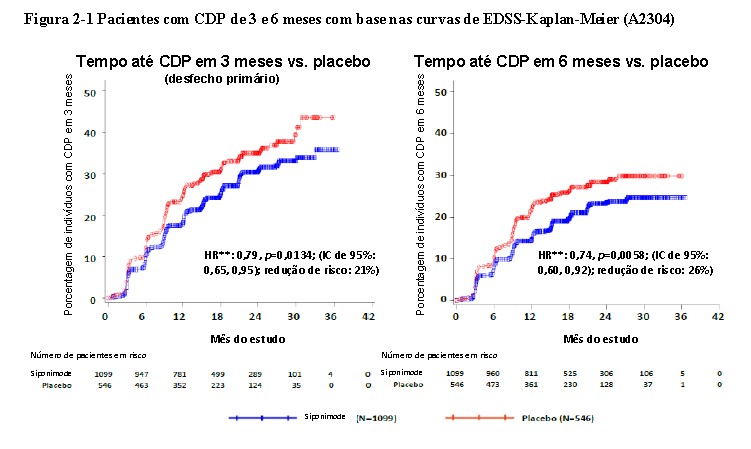
Os resultados do estudo mostraram uma redução variável, mas consistente, do risco no tempo até a progressão da incapacidade confirmada (CDP) em 3 e 6 meses com Kiendra® em comparação com o placebo em subgrupos definidos com base no sexo, idade, atividade de recidiva pré-estudo, atividade da doença por RM no baseline, duração da doença e níveis de incapacidade no baseline3.
No subgrupo de doentes (n=779) com doença ativa (definida como doentes com surtos nos 2 anos anteriores ao estudo e/ou presença de lesões T1 realçadas por Gd na baseline) as características basais foram semelhantes às da população global. A mediana da idade foi 47 anos, a duração mediana da doença foi de 15 anos e o valor mediano de EDSS inicial foi 6,0.
O tempo até início da CDP aos 3 meses e 6 meses foi retardado de forma significativa nos pacientes com doença ativa tratados com siponimode, em 31% em comparação com placebo (HR 0.69; 95% IC: 0,53; 0,91) e em 37% em comparação com placebo (HR 0,63; 95% IC: 0,47; 0,86), respetivamente. A ARR (surtos confirmados) foi reduzida em 46% (taxa de ARR 0,54; 95% IC: 0,39; 0,77) em comparação com placebo. A redução da taxa relativa do número cumulativo de lesões T1 realçadas por Gd avaliadas durante 24 meses foi de 85% (razão da taxa 0,155; 95% IC: 0,104; 0,231) em comparação com placebo. As diferenças na alteração do volume das lesões T2 e na percentagem de alteração do volume cerebral (média ao longo dos meses 12 e 24) em comparação com placebo foram -1163 mm3 (95% IC: -1484, -843 mm3) e 0,141% (95% IC: 0,020; 0,261%), respetivamente.
Os resultados no subgrupo de pacientes com EMSP ativa estão resumidos na Figura 2-2.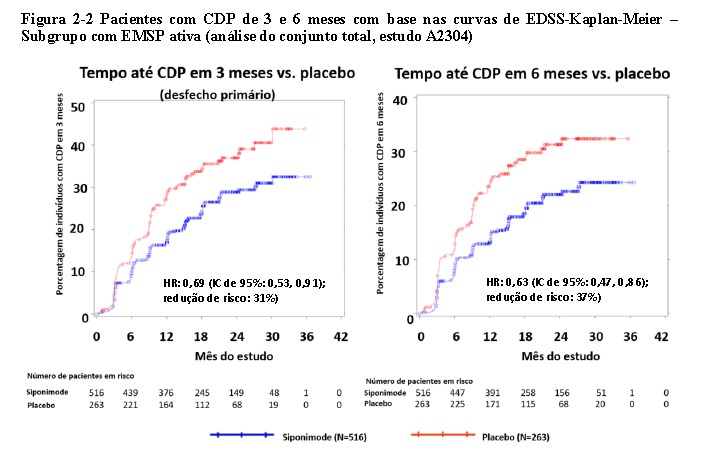
No subgrupo de pacientes (n=827) sem sinais e sintomas de atividade da doença (definidos como pacientes sem surtos nos 2 anos anteriores ao estudo e sem presença de lesões T1 com aumento de Gd na linha de base), efeitos em 3 meses e 6 meses CDP foram pequenos (reduções de risco foram de 7% e 13%, respectivamente).
Referências bibliográficas
1. Clinical Overview -BAF312 -2.5 Clinical Overview. Novartis. 2018.
2. Summary of Clinical Efficacy -BAF312 -2.7.3 Summary of Clinical Efficacy. Novartis. 2018.
3. Summary of Clinical Safety -BAF312 -2.7.4 Summary of Clinical Safety. Novartis. 2018.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Imunossupressores seletivos Código ATC: L04AA42.
Mecanismo de ação
O siponimode é um modulador do receptor de esfingosina-1-fosfato (S1P). O siponimode liga-se seletivamente em dois dos cinco receptores acoplados à proteína G (GPCRs) para S1P, ou seja, S1P1 e S1P5. Ao atuar como antagonista funcional dos receptores S1P1 nos linfócitos, o siponimode evita a saída dos linfócitos dos linfonodos. Isso reduz a recirculação de células T no sistema nervoso central (SNC) para limitar a inflamação central.
O siponimode atravessa facilmente a barreira hematoencefálica.
Em estudos com animais, foram demonstrados efeitos diretos para o siponimode nas células neurais, via S1P1 em astrócitos e S1P5 em oligodendrócitos. Em um modelo de camundongo com encefalomielite autoimune experimental, também foi demonstrado um efeito neuroprotetor direto, independente dos efeitos nos linfócitos, para o siponimode aplicado centralmente (por meio de infusões intracerebroventriculares).
Farmacodinâmica
Sistema imunológico
Kiendra® induz uma redução dependente da dose da contagem de linfócitos no sangue periférico dentro de 6 horas após a primeira dose, devido ao sequestro reversível de linfócitos nos tecidos linfoides.
Com a administração diária contínua, a contagem de linfócitos continua a diminuir, atingindo uma contagem mediana de nadir (IC de 90%) de aproximadamente 0,560 (0,271 a 1,08) células/nL em um paciente não japonês com EMSP com genótipo CYP2C9 *1*1 ou *1*2 típico, que corresponde a 20 a 30% do valor de baseline. As baixas contagens de linfócitos são mantidas com doses diárias crônicas.
A contagem de linfócitos normalmente retorna ao intervalo normal na grande maioria (90%) dos pacientes com EMSP dentro de 10 dias após a interrupção da terapia. Após a interrupção do tratamento com Kiendra®, os efeitos residuais de redução na contagem de linfócitos periféricos podem persistir por até 3 a 4 semanas após a última dose.
Eletrofisiologia cardíaca
Frequência cardíaca e ritmo
Kiendra® causa uma redução transitória da frequência cardíaca e condução atrioventricular no início do tratamento (ver seção 9. REAÇÕES ADVERSAS). O declínio máximo na frequência cardíaca é observado nas primeiras 6 horas após a dose. Respostas autonômicas do coração, incluindo variação diurna da frequência cardíaca e resposta ao exercício físico, não são afetadas pelo tratamento com siponimode.
Foi observada uma diminuição transitória e dependente da dose na frequência cardíaca durante a fase de dosagem inicial de Kiendra®, que atingiu platô em doses ≥5 mg e eventos bradiarrítmicos (bloqueios AV e pausas sinusais) foram detectados com maior incidência no tratamento com Kiendra® em comparação ao placebo.
Não foram observados bloqueios AV de segundo grau de Mobitz tipo II ou grau superior. A maioria dos bloqueios AV e pausas sinusais ocorreram acima da dose terapêutica de 2 mg, com incidência notavelmente mais alta em condições não tituladas em comparação com as condições de titulação da dose.
A diminuição da frequência cardíaca induzida por Kiendra® pode ser revertida por atropina ou isoprenalina.
Potencial de prolongar o intervalo QT
Os efeitos de doses terapêuticas (2 mg) e supraterapêuticas (10 mg) de siponimode na repolarização cardíaca foram investigados em um estudo completo do QT. Os resultados não sugeriram um potencial arritmogênico relacionado ao prolongamento do intervalo QT com siponimode. O siponimode aumentou o QTcF ajustado basal corrigido pelo placebo (??QTcF) em mais de 5 ms, com um efeito médio máximo de 7,8 ms (2 mg) e 7,2 ms (10 mg), respectivamente, três horas após a dose. O limite superior do IC de 95% unilateral para o ??QTcF em todos os momentos permaneceu abaixo de 10 ms. A análise categórica não revelou valores de QTc emergentes do tratamento acima de 480 ms, nenhum aumento de QTc em relação ao valor de baseline superior a 60 ms e nenhum valor de QT/QTc corrigido ou não corrigido excedeu 500 m.
Função pulmonar
O tratamento com Kiendra® com doses únicas ou múltiplas por 28 dias não está associado a aumentos clinicamente relevantes na resistência das vias aéreas, medidos pelo volume expiratório forçado em 1 segundo (VEF1) e fluxo expiratório forçado (FEF) durante a expiração de 25 a 75% da capacidade vital forçada (FEF25-75%). Foi detectada uma ligeira tendência de redução do VEF1 em doses únicas não terapêuticas ( > 10 mg). As doses múltiplas de Kiendra® foram associadas a alterações leves a moderadas no VEF1 e no FEF25-75%, que não eram dependentes da dose e do dia e não estavam associadas a nenhum sinal clínico de aumento da resistência das vias aéreas.
O tratamento concomitante de Kiendra® com propranolol resultou em diminuição mínima do VEF1 em comparação ao propranolol em monoterapia. As alterações com os medicamentos individuais ou com a combinação estavam dentro da variabilidade fisiológica do VEF1 e não eram clinicamente significativas.
Farmacocinética
Absorção
O tempo (Tmáx) até alcançar as concentrações plasmáticas máximas (Cmáx) após a administração oral múltipla de siponimode foi de cerca de 4 horas (intervalo de 2 a 12 horas). A absorção de siponimode é extensa (≥ 70%, com base na quantidade de radioatividade excretada na urina e na quantidade de metabólitos nas fezes extrapoladas para o infinito). A biodisponibilidade oral absoluta do siponimode é de aproximadamente 84%. Para 2 mg de siponimode administrado uma vez ao dia durante 10 dias, foram observadas uma Cmáx média de 30,4 ng / mL e uma ASCtau média de 558 h*ng/mL no dia 10. O estado de equilíbrio foi alcançado após aproximadamente 6 dias de administração múltipla uma vez ao dia de siponimode.
Efeito do alimento
A ingestão de alimentos não teve efeito na exposição sistêmica do siponimode (Cmáx e ASC). Portanto, Kiendra® pode ser tomado sem levar em consideração as refeições (consulte a seção 8. POSOLOGIA E MODO DE USAR).
Distribuição
O siponimode é distribuído para os tecidos do corpo com um volume médio moderado de distribuição de 124 L. A fração de siponimode encontrada no plasma é de 68% em humanos. Estudos em animais mostram que o siponimode atravessa facilmente a barreira hematoencefálica. A ligação proteica do siponimode é > 99,9% em indivíduos saudáveis e em pacientes com insuficiência hepática e renal.
Biotransformação/metabolismo
O siponimode é extensamente metabolizado, principalmente pelo CYP2C9 (79,3%), seguido pelo CYP3A4 (18,5%). Não se espera que a atividade farmacológica dos principais metabólitos M3 e M17 contribua para o efeito clínico e a segurança do siponimode em humanos.
Eliminação
Um clearance sistêmico aparente (CL/F) de 3,11 L/h foi estimado em pacientes com EM (veja abaixo subseção Farmacogenômica). A meia-vida de eliminação aparente é de aproximadamente 30 horas. O siponimode é eliminado da circulação sistêmica principalmente devido ao metabolismo e subsequente excreção biliar/fecal. O siponimode inalterado não foi detectado na urina.
Linearidade
A concentração de siponimode aumenta de maneira proporcional à dose aparente após doses múltiplas uma vez ao dia de siponimode de 0,3 mg a 20 mg.
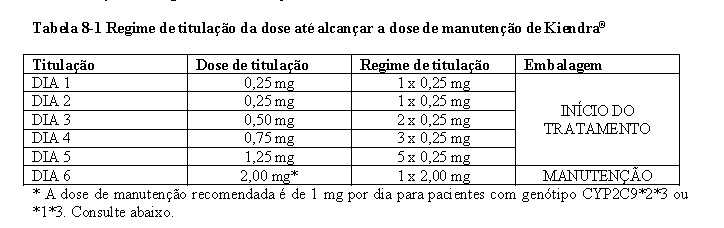
As concentrações plasmáticas no estado de equilíbrio são alcançadas após aproximadamente 6 dias de administração uma vez ao dia e os níveis no estado de equilíbrio são aproximadamente 2 a 3 vezes maiores que após a dose inicial. É utilizado um regime de titulação ascendente para alcançar gradualmente a dose terapêutica clínica de siponimode de 2 mg após 6 dias e são necessários mais 4 dias de dosagem para atingir as concentrações plasmáticas no estado de equilíbrio.
Avaliação in vitro e in vivo do potencial de interação medicamentosa Siponimode (e metabólitos M3, M17) como agente causador da interação
Investigações in vitro indicaram que o siponimode e seus principais metabólitos sistêmicos M3 e M17 não apresentam potencial de interação medicamentosa clinicamente relevante na dose terapêutica de 2 mg uma vez ao dia para todas as enzimas do CYP e transportadores investigados e não necessitam de investigação clínica.
Siponimode como objeto de interação
O CYP2C9 é polimórfico e o genótipo influencia as contribuições fracionárias das duas vias do metabolismo oxidativo para a eliminação geral. A modelagem farmacocinética de base fisiológica indica uma inibição diferencial dependente do genótipo do CYP2C9 e indução das vias do CYP3A4. Com a diminuição da atividade metabólica do CYP2C9 nos respectivos genótipos, é esperado um efeito maior dos perpetradores do CYP3A4 na exposição ao siponimode.
Administração concomitante de siponimode com inibidores de CYP2C9 e CYP3A4
A administração concomitante de fluconazol (inibidor moderado duplo do CYP2C9/CYP3A4) 200 mg diariamente no estado de equilíbrio e uma dose única de siponimode 4 mg em voluntários saudáveis com CYP2C9 *1*1 levou a um aumento de duas vezes na ASC do siponimode. A meia-vida média terminal do siponimode aumentou 50%.
Administração concomitante de siponimode com indutores de CYP2C9 e CYP3A4
Indutores fortes do CYP3A4/moderado do 2C9 (por exemplo, carbamazepina) em todos os pacientes, independentemente do genótipo; e indutores moderados do CYP3A4 (por exemplo, modafinil) em pacientes com genótipo CYP2C9*1*3 ou *2*3 reduziram significativamente a exposição ao siponimode em até 76% e até 51%, respectivamente, de acordo com estudos clínicos de interação medicamentosa e avaliação in silico do potencial de interação do medicamento. A administração concomitante de siponimode 2 mg diariamente na presença de doses diárias de 600 mg de rifampicina (forte indutor do CYP3A4/indutor moderado do CYP2C9) diminuiu a ASCtau,ss e a Cmáx,ss do siponimode em 57% e 45%, respectivamente, em indivíduos com CYP2C9 *1*1.
Populações especiais
Pacientes geriátricos (65 anos ou mais)
Os resultados da farmacocinética da população sugerem que o ajuste da dose não seria necessário em pacientes idosos. Pacientes acima de 61 anos não foram incluídos nos estudos clínicos. Siponimode deve ser usado com cautela em pacientes idosos..
Sexo
O sexo não tem influência na farmacocinética do siponimode.
Raça/Etnia
Os parâmetros de farmacocinética da dose única não foram diferentes entre indivíduos saudáveis japoneses e caucasianos, indicando ausência de sensibilidade étnica na farmacocinética do siponimode.
Comprometimento renal
Não são necessários ajustes da dose de siponimode em pacientes com insuficiência renal leve, moderada ou grave. A meia-vida média do siponimode e a Cmáx (total e não ligada) foram comparáveis entre indivíduos com comprometimento renal grave e indivíduos saudáveis. As ASCs totais e não ligadas aumentaram apenas ligeiramente (em 23 a 33%), em comparação com indivíduos saudáveis. Os efeitos da doença renal em fase terminal ou da hemodiálise na farmacocinética do siponimode não foram estudados. Devido à alta ligação às proteínas plasmáticas ( > 99,9%) do siponimode, não se espera que a hemodiálise altere a concentração total e não ligada de siponimode e não são previstos ajustes de dose com base nessas considerações.
Comprometimento hepático
Não são necessários ajustes de dose para siponimode em doentes com comprometimento hepático. A ASC da farmacocinética do siponimode não ligada é 15% e 50% superior em indivíduos com comprometimento hepático moderado e grave, respectivamente, em comparação com indivíduos saudáveis para a dose única de 0,25 mg estudada. A meia-vida média do siponimode permaneceu inalterada no comprometimento hepático.
Farmacogenômica
O genótipo CYP2C9 tem um impacto significativo no metabolismo do siponimode. Pacientes homozigotos para o CYP2C9*3 (genótipo CYP2C9*3*3: aproximadamente 0,3 a 0,4% dos caucasianos e menos em outros) são contraindicados com Kiendra® (ver seção 4. CONTRAINDICAÇÕES). O uso de Kiendra® nesses pacientes resulta em níveis plasmáticos de siponimode substancialmente elevados. A dose de manutenção recomendada de Kiendra® é de 1 mg por dia em pacientes com genótipo CYP2C9 *2*3 ou *1*3 para evitar um aumento da exposição ao siponimode (ver seção 8. POSOLOGIA E MODO DE USAR).
Existem outros polimorfismos menos frequentes para CYP2C9. A farmacocinética do siponimode não foi avaliada nesses indivíduos. Alguns polimorfismos como *5, *6, *8 e *11 estão associados à diminuição ou perda da função enzimática. Estima-se que os alelos CYP2C9 *5, *6, *8 e *11 tenham uma frequência combinada de aproximadamente 10% em populações com ascendência africana, 2% em latinos/hispânicos e < 0,4% em caucasianos e asiáticos.
Após uma dose única de 0,25 mg de siponimode, ambos ASCinf e ASClast foram aproximadamente 2 e 4 vezes mais altas em indivíduos com os genótipos CYP2C9*2*3 e CYP2C9*3*3, respectivamente, enquanto houve apenas um pequeno aumento da Cmáx de 21% e 16%, respectivamente, em comparação com metabolizadores extensos (CYP2C9*1*1). A meia-vida média foi prolongada nos portadores de CYP2C9*2*3 e CYP2C9*3*3 (51 e 126 h).
Foi estimado um clearance sistêmico aparente (CL/F) de 3,11 L/h em pacientes com EMSP com metabolismo extenso do CYP2C9 (CYP2C9*1*1 e CYP2C9*1*2) após múltiplas administrações orais de siponimode. O Cl/F é de 2,5, 1,9, 1,6 e 0,9 L/h em indivíduos com os genótipos CYP2C9*2*2, CYP2C9*1*3, CYP2C9*2*3 e CYP2C9*3*3, respectivamente. Como o clearance aparente estimado para indivíduos com o genótipo CYP2C9*1*2 foi comparável ao dos indivíduos com o genótipo CYP2C9*1*1, é esperada uma exposição semelhante ao siponimode para ambos os genótipos. O aumento resultante na ASC do siponimode foi de 25, 61, 91, 285% em indivíduos com os genótipos CYP2C9*2*2, CYP2C9*1*3, CYP2C9*2*3 e CYP2C9*3*3, respectivamente, em comparação com o genótipo CYP2C9*1*1.
Dados de segurança não clínicos
O siponimode foi avaliado em estudos de farmacologia de segurança e de toxicidade de dose repetida em camundongos, ratos e macacos cynomolgus, bem como em estudos para avaliar a genotoxicidade, carcinogenicidade, toxicidade reprodutiva e de desenvolvimento, tolerabilidade local, potencial fotorreativo, imunotoxicidade, potencial de dependência e abuso e uma avaliação para qualificar impurezas. Os dados pré-clínicos não revelaram riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida e genotoxicidade. Efeitos adversos em estudos pivotais de dose repetida foram observados em animais com exposição cem vezes maior que os níveis de exposição clínica ou tinham relevância humana limitada. No total, os dados de segurança não clínica não revelaram riscos especiais relevantes para o ser humano, exceto no desenvolvimento embrionário-fetal (vide 5 Advertências e precauções -Gravidez, lactação, mulheres e homens com potencial reprodutivo -Dados em animais).
Farmacologia de segurança e toxicidade de dose repetida
As investigações de farmacologia de segurança no sistema respiratório e no SNC no rato demonstraram apenas efeitos menores na função respiratória e nenhum efeito neurofarmacológico adverso. A avaliação da farmacologia de segurança cardiovascular em ratos, cobaias e macacos mostrou redução transitória da frequência cardíaca.
Foram realizados estudos de toxicidade de dose única e repetida por via oral em camundongos (até 13 semanas), ratos (até 26 semanas) e macacos (até 52 semanas). Reduções relacionadas ao siponimode na contagem total de linfócitos foram evidenciadas em todos os níveis de dose em estudos de toxicidade de dose repetida entre espécies. Os efeitos foram reversíveis ou parcialmente reversíveis e de acordo com o modo de ação farmacológico do siponimode. As toxicidades limitantes da dose em espécies animais foram nefrotoxicidade em camundongos, desenvolvimento de peso corporal em ratos e efeitos adversos no SNC e efeitos gastrointestinais em macacos. Os principais órgãos alvo de toxicidade identificados pela histopatologia em roedores incluíram pulmão, fígado, tireoide, rim e útero/vagina. Nos macacos, foram observados efeitos nos músculos e na pele em animais individuais.
Os NOAELs em ratos foram definidos como 50 e 15 mg/kg/dia para machos e fêmeas, respectivamente, e em macacos como 10 mg/kg/dia para ambos os sexos. Múltiplos de exposição baseados na Cmáx e ASC de 190 a 342 em ratos e de 171 a 222 em macacos para efeitos sistêmicos foram calculados em relação à dose de manutenção de 2 mg/dia.
O siponimode não tem potencial fototóxico nem potencial de abuso e dependência.
Carcinogenicidade e genotoxicidade
Os testes de genotoxicidade in vitro (mutação bacteriana, teste de micronúcleo e teste de aberração cromossômica com linfócitos humanos) e um estudo in vivo de micronúcleo em ratos não revelaram potencial genotóxico do siponimode.
Consistente com um efeito imunomodulador, o siponimode induziu aumento da incidência de linfoma maligno em camundongos; a relevância humana é desconhecida. Em um estudo de carcinogenicidade em camundongos, foram observadas incidências aumentadas de hemangiossarcomas e hemangiomas em todas as doses dos níveis de dose em ambos os sexos. Estudos mecanísticos mostraram ativação das células endoteliais vasculares, levando à indução de angiogênese anormal e, finalmente, hemangiossarcomas. Não foram encontradas ativações sustentadas das células endoteliais vasculares nem aumento da incidência de hemangiossarcomas em ratos. Culturas de células endoteliais de camundongo, rato e humano demonstraram respostas diferentes após o tratamento com siponimode. As células humanas e de rato não apresentaram respostas proliferativas em oposição às células de camundongo. Portanto, os hemangiossarcomas induzidos por siponimode em camundongos são considerados específicos da espécie e não há evidências que sugiram um risco associado ao ser humano.
Em ratos, considera-se que alterações neoplásicas relacionadas ao siponimode (adenoma/carcinoma de células foliculares) na glândula tireoide apenas nos machos e alterações proliferativas não neoplásicas na glândula tireoide (apenas machos) e no fígado (ambos os sexos) decorrentes de um conhecido efeito específico do roedor ('eixo fígado-tireoide'). Considera-se que essas alterações representem efeitos adaptativos em roedores com relevância humana limitada.
Toxidade reprodutiva
Para detalhes, consulte 5. ADVERTÊNCIAS E PRECAUÇÕES
Fertilidade
Nos estudos de fertilidade em ratos machos e fêmeas, os animais receberam doses orais de siponimode até 200 mg/kg/dia e 1 mg/kg/dia, respectivamente, antes do acasalamento e até 2 semanas após o acasalamento para machos e até o dia 6 da gestação para fêmeas.
Não houve efeito nos parâmetros de acasalamento ou espermatozoides nos machos e no acasalamento e desenvolvimento embrionário precoce até a implantação em fêmeas de ratos, indicando que o siponimode não está associado a um risco aumentado de efeito sobre a fertilidade.
Não houve alterações relevantes nos órgãos reprodutivos de ratos e macacos após administração crônica.
4. CONTRAINDICAÇÕES
• Esse medicamento é contraindicado para paciente com hipersensibilidade à substância ativa ou qualquer componente da fórmula, ver "Composição".
• Pacientes com genótipo CYP2C9 * 3 * 3.
• Pacientes que nos últimos 6 meses tiveram infarto do miocárdio (MI), angina pectoris instável, acidente vascular cerebral / ataque isquêmico transitório (AIT), insuficiência cardíaca descompensada (que requer tratamento hospitalar) ou insuficiência cardíaca Classe III / IV da New York Heart Association.
• Pacientes com bloqueio atrioventricular (AV) de segundo grau Mobitz tipo II ou bloqueio AV de terceiro grau, ou síndrome do nódulo sinusal, se não tiverem marcapasso (consulte a seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
• Síndrome de imunodeficiência.
• História de leucoencefalopatia multifocal progressiva ou meningite criptocócica.
• Neoplasias malignas ativas.
• Comprometimento hepático grave (classe C de Child-Pugh).
• Durante a gravidez e em mulheres com potencial para engravidar que não utilizam métodos contraceptivos eficazes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Infecções
Um efeito farmacodinâmico principal de Kiendra® é uma redução dependente da dose da contagem de linfócitos periféricos para 20 a 30% dos valores de baseline. Isto é devido ao sequestro reversível delinfócitos nos tecidos linfoides (consulte a seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Os efeitos do sistema imunológico de Kiendra® podem aumentar o risco de infecções (consulte a seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Antes de iniciar o tratamento com Kiendra®, um hemograma completo recente (HG) (ou seja, nos últimos 6 meses ou após a descontinuação da terapia anterior) deve estar disponível. Avaliações de hemograma completo também são recomendadas periodicamente durante o tratamento. Contagens absolutas de linfócitos < 0,2 x 109/l, se confirmadas, devem levar à redução da dose para 1 mg, porque em estudos clínicos a dose de siponimode foi reduzida em pacientes com contagens absolutas de linfócitos < 0,2 x 109/l. A contagem absoluta de linfócitos confirmada < 0,2 x 109/l em um paciente que já está recebendo siponimode 1 mg deve levar à interrupção da terapia com siponimode até que o nível atinja 0,6 x 109/l, quando o reinício do siponimode pode ser considerado.
O início do tratamento com Kiendra® deve ser postergado em pacientes com infecção ativa grave até a resolução. Considerando que os efeitos farmacodinâmicos residuais, como efeitos redutores na contagem de linfócitos periféricos, podem persistir por até 3 a 4 semanas após a descontinuação de Kiendra®, a vigilância da infecção deve continuar durante esse período (veja abaixo: Interrompendo a terapia com Kiendra®).
Os pacientes que recebem Kiendra® devem ser orientados a relatar sintomas de infecções ao seu médico. Estratégias terapêuticas e de diagnóstico eficazes devem ser empregadas em pacientes com sintomas de infecção durante o tratamento. A suspensão do tratamento com Kiendra® deve ser considerada se um paciente desenvolver uma infecção grave.
Casos de meningite criptocócica (MC) foram relatados com Kiendra®. Os médicos devem estar atentos a sintomas clínicos ou sinais de MC. Pacientes com tais sintomas e sinais devem ser submetidos a uma avaliação diagnóstica imediata. O tratamento com Kiendra® deve ser suspenso até que a MC seja excluída. Se a MC for diagnosticada, deve-se iniciar o tratamento adequado.
Não foram relatados casos de leucoencefalopatia multifocal progressiva (LMP) para Kiendra® no programa de desenvolvimento, no entanto, casos de LMP foram relatados para outro modulador de receptor S1P. Os médicos devem estar atentos a sintomas clínicos ou achados de ressonância magnética que possam sugerir LMP. Se houver suspeita de LMP, o tratamento com Kiendra® deve ser suspenso até que a LMP seja resolvida.
Foram relatados casos de infecção viral por herpes, incluindo casos de meningite ou meningoencefalite causada pelo vírus varicela-zoster, com Kiendra®. Pacientes sem um histórico de varicela (catapora) confirmado por um profissional de saúde ou sem documentação de um ciclo completo de vacinação contra o vírus da varicela-zoster (VZV) devem ser testados quanto a anticorpos para o VZV antes de iniciar o Kiendra® (consulte a subseção Vacinação).
As terapias antineoplásicas, imunomoduladoras ou imunossupressoras (incluindo corticosteroides) devem ser administradas concomitantemente com cautela devido ao risco de efeitos aditivos do sistema imunológico durante essa terapia (consulte a seção 6 Interações medicamentosas)1.
Vacinação
Recomenda-se um ciclo completo de vacinação para pacientes negativos para anticorpos com vacina contra varicela antes do início do tratamento com Kiendra®, após o qual o início do tratamento com Kiendra® deve ser adiado por 1 mês para permitir que ocorra o efeito total da vacinação (consulte a seção
9. REAÇÕES ADVERSAS).
Vacinas vivas atenuadasO uso de vacinas vivas atenuadas deve ser evitado enquanto os pacientes estiverem tomando Kiendra® e por 4 semanas após a interrupção do tratamento com Kiendra® (consulte a seção 6. INTERAÇÕES MEDICAMENTOSAS).
Vacinas não vivas atenuadas As vacinas não vivas atenuadas podem ser menos eficazes se administradas durante o tratamento com Kiendra®. A decisão de continuar ou pausar o tratamento com Kiendra® deve ser baseada na avaliação risco-benefício individual do paciente (consulte abaixo o item "Interrompendo a terapia" e a seção 6.INTERAÇÕES MEDICAMENTOSAS).
Edema macular
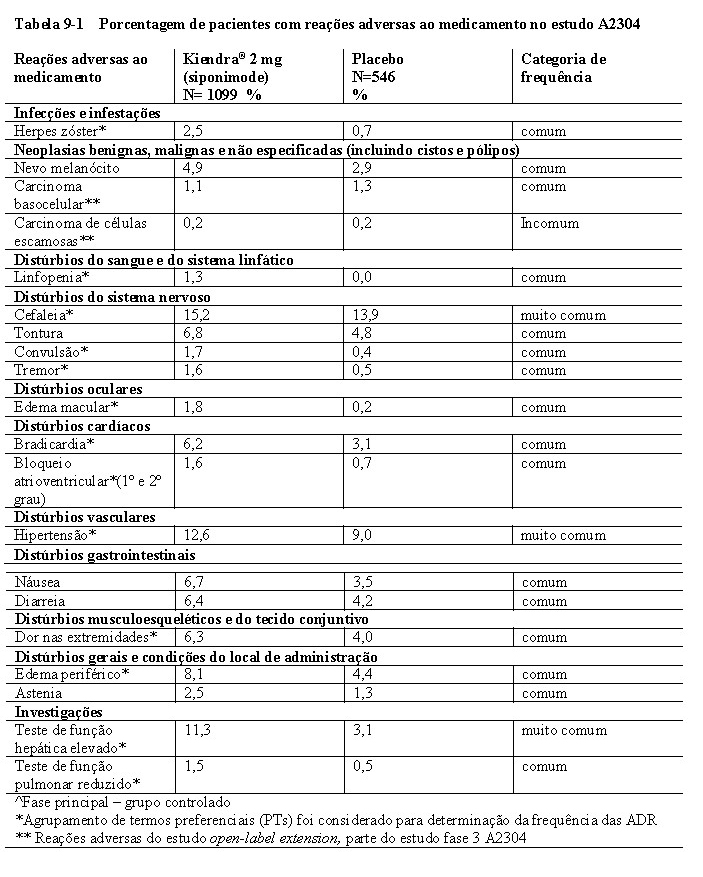
O edema macular (consulte a seção 9. REAÇÕES ADVERSAS) com ou sem sintomas visuais foi mais frequentemente relatado com siponimode (1,8%) do que com placebo (0,2%) no estudo clínico de fase 3 (A2304). A maioria dos casos ocorreu nos primeiros 3 a 4 meses de terapia. Portanto, recomenda-se uma avaliação oftalmológica 3 a 4 meses após o início do tratamento. Como também ocorreram casos de edema macular no tratamento de longo prazo, os pacientes devem relatar distúrbios visuais a qualquer momento durante a terapia com Kiendra® e recomenda-se uma avaliação do fundo, incluindo a mácula.
A terapia com Kiendra® não deve ser iniciada em pacientes com edema macular até a resolução.
Pacientes com histórico de diabetes mellitus, uveíte ou doenças subjacentes/coexistentes da retina apresentam maior risco de edema macular. Recomenda-se que os pacientes com diabetes mellitus, uveíte ou história de distúrbios da retina sejam submetidos a uma avaliação oftálmica antes de iniciar a terapia com Kiendra® e façam avaliações de acompanhamento durante o tratamento com Kiendra®.
A continuação da terapia com Kiendra® em pacientes com edema macular não foi avaliada. Recomendase que a terapia com Kiendra® seja descontinuada caso o paciente desenvolva edema macular. Uma decisão sobre o reinício ou não da terapia com Kiendra® deve levar em consideração os possíveis benefícios e riscos potenciais para cada paciente.
Bradiarritmia
Frequência cardíaca
Uma vez que o início do tratamento com Kiendra® resulta numa diminuição transitória da frequênciacardíaca (consulte a seção 3. CARACTERÍSTICAS FARMACOLÓGICAS), é aplicado um esquema de titulação ascendente para atingir a dose de manutenção de Kiendra® no dia 6 no início do tratamento (consulte a seção 8. POSOLOGIA E MODO DE USAR).
Após a primeira dose de titulação, a diminuição da frequência cardíaca começa dentro de uma hora e o declínio do dia 1 é máximo em aproximadamente 3 a 4 horas. Com a titulação ascendente contínua, são observadas reduções adicionais da frequência cardíaca nos dias subsequentes, com redução máxima do dia 1-baseline alcançada no dia 5 ao 6. A maior redução diária pós-dose na frequência cardíaca média horária absoluta é observada no dia 1 com uma diminuição média no pulso de 5 a 6 batimentos por minuto (bpm). Os declínios pós-dose nos dias seguintes são menos pronunciados. Com a administração contínua, a frequência cardíaca começa a aumentar após o dia 6 e atinge os níveis de placebo 10 dias após o início do tratamento.
Frequências cardíacas abaixo de 40 bpm foram raramente observadas. Os pacientes que apresentaram bradicardia eram geralmente assintomáticos. Poucos pacientes apresentaram sintomas leves a moderados, incluindo tontura ou dor no peito não cardíaca que se resolveram dentro de 24 horas sem intervenção (consulte a seção 9 Reações adversas). A diminuição da frequência cardíaca induzida pelo siponimode pode ser revertida por atropina ou isoprenalina.
Condução Atrioventricular
O início do tratamento com Kiendra® foi associado a atrasos transitórios da condução atrioventricular que seguem um padrão temporal semelhante ao observado na diminuição da frequência cardíaca durante a titulação da dose. Os atrasos na condução atrioventricular manifestam-se, na maioria dos casos, como bloqueios atrioventriculares (AV) de primeiro grau (intervalo PR prolongado no eletrocardiograma). Bloqueios AV de segundo grau, geralmente Mobitz tipo I (Wenckebach), foram observados em menos de 1,7% dos pacientes em estudos clínicos no momento do início do tratamento com Kiendra®. As anormalidades de condução eram normalmente transitórias, assintomáticas, resolvidas em 24 horas e não exigiam a descontinuação do tratamento com Kiendra®.
Recomendações de início do tratamento
O início do tratamento com Kiendra® com uma titulação da dose é geralmente bem tolerado (consulte aseção 8. POSOLOGIA E MODO DE USAR e seção 4. CONTRAINDICAÇÕES). Como medida de precaução, pacientes com:
• bradicardia sinusal (frequência cardíaca (FC) < 55 bpm),
• bloqueio atrioventricular (bloqueio AV) de primeiro ou segundo grau [Mobitz tipo I], • ou histórico de infarto do miocárdio ou insuficiência cardíaca (pacientes com NYHA classe I e II), se não contraindicado, devem ser observados um período de 6 horas após a primeira dose de Kiendra® quanto a sinais e sintomas de bradicardia. É recomendável fazer um eletrocardiograma (ECG) antes da dosagem e no final do período de observação nesses pacientes. Se ocorrer bradiarritmia pós-dose ou sintomas relacionados à condução ou se o ECG 6 horas após a dose mostrar novo bloqueio AV de segundo grau ou mais alto ou QTc ≥ 500 ms, um tratamento adequado deve ser iniciado e a observação deve continuar até que os sintomas/achados sejam resolvidos. Se for necessário tratamento farmacológico, a monitorização deveria ser continuada durante a noite e a monitorização de 6 horas deveria ser repetida após a segunda dose.
Devido ao risco de distúrbios graves do ritmo cardíaco ou bradicardia significativa, Kiendra® não deve ser usado em pacientes com síndrome do nódulo sinusal ou bloqueio cardíaco sinoatrial.
Como a bradicardia significativa pode ser mal tolerada em pacientes com histórico de parada cardíaca com início > 6 meses antes do início do tratamento com Kiendra®, doença cerebrovascular, hipertensão não controlada ou apneia do sono não tratada grave, Kiendra® não deve ser usado nesses pacientes. Se o tratamento for considerado, deve-se procurar aconselhamento de um cardiologista antes do início do tratamento, a fim de determinar a estratégia de monitoramento mais apropriada.
O uso de Kiendra® em pacientes com histórico de síncope recorrente ou bradicardia sintomática, hipertensão não controlada ou apneia do sono grave não tratada deve basear-se em uma avaliação geral de risco/benefício. Se o tratamento for considerado, deve-se procurar aconselhamento de um cardiologista antes do início do tratamento para determinar o monitoramento mais apropriado.
Um estudo completo do QT demonstrou que o Kiendra® não possui efeito significativo no prolongamento direto do intervalo QT, e o Kiendra® não está associado a um potencial arritmogênico relacionado ao prolongamento do intervalo QT. O início do tratamento com Kiendra® pode resultar em diminuição da frequência cardíaca e prolongamento indireto do intervalo QT durante a fase de titulação. Kiendra® não foi estudado em pacientes com prolongamento significativo do intervalo QT (QTc > 500 ms) ou que foram tratados com medicamentos que prolongam o intervalo QT. Se o tratamento com Kiendra® for considerado em pacientes com prolongamento significativo do intervalo QT preexistente ou que sejam tratados com medicamentos que prolongam o intervalo QT com propriedades arritmogênicas conhecidas, deve-se procurar aconselhamento de um cardiologista antes do início do tratamento para determinar a estratégia de monitoramento mais apropriada durante início do tratamento.
Kiendra® não foi estudado em pacientes com arritmias que precisam de tratamento com medicamentos antiarrítmicos de Classe Ia (por exemplo, quinidina, procainamida) ou Classe III (por exemplo, amiodarona, sotalol). Os fármacos antiarrítmicos de Classe Ia e Classe III têm sido associados a casos de Torsades