KEYTRUDA
MSD
pembrolizumabe
Anticorpo monoclonal.
Apresentações.
KEYTRUDATM
Solução injetável de -100 mg de pembrolizumabe em embalagem com 1 frasco-ampola com 4 mL de solução (25 mg/mL).
VIA INTRAVENOSA
USO ADULTO
Composição.
KEYTRUDATM Cada frasco-ampola contém 100 mg de pembrolizumabe em 4 mL de solução (25 mg/mL).
Excipientes: histidina, cloridrato de histidina monoidratado, sacarose, polissorbato 80 e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Melanoma
KEYTRUDATM (pembrolizumabe) é indicado como monoterapia para o tratamento de pacientes com melanoma metastático ou irressecável.
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
Melanoma
KEYNOTE-006: Estudo controlado em pacientes com melanoma sem tratamento prévio com ipilimumabe
A segurança e a eficácia de KEYTRUDATM foram avaliadas no KEYNOTE-006, um estudo de Fase III, multicêntrico e controlado do tratamento de melanoma metastático ou irressecável em pacientes sem tratamento prévio com ipilimumabe e que haviam recebido uma ou nenhuma terapia sistêmica prévia. Os pacientes foram distribuídos de forma randômica (1:1:1) para receber KEYTRUDATM, na dose de 10 mg/kg a cada duas (n = 279) ou três (n = 277) semanas, ou ipilimumabe (n = 278). A randomização foi estratificada por linha de terapia, ECOG performance status (PS) e status de expressão de PD-L1. O estudo excluiu pacientes com doença autoimune ou que recebiam imunossupressores; com hipersensibilidade grave prévia a outros anticorpos monoclonais; e com infecção por HIV, hepatite B ou hepatite C. Não se exigiu que os pacientes com melanoma com mutação BRAF V600E tivessem recebido terapia anterior com um inibidor de BRAF.
Os pacientes foram tratados com KEYTRUDATM até que houvesse progressão da doença ou toxicidade inaceitável. Pacientes clinicamente estáveis com evidência inicial de progressão da doença foram autorizados a permanecer sob tratamento até que a progressão da doença fosse confirmada. A avaliação do status do tumor foi realizada em 12 semanas, depois a cada 6 semanas até a semana 48, e, a partir de então, a cada 12 semanas.
Dos 834 pacientes no KEYNOTE-006, 60% eram homens, 44% tinham ≥ 65 anos (a mediana de idade foi de 62 anos [faixa de 18 a 89]) e 98% eram brancos. Sessenta e seis por cento não tinham recebido terapias sistêmicas prévias e, portanto, receberam a terapia do estudo como tratamento de primeira linha, enquanto 34% haviam recebido uma terapia prévia e, portanto, receberam a terapia do estudo como tratamento de segunda linha. Trinta e um por cento tinham um ECOG PS de 1 e 69% tinham um ECOG PS de 0. Oitenta por cento dos pacientes tiveram resultado positivo para PD-L1 (expressão de PD-L1 de membrana por ≥ 1% das células tumorais e das imuno-associadas, conforme avaliado prospectivamente por um ensaio imuno-histoquímico com o anticorpo anti-PDL1 22C3) e 18% tiveram resultado negativo para PD-L1. Sessenta e cinco por cento dos pacientes eram estadio M1c, 32% apresentavam LDH elevada e 9% tinham metástases cerebrais. As mutações do BRAF foram relatadas em 302 (36%) dos pacientes. Entre os pacientes com tumor com mutação BRAF, 139 (46%) haviam sido tratados previamente com um inibidor de BRAF. As características basais foram bem equilibradas entre os braços de tratamento.
As medidas do desfecho primário de eficácia foram sobrevida global (SG) e sobrevida livre de progressão (SLP; conforme avaliado por revisão da Integrated Radiology and Oncology Assessment (IRO) utilizando o Response Evaluation Criteria in Solid Tumor [RECIST 1.1]). As medidas do desfecho secundário de eficácia foram taxa global de resposta (TGR) e duração da resposta. A Tabela 1 resume as principais medidas de eficácia.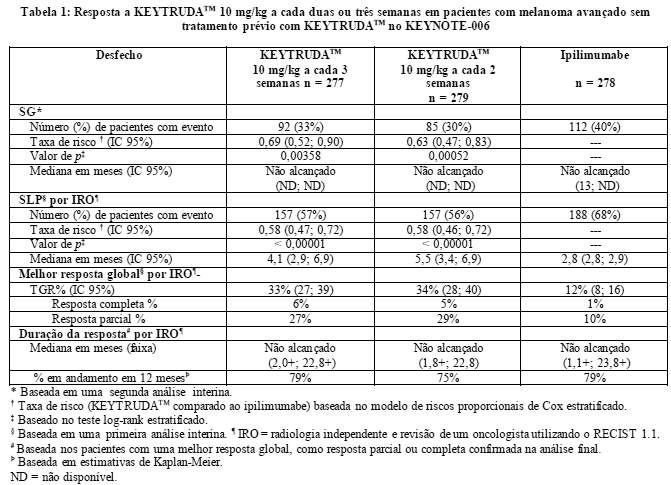
A análise final foi realizada após todos os pacientes terem concluído, pelo menos, 21 meses de acompanhamento. A análise final da SG foi realizada após a ocorrência de eventos com 383 pacientes, sendo 119 no grupo de KEYTRUDATM 10 mg/kg, a cada 3 semanas, 122 no de KEYTRUDATM 10 mg/kg, a cada 2 semanas, e 142 no do ipilimumabe. A TR da SG, versus a com o ipilimumabe, foi de 0,68 (IC 95%: 0,53, 0,86; p < 0,001) entre os pacientes tratados com KEYTRUDATM 10 mg/kg, a cada 3 semanas, e de 0,68 (IC 95%: 0,53, 0,87; p < 0,001) entre os tratados com KEYTRUDATM 10 mg/kg, a cada 2 semanas. As taxas de SG em 18 e 24 meses foram de 62% e 55%, respectivamente, com KEYTRUDATM 10 mg/kg, a cada 3 semanas; de 60% e 55%, respectivamente, com KEYTRUDATM 10 mg/kg, a cada 2 semanas, e de 47% e 43%, respectivamente, com o ipilimumabe. Na análise final, realizou-se uma avaliação de longa duração da SLP a partir de eventos com 566 pacientes, sendo 183 no grupo de KEYTRUDATM 10 mg/kg, a cada 3 semanas, 181 no de KEYTRUDATM 10 mg/kg, a cada 2 semanas, e 202 no do ipilimumabe). A TR da SLP, versus a com o ipilimumabe, foi de 0,61 (IC 95%: 0,50, 0,75) entre os pacientes tratados com KEYTRUDATM 10 mg/kg, a cada 3 semanas, e de 0,61 (IC 95%: 0,50, 0,75) entre os tratados com KEYTRUDATM10 mg/kg, a cada 2 semanas (veja as figuras 1 e 2). A porcentagem de respondedores com duração da resposta em 18 meses foi de 68% no grupo de KEYTRUDATM 10 mg/kg, a cada 3 semanas, de 71% no de KEYTRUDATM 10 mg/kg, a cada 2 semanas, e de 70% no do ipilimumabe.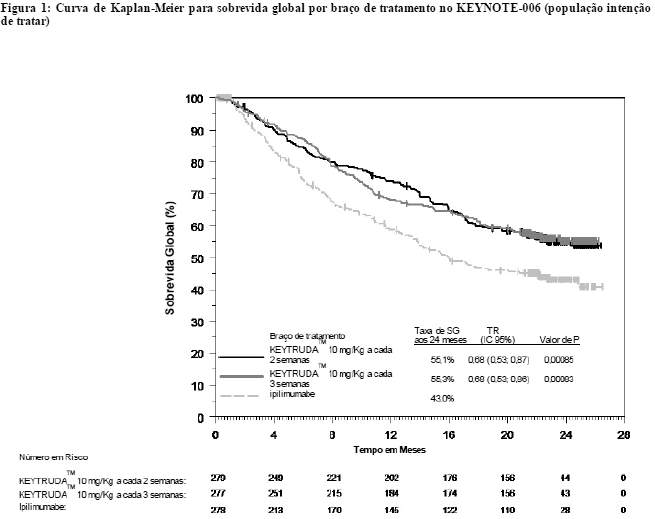
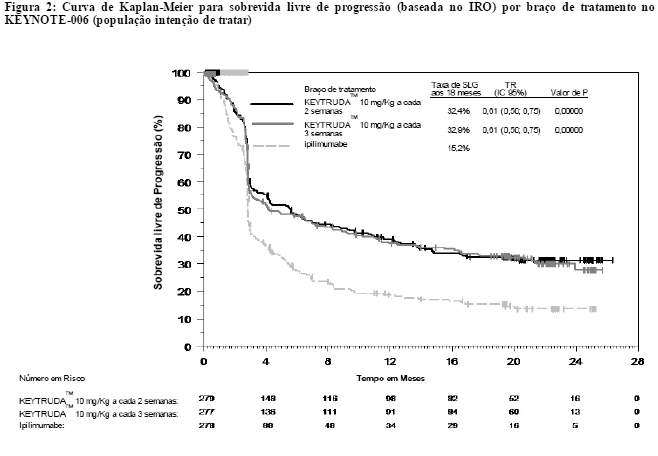
Análise de subpopulação por status de mutação BRAF
Realizou-se uma análise de subgrupo como parte da análise final do KEYNOTE-006 enter os pacientes com BRAF do tipo selvagem, os com mutação BRAF sem tratamento prévio com um inibidor de BRAF e os com mutação BRAF e tratamento prévio com um inibidor de BRAF. As taxas de risco (TRs) de SLP (KEYTRUDATM agrupado [10 mg/kg a cada duas ou três semanas] versus ipilimumabe) foram de 0,61 (IC 95%: 0,49; 0,76) para BRAF do tipo selvagem, 0,52 (IC 95%: 0,35; 0,78) para mutação BRAF sem tratamento prévio com inibidor de BRAF e 0,76 (IC 95%: 0,51; 1,14) para mutação BRAF com tratamento prévio com um inibidor de BRAF. As TRs de SG com KEYTRUDATM agrupado versus ipilimumabe foram de 0,68 (0,52; 0,88) para BRAF do tipo selvagem, 0,70 (IC 95%: 0,40; 1,22) para mutação BRAF sem tratamento prévio com inibidor de BRAF e 0,66 (IC 95%: 0,41; 1,04) para mutação BRAF com tratamento prévio com inibidor de BRAF. A TGR com KEYTRUDATM agrupado versus ipilimumabe foi de 38% versus 14% para BRAF do tipo selvagem, 41% versus 15% para mutação BRAF sem tratamento prévio com inibidor de BRAF e 24% versus 10% para mutação BRAF com tratamento prévio com inibidor de BRAF.
Análise de subpopulação por status PD-L1
Realizou-se uma análise de subgrupo como parte da análise final do KEYNOTE-006 em pacientes que eram PD-L1 positivo versus pacientes que eram PD-L1 negativo. As TRs de SLP (KEYTRUDATM agrupado [10 mg/kg a cada duas ou três semanas] versus ipilimumabe) foram de 0,53 (IC 95%: 0,44; 0,65) para pacientes que eram PD-L1 positivo e 0,87 (IC 95%: 0,58; 1,30) para pacientes que eram PD-L1 negativo. As TRs de SG com KEYTRUDATM agrupado versus ipilimumabe foram de 0,63 (IC 95%: 0,50; 0,80) para pacientes que eram PD-L1 positivo e 0,76 (IC 95%: 0,48; 1,19) para pacientes que eram PD-L1 negativo.
KEYNOTE-002: Estudo controlado de pacientes com melanoma previamente tratados com ipilimumabe
A segurança e a eficácia de KEYTRUDATM foram analisadas no KEYNOTE-002, um estudo multicêntrico e controlado do tratamento de melanoma metastático ou irressecável em pacientes previamente tratados com ipilimumabe e, se houvesse mutação BRAF V600 positiva, com um inibidor de BRAF ou MEK. Os pacientes foram distribuídos de forma randômica (1:1:1) para receber KEYTRUDATM na dose de 2 mg/kg (n = 180) ou 10 mg/kg (n = 181) a cada três semanas ou quimioterapia (n = 179; incluindo dacarbazina, temozolomida, carboplatina, paclitaxel ou carboplatina + paclitaxel). O estudo excluiu pacientes com doença autoimune ou que recebiam imunossupressores; pacientes com histórico de reações adversas imunomediadas graves ou com risco de morte com o tratamento com ipilimumabe, definido como qualquer toxicidade Grau 4 ou Grau 3 que requeira tratamento com corticosteroide (dose superior a 10 mg/dia de prednisona ou equivalente) por mais de 12 semanas; hipersensibilidade grave prévia a outros anticorpos monoclonais; histórico de pneumonia ou doença pulmonar intersticial; infecção por HIV, hepatite B ou hepatite C.
Os pacientes foram tratados com KEYTRUDATM até que houvesse progressão da doença ou toxicidade inaceitável. Pacientes clinicamente estáveis com evidência inicial de progressão da doença foram autorizados a permanecer sob tratamento até que a progressão da doença fosse confirmada. A avaliação do status do tumor foi realizada em 12 semanas, depois a cada 6 semanas até a semana 48, e, a partir de então, a cada 12 semanas. Pacientes em quimioterapia que tiveram progressão da doença verificada independentemente após a primeira avaliação agendada estavam aptos para participar da análise cruzada e receber 2 mg/kg ou 10 mg/kg de KEYTRUDATM a cada 3 semanas de forma duplo-cega.
Dos 540 pacientes no KEYNOTE-002, 61% eram homens, 43% tinham ≥ 65 anos (a mediana de idade foi de 62 anos [faixa de 15 a 89]) e 98% eram brancos. Oitenta e dois por cento dos pacientes eram estadio M1c, 73% tinham recebido, no mínimo, duas e 32% tinham recebido três ou mais terapias sistêmicas prévias para melanoma avançado. Quarenta e cinco por cento tinham um ECOG PS de 1, 40% apresentavam LDH elevada e 23% tinham um tumor com mutação BRAF. As características basais foram bem equilibradas entre os braços de tratamento.
As medidas do desfecho primário de eficácia foram SLP (conforme avaliado por revisão IRO utilizando RECIST 1.1) e SG. As medidas do desfecho secundário de eficácia foram SLP (de acordo com o avaliado pelo pesquisador utilizando RECIST 1.1), TGR e duração da resposta. A Tabela 2 resume as principais medidas de eficácia em pacientes previamente tratados com ipilimumabe, e a curva de Kaplan-Meier para SLP é mostrada na Figura 3. Não houve diferença estatisticamente significante entre KEYTRUDATM e a quimioterapia na análise finalda SG, que não foi ajustada para potenciais efeitos de confusão da análise cruzada. Dos pacientes distribuídos randomicamente para o braço de quimioterapia, 55% passaram por análise cruzada e receberam tratamento subsequente com KEYTRUDATM.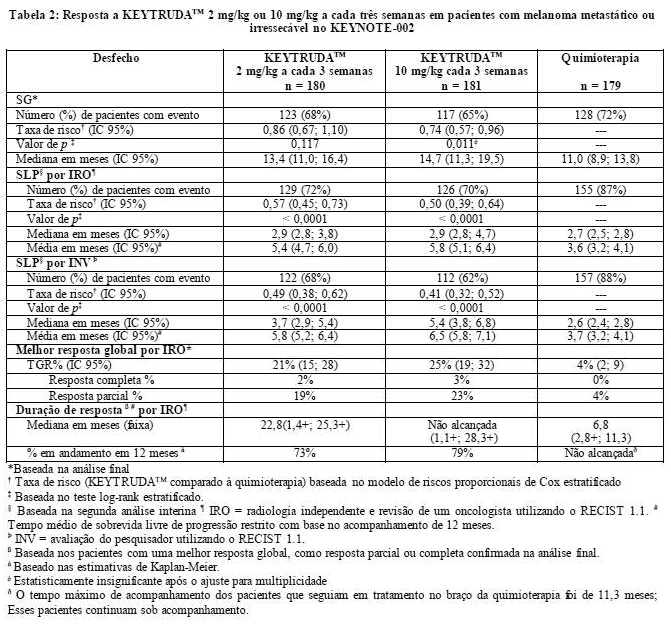
Na análise final, realizou-se uma avaliação de longa duração da SLP a partir de 466 eventos, sendo (150 com KEYTRUDATM 2 mg/kg a cada 3 semanas, 144 com KEYTRUDATM 10 mg/kg a cada 3 semanas e 172 com a quimioterapia). A TR da SLP, versus quimioterapia, foi de 0,58 (IC 95%: 0,46, 0,73) entre os pacientes tratados com KEYTRUDATM 2 mg/kg a cada 3 semanas e 0,47 (IC 95%: 0,37, 0,60) para pacientes tratados com KEYTRUDATM 10 mg/kg a cada 3 semanas (Figura 3).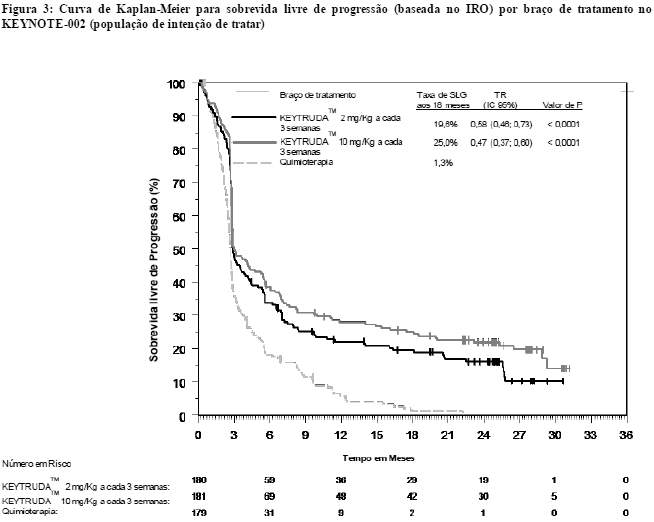
Status de mutação BRAF
Realizou-se uma análise de subgrupo do KEYNOTE-002 em pacientes com BRAF do tipo selvagem (n = 415; 77%) ou mutação BRAF com tratamento prévio com inibidor de BRAF (n = 125; 23%). As taxas de risco (TRs) de SLP (pembrolizumabe agrupado [2 mg/kg ou 10 mg/kg a cada três semanas] versus quimioterapia) foram de 0,51 (IC 95%: 0,41; 0,65) para BRAF do tipo selvagem e de 0,56 (IC 95%: 0,37; 0,85) para mutação BRAF com tratamento prévio com inibidor de BRAF. As TRs de SLP para pembrolizumabe 2 mg/kg a cada 3 semanas versus quimioterapia foram de 0,51 (IC 95%: 0,39; 0,67) para BRAF do tipo selvagem e 0,74 (IC 95%: 0,46; 1,18) para mutação BRAF com tratamento prévio com inibidor de BRAF. As TRs de SG para pembrolizumabe agrupado versus quimioterapia foram de 0,83 (IC 95%: 0,60; 1,15) para BRAF do tipo selvagem e 0,82 (IC 95%: 0,47; 1,43) para mutação BRAF com tratamento prévio com inibidor de BRAF. As TRs de SG para pembrolizumabe 2 mg/kg a cada 3 semanas versus quimioterapia foram de 0,80 (IC 95%: 0,55; 1,18) para BRAF do tipo selvagem e 1,03 (IC 95%: 0,55; 1,91) para mutação KEYTRUDA_BU13_022017a_VPS BRAF com tratamento prévio com inibidor de BRAF. A TGR para pembrolizumabe agrupado e pembrolizumabe 2 mg/kg a cada 3 semanas versus quimioterapia foi de 27% e 25% versus 6% para BRAF do tipo selvagem e 12% e 9% versus 0% para mutação BRAF com tratamento prévio com inibidor de BRAF.
Status de PD-L1
Realizou-se uma análise de subgrupo do KEYNOTE-002 em pacientes que eram PD-L1 positivo (escore de proporção Allred ≥ 2, representando expressão de membrana de PD-L1 em ≥ 1% das células tumorais) versus pacientes que eram PD-L1 negativo (escore de proporção Allred de 0 ou 1). A expressão de PD-L1 foi testada retrospectivamente por ensaio de pesquisa imuno-histoquímica com o anticorpo 22C3 anti-PD-L1. Entre os pacientes que eram avaliáveis para expressão de PD-L1 (78%), 69% (n = 291) eram PD-L1 positivo e 31% (n = 130) eram PD-L1 negativo. As TRs de SLP (pembrolizumabe agrupado [2 mg/kg ou 10 mg/kg a cada 3 semanas] versus quimioterapia) foram de 0,52 (IC 95%: 0,39; 0,68) para pacientes PD-L1 positivo e 0,60 (IC 95%: 0,38; 0,94) para pacientes PD-L1 negativo. As TRs de SLP para pembrolizumabe 2 mg/kg a cada 3 semanas versus quimioterapia foram de 0,54 (IC95%: 0,39;0,75) para pacientes PD-L1 positivo e0,89 (IC 95%: 0,53; 1,50) para pacientes PD-L1 negativo. As TRs de SG para pembrolizumabe agrupado versus quimioterapia foram de 0,82 (IC 95%: 0,55; 1,23) para pacientes PD-L1 positivo e 0,77 (IC 95%: 0,43; 1,37) para pacientes PD-L1 negativo. As TRs de SG para pembrolizumabe 2 mg/kg a cada 3 semanas versus quimioterapia foram de 0,93 (IC 95%: 0,58; 1,49) para pacientes PD-L1 positivo e 1,19 (IC 95%: 0,58; 2,46) para pacientes PD-L1 negativo. As TRGs para pembrolizumabe agrupado e para pembrolizumabe 2 mg/kg a cada 3 semanas versus quimioterapia foram de 26% e 23% versus 4% para pacientes PD-L1 positivo e 15% e 11% versus 8% para pacientes PD-L1 negativo.
KEYNOTE-001: Estudo aberto em pacientes com melanoma
A segurança e a eficácia de KEYTRUDATM foram também avaliadas em um estudo aberto não controlado do tratamento de melanoma metastático ou irressecável. A eficácia foi avaliada em 276 pacientes de duas coortes definidas do KEYNOTE-001, uma das quais incluiu pacientes previamente tratados com ipilimumabe (e, se houvesse mutação BRAF V600 positiva, com um inibidor de BRAF ou de MEK) e a outra que incluiu pacientes sem tratamento prévio com ipilimumabe. Os pacientes foram distribuídos de forma randômica para receber KEYTRUDATM na dose de 2 mg/kg a cada três semanas ou 10 mg/kg a cada três semanas. Os pacientes foram tratados com KEYTRUDATM até que houvesse progressão da doença ou toxicidade inaceitável. Pacientes clinicamente estáveis com evidência inicial de progressão da doença foram autorizados a permanecer sob tratamento até que a progressão da doença fosse confirmada. Os critérios de exclusão foram semelhantes aos do KEYNOTE-002.
Dos 89 pacientes recebendo 2 mg/kg de KEYTRUDATM que haviam sido previamente tratados com ipilimumabe, 53% eram homens e 33% tinham ≥ 65 anos de idade. A mediana de idade foi de 59 anos (faixa de 18 a 88). Todos, com exceção de dois pacientes, eram brancos. Oitenta e quatro por cento dos pacientes eram estadio M1c e 8% tinham histórico de metástases cerebrais. Setenta e oito por cento dos pacientes haviam recebido, no mínimo, duas e 35% haviam recebido três ou mais terapias sistêmicas prévias para melanoma avançado. As mutações do BRAF foram relatadas em 13% da população do estudo.
Dos 51 pacientes recebendo 2 mg/kg de KEYTRUDATM sem tratamento prévio com ipilimumabe, 63% eram homens e 35% tinham ≥ 65 anos de idade. A mediana de idade foi de 60 anos (faixa de 35 a 80). Todos, com exceção de um paciente, eram brancos. Sessenta e três por cento dos pacientes eram estadio M1c e 2% tinham histórico de metástases cerebrais. Quarenta e cinco por cento não haviam recebido terapias anteriores para melanoma avançado. As mutações do BRAF foram relatadas em 39% da população do estudo.
A medida do desfecho primário de eficácia foi TGR, conforme avaliada por revisão independente que utilizou respostas confirmadas e o RECIST 1.1. As medidas do desfecho secundário de eficácia foram taxa de controle de doença (TCD; incluindo resposta completa, resposta parcial e doença estável), duração da resposta, SLP e SG. A resposta tumoral foi avaliada em intervalos de 12 semanas. A Tabela 3 resume as principais medidas de eficácia em pacientes previamente tratados ou sem tratamento prévio com ipilimumabe que receberam KEYTRUDATM na dose recomendada com base em um período mínimo de acompanhamento de 30 meses para todos os pacientes.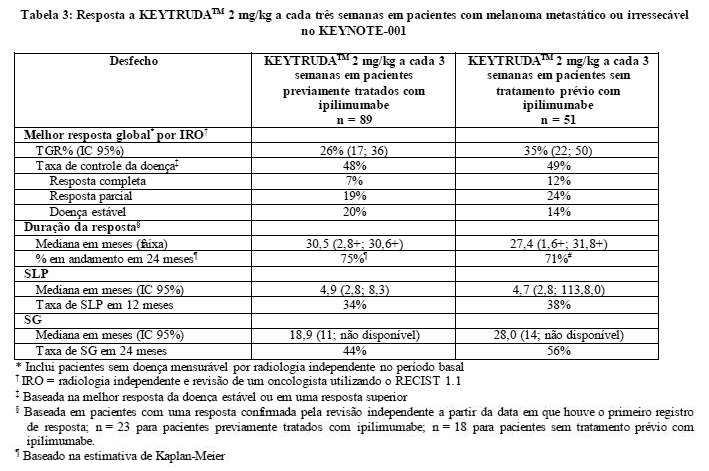
Os resultados para os pacientes previamente tratados com ipilimumabe (n = 84) e os sem tratamento prévio com ipilimumabe (n = 52) que receberam 10 mg/kg de KEYTRUDATM a cada três semanas foram semelhantes aos observados em pacientes que receberam 2 mg/kg de KEYTRUDATM a cada três semanas.
Melanoma ocular
Existem dados limitados sobre a segurança e a eficácia de KEYTRUDA™ em pacientes com melanoma ocular. Em 20 indivíduos com melanoma ocular incluídos no KEYNOTE-001, não foram relatadas respostas objetivas; doença estável foi relatada em 6 pacientes.
Imunogenicidade
Nos estudos clínicos em 1.819 pacientes tratados com pembrolizumabe em uma dose de 2 mg/kg a cada três semanas ou de 10 mg/kg a cada duas ou três semanas, um (0,3%) dos 392 pacientes avaliáveis teve resultado positivo em exame para anticorpos contra pembrolizumabe emergentes do tratamento com KEYTRUDATM. Neste caso específico, descobriu-se que os anticorpos agiam como neutralizadores contra o pembrolizumabe, sem sequela clínica aparente.
No subgrupo de pacientes tratados com o regime de dose de 2 mg/kg a cada três semanas, nenhum dos 225 pacientes avaliáveis teve resultado positivo em exame para anticorpos contra pembrolizumabe emergentes do tratamento durante a terapia com KEYTRUDATM.
3. CARACTERÍSTICAS FARMACOLÓGICAS
KEYTRUDATM (pembrolizumabe) é um anticorpo monoclonal humanizado seletivo desenhado para bloquear a interação entre a PD-1 e os seus ligantes, PD-L1 e PD-L2. O pembrolizumabe é uma imunoglobulina kappa IgG4 com um peso molecular aproximado de 149 kDa.
Farmacologia clínica
Classe terapêutica
KEYTRUDATM (pembrolizumabe) é um agente antineoplásico, um anticorpo monoclonal.
Mecanismo de ação
O PD-1 é um checkpoint (receptor) imunológico que limita a atividade das células (linfócitos) T nos tecidos periféricos. A via PD-1 é um checkpoint de controle imunológico que pode ser acoplado pelas células tumorais para inibir a vigilância imunológica da célula T ativa. KEYTRUDATM é um anticorpo de alta afinidade contra a PD-1, que exerce bloqueio ligante duplo da via PD-1, inclusive dos PD-L1 e PD-L2, no antígeno existente ou nas células tumorais. Ao bloquear a ligação entre PD-1 e seus ligantes, KEYTRUDATM reativa os linfócitos T citotóxicos específicos do tumor no microambiente tumoral e a imunidade antitumoral.
Farmacodinâmica
No sangue periférico de pacientes que receberam KEYTRUDATM 2 mg/kg a cada três semanas ou 10 mg/kg a cada duas ou três semanas, uma percentagem aumentada de células T CD4+ e CD8+ ativadas (por exemplo, HLA-DR+) foi observada após tratamento com todas as doses e em todos os cronogramas, sem aumento no número de linfócitos T circulantes.
Farmacocinética
A farmacocinética do pembrolizumabe foi estudada em 2.195 pacientes com melanoma irressecável ou metastático, CPNPC (câncer de pulmão não pequenas células) ou outros carcinomas, que receberam doses na faixa de 1 a 10 mg/kg a cada duas ou três semanas.
Absorção
KEYTRUDATM é administrado por via intravenosa e, portanto, é biodisponível imediata e completamente.
Distribuição
O volume de distribuição de pembrolizumabe no estado de equilíbrio é pequeno (~7 L; coeficiente de variação [CV]: 19%), o que é consistente com uma distribuição extravascular limitada. Como esperado de um anticorpo, o pembrolizumabe não se liga às proteínas plasmáticas de modo específico.
Metabolismo
O pembrolizumabe é catabolizado por vias não específicas; o metabolismo não contribui para a sua depuração.
Eliminação
A depuração sistêmica do pembrolizumabe é de ~0,2 L/dia (CV: 37%) e a meia-vida terminal (t1/2) é de ~27 dias (CV: 38%).
A exposição ao pembrolizumabe tal como expressada pelo pico de concentração (Cmáx) ou pela área sob a curva (AUC) de tempo de concentração plasmática aumentou proporcionalmente a dosagem dentro da faixa de dose para eficácia. Em dosagem repetida, a depuração do pembrolizumabe foi considerada independente do tempo, e o acúmulo sistêmico foi de aproximadamente 2,2 vezes quando pembrolizumabe foi administrado a cada três semanas. Concentrações de pembrolizumabe próximas ao estado de equilíbrio foram alcançadas em 19 semanas; a Cmin média em 19 semanas foi de aproximadamente 26 mcg/mL na dose de 2 mg/kg a cada três semanas.
Populações especiais
Os efeitos de várias covariáveis na farmacocinética do pembrolizumabe foram avaliados em análises de farmacocinética populacional. A depuração do pembrolizumabe elevou com o aumento do peso corpóreo; diferenças na exposição resultante são tratadas pela administração na base de mg/kg. Os seguintes fatores não tiveram efeito clinicamente importante na depuração do pembrolizumabe: idade (faixa de 15 a 94 anos), sexo, raça, insuficiência renal leve ou moderada, insuficiência hepática leve e carga tumoral.
Insuficiência renal
O efeito da insuficiência renal na depuração do pembrolizumabe foi avaliado pela análise de farmacocinética populacional em pacientes com insuficiência renal leve (TFG < 90 e ≥ 60 mL/min/1,73 m2; n = 937) ou moderada (TFG < 60 e ≥ 30 mL/min/1,73 m2; n = 201) comparados a pacientes com função renal normal (TFG ≥ 90 mL/min/1,73 m2; n = 1.027). Diferenças não clinicamente importantes na depuração do pembrolizumabe foram identificadas entre pacientes com insuficiência renal leve ou moderada e pacientes com função renal normal em ambas as populações. KEYTRUDATM não foi estudado em pacientes com insuficiência renal grave (TFG < 30 e ≥ 15 mL/min/1,73 m2) (veja 8. POSOLOGIA E MODO DE USAR).
Insuficiência hepática
O efeito da insuficiência hepática na depuração do pembrolizumabe foi avaliado pela análise de farmacocinética populacional em pacientes com insuficiência hepática leve (bilirrubinatotal (BT) 1,0 a 1,5 x LSN ou AST > LSN conforme definido usando o critério de disfunção hepática do National Cancer Institute; n = 269) comparados a pacientes com função hepática normal (BT e AST ≤ LSN; n = 1.871). Diferenças não clinicamente importantes na depuração do pembrolizumabe foram identificadas entre pacientes com insuficiência hepática leve e função hepática normal em ambas as populações. KEYTRUDATM não foi estudado em pacientes com insuficiência hepática moderada (BT > 1,5 a 3 x LSN e qualquer AST) ou grave (BT > 3 x LSN e qualquer AST) (veja 8. POSOLOGIA E MODO DE USAR).
Toxicologia animal
Toxicidade crônica
A segurança do pembrolizumabe foi avaliada em um estudo de toxicidade de dose repetida de 1 mês e em um de 6 meses em macacos Cynomolgus, com doses de 6, 40 ou 200 mg/kg administradas por via IV, uma vez por semana, no estudo de um mês, e uma vez a cada duas semanas no estudo de 6 meses, seguidas por um período de 4 meses sem tratamento. Não foram observados achados de significância toxicológica e o nível de efeito adverso não observado (No Observed Adverse Effect Level - NOAEL) em ambos os estudos foi de ≥ 200 mg/kg, o que é 19 vezes a exposição em humanos na dose mais alta testada clinicamente (10 mg/kg) e 94 vezes a exposição em humanos na dose recomendada (2 mg/kg).
Carcinogênese
O potencial carcinogênico do pembrolizumabe não foi avaliado em estudos animais de longa duração.
Mutagênese
A potencial genotoxicidade do pembrolizumabe não foi avaliada.
Reprodução
Estudos de reprodução animal não foram conduzidos com KEYTRUDATM. A função central da via anti PD-1/PD-L1 é preservar a gravidez pela manutenção da tolerância imunológica ao feto. O bloqueio da sinalização do PD-L1 demonstrou interromper a tolerância ao feto e resultar no aumento de perda fetal em modelos murídeos de gravidez. Esses resultados indicam um risco potencial de que a administração de KEYTRUDATM durante a gravidez possa causar danos fetais, incluindo aumento nas taxas de aborto ou de natimorto.
Desenvolvimento
Estudos de toxicidade do desenvolvimento não foram conduzidos com o pembrolizumabe. Com base em estudos de toxicidade com doses repetidas de 1 mês e 6 meses em macacos, não houve efeitos notáveis em órgãos reprodutivos masculinos e femininos.
4. CONTRAINDICAÇÕES
KEYTRUDATM é contraindicado para pacientes com hipersensibilidade grave ao pembrolizumabe ou a qualquer um de seus ingredientes inativos.
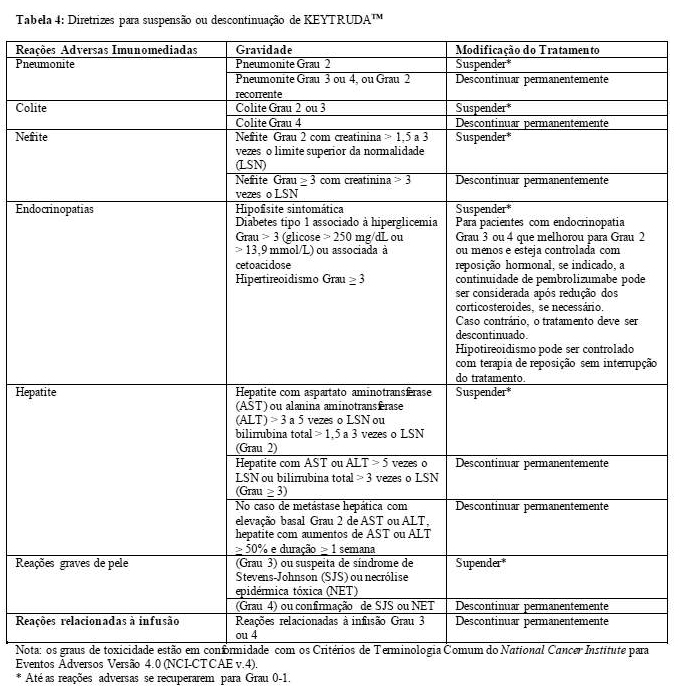
5. ADVERTÊNCIAS E PRECAUÇÕES
Reações adversas imunomediadas
Reações adversas imunomediadas ocorreram em pacientes que receberam KEYTRUDATM. Em ensaios clínicos, a maioria das reações adversas imunomediadas foi reversível e controlada com interrupção de KEYTRUDATM, administração de corticosteroides e/ou cuidados de suporte. Reações adversas imunomediadas que afetam mais de um sistema corporal podem ocorrer simultaneamente.
Em caso de suspeita de reações adversas imunomediadas, deve-se assegurar uma avaliação adequada para confirmar etiologia ou excluir outras causas. Dependendo da gravidade da reação adversa, não ministre KEYTRUDATM e considere a administração de corticosteroides. Ao haver melhora para Grau 1 ou inferior, deve-se iniciar a redução do corticosteroide e continuar a diminuição ao longo de pelo menos um mês. De acordo com os dados limitados dos estudos clínicos, em pacientes cujas reações adversas imunorrelacionadas não puderam ser controladas com uso de corticosteroides, a administração de outros imunossupressores sistêmicos pode ser considerada. Reiniciar KEYTRUDATM se a reação adversa permanecer no Grau 1 ou inferior. Se outro episódio de uma reação adversa grave ocorrer, deve-se descontinuar KEYTRUDATM permanentemente (veja 8. POSOLOGIA E MODO DE USAR e 9. REAÇÕES
ADVERSAS). KEYTRUDATM pode ser reiniciado no prazo de 12 semanas após sua última dose se a reação adversa permanecer ≤ Grau 1 e a dose de corticosteroide for reduzida para ≤ 10 mg de prednisona ou equivalente por dia.
Pneumonite imunomediada
Relatou-se pneumonite (incluindo um caso fatal [Grau 5] em um estudo envolvendo 550 pacientes com carcinoma de pulmão de células não pequenas) em pacientes recebendo KEYTRUDATM (veja 9. REAÇÕES ADVERSAS). Monitorar os pacientes quanto a sinais e sintomas de pneumonite. Se houver suspeita de pneumonite, avaliar com imagem radiográfica e excluir outras causas. Administrar corticosteroides em caso de Grau 2 ou maior (dose inicial de 1 a 2 mg/kg/dia de prednisona ou equivalente seguida de redução), suspender KEYTRUDATM em caso de pneumonite moderada (Grau 2) e descontinuar KEYTRUDATM permanentemente em caso de pneumonite grave (Grau 3), com risco de morte (Grau 4) ou moderada recorrente (Grau 2) (veja 8. POSOLOGIA E MODO DE USAR e 5. ADVERTÊNCIAS E PRECAUÇÕES -Reações adversas imunomediadas).
Colite imunomediada
Relatou-se colite em pacientes que receberam KEYTRUDATM (veja 9. REAÇÕES ADVERSAS). Monitorar os pacientes quanto a sinais e sintomas de colite e excluir outras causas. Administrar corticosteroides em caso de Grau 2 ou maior (dose inicial de 1 a 2 mg/kg/dia de prednisona ou equivalente seguida de uma redução), suspender KEYTRUDATM em caso de colite moderada (Grau 2) ou grave (Grau 3) e descontinuar KEYTRUDATM permanentemente em caso de colite com risco de morte (Grau 4) (veja 8. POSOLOGIA E MODO DE USAR e 5. ADVERTÊNCIAS E PRECAUÇÕES -Reações adversas imunomediadas). O risco de perfuração gastrintestinal deve ser considerado.
Hepatite imunomediada
Relatou-se hepatite em pacientes que receberam KEYTRUDATM (veja 9. REAÇÕES ADVERSAS). Monitorar os pacientes quanto a alterações na função hepática (no início do tratamento, periodicamente durante o tratamento e conforme indicado com base na avaliação clínica) e sintomas de hepatite, e descartar outras causas. Administrar corticosteroides (dose inicial de 0,5 a 1 mg/kg/dia [para eventos Grau 2] e 1 a 2 mg/kg/dia [para Grau 3 ou eventos maiores] de prednisona ou equivalente seguida de uma redução) e, com base na gravidade das elevações da enzima hepática, suspender ou descontinuar KEYTRUDATM (veja 8. POSOLOGIA E MODO DE USAR e 5. ADVERTÊNCIAS E PRECAUÇÕES -Reações adversas imunomediadas).
Nefrite imunomediada
Relatou-se nefrite em pacientes que receberam KEYTRUDATM (veja 9. REAÇÕES ADVERSAS). Monitorar os pacientes quanto a alterações na função renal e excluir outras causas. Administrar corticosteroides em caso de Grau 2 ou maior (dose inicial de 1 a 2 mg/kg/dia de prednisona ou equivalente seguida de uma redução), suspender KEYTRUDATM em caso de nefrite moderada (Grau 2) e descontinuar KEYTRUDATM permanentemente em caso de nefrite grave ou com risco de morte (Grau 4) (veja 8. POSOLOGIA E MODO DE USAR e 5. ADVERTÊNCIAS E PRECAUÇÕES -Reações adversas imunomediadas).
Endocrinopatias imunomediadas
Relatou-se hipofisite em pacientes que receberam KEYTRUDATM (veja 9. REAÇÕES ADVERSAS). Monitorar os pacientes quanto a sinais e sintomas de hipofisite (incluindo hipopituitarismo e insuficiência adrenal secundária) e excluir outras causas. Administrar corticosteroides para tratar insuficiência adrenal secundária e reposição hormonal adicional conforme indicado clinicamente, suspender KEYTRUDATM em caso de hipofisite moderada (Grau 2), suspender ou descontinuar KEYTRUDATM em caso de hipofisite grave (Grau 3) ou com risco de morte (Grau 4) (veja 8. POSOLOGIA E MODO DE USAR e 5. ADVERTÊNCIAS E PRECAUÇÕES -Reações adversas imunomediadas).
Relatou-se diabetes mellitus tipo 1, incluindo cetoacidose diabética, em pacientes que receberam KEYTRUDATM (veja 9. REAÇÕES ADVERSAS). Monitorar os pacientes quanto a hiperglicemia ou outros sinais e sintomas de diabetes. Administrar insulina em caso de diabetes tipo 1 e suspender KEYTRUDATM em caso de hiperglicemia grave até atingir o controle metabólico.
Distúrbios tireoideanos foram relatados em pacientes que receberam KEYTRUDATM, e podem ocorrer a qualquer momento no tratamento; portanto, monitorar os pacientes quanto a alterações na função tireoideana (no início do tratamento, periodicamente durante o tratamento e conforme indicado com base na avaliação clínica) e sinais e sintomas clínicos de distúrbios tireoideanos. O hipotireoidismo pode ser controlado com terapia de reposição sem interrupção do tratamento e sem corticosteroides. O hipertireoidismo pode ser controlado sintomaticamente. Suspender ou descontinuar KEYTRUDATM em caso de hipertireoidismo grave (Grau 3) ou com risco de morte (Grau 4) (veja 8. POSOLOGIA E MODO DE USAR, 9. REAÇÕES ADVERSAS, 5. ADVERTÊNCIAS E PRECAUÇÕES -Reações adversas imunomediadas).
A continuação do uso de KEYTRUDATM pode ser considerada para pacientes com endocrinopatia grave (Grau 3) ou com risco de morte (Grau 4) que melhorar para Grau 2 ou inferior e que estiver controlada com reposição hormonal.
Reações graves de pele
Reações graves de pele imunomediadas foram reportadas em pacientes tratados com KEYTRUDATM. Pacientes com suspeita de reações graves de pele devem ser monitorados e outras causas devem ser excluídas. Baseada na gravidade da reação adversa, suspender ou descontinuar permanentemente o tratamento com KEYTRUDATM e administrar corticosteróides (veja 8. POSOLOGIA E MODO DE USAR).
Foram reportados casos de síndrome de Stevens-Johnson (SJS) e necrólise epidérmica tóxica (NET), alguns casos com desfechos fatais, em pacientes tratados com KEYTRUDATM. Em caso de sinais ou sintomas de SJS ou NET, suspender o tratamento com KEYTRUDATM e encaminhar o paciente para atendimento especializado para avaliação e tratamento. Em caso de confirmação de SJS ou NET, descontinuar permanentemente o tratamento com KEYTRUDATM (veja 8. POSOLOGIA E MODO DE USAR).
Outras reações adversas imunomediadas
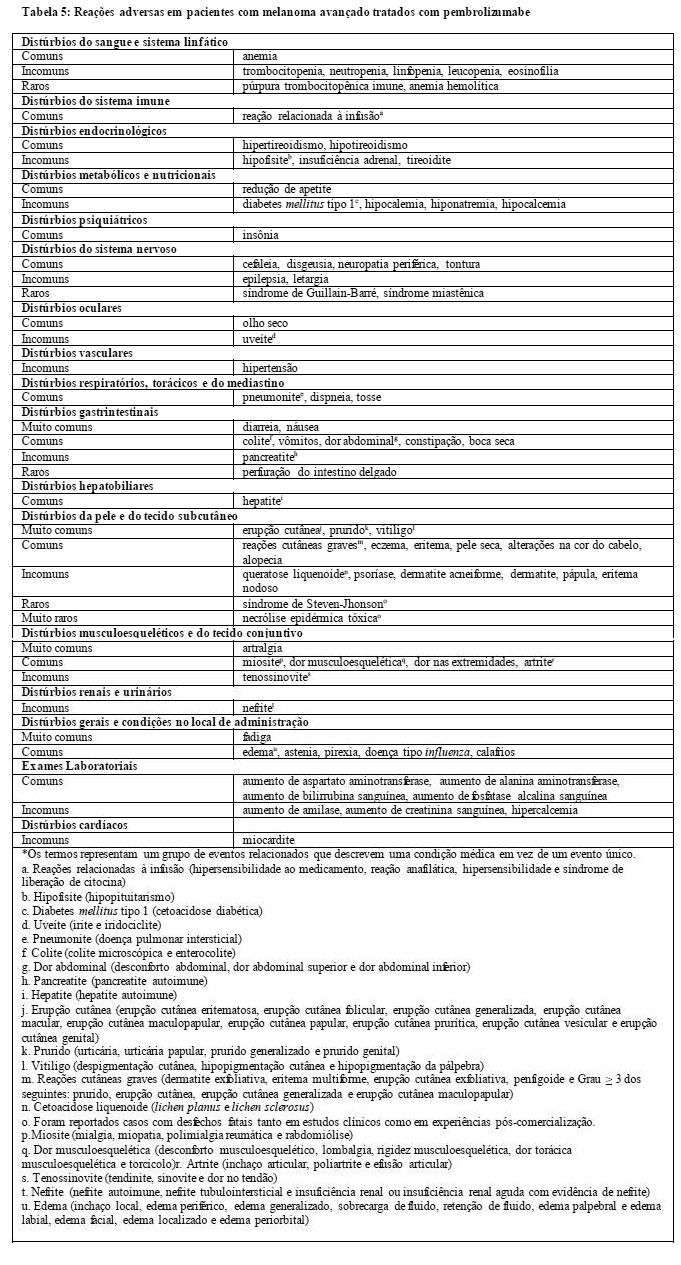
As seguintes reações adversas imunomediadas clinicamente significantes adicionais foram relatadas em menos de 1% de pacientes tratados com KEYTRUDATM nos estudos KEYNOTE-001, KEYNOTE-002 e KEYNOTE-006: uveíte, miosite, síndrome de Guillain-Barré e pancreatite.
Têm sido relatados casos dessas reações adversas imunomediadas, alguns dos quais graves, em estudos clínicos ou no uso pós comercialização. A seguinte reação adversa foi reportada em outros estudos clínicos com KEYTRUDA™ ou em uso pós comercialização: miocardite.
Rejeição de transplante de órgãos sólidos foi relatada no uso pós-comercialização em pacientes tratados com KEYTRUDATM. O tratamento com KEYTRUDATM pode aumentar o risco de rejeição em receptores de transplante de órgãos sólidos. Considere o benefício do tratamento com KEYTRUDATM versus o risco da possibilidade de rejeição de órgãos nesses pacientes
Reações relacionadas com a infusão
Reações graves à infusão, incluindo hipersensibilidade e anafilaxia, foram relatadas em 2 (0,1%) de 1.567 pacientes que receberam KEYTRUDATM nos estudos KEYNOTE-001, KEYNOTE-002 e KEYNOTE-006. Em caso de reações graves à infusão, parar a infusão e descontinuar KEYTRUDATM permanentemente (veja 8. POSOLOGIA E MODO DE USAR). Os pacientes com reação leve ou moderada à infusão podem continuar a receber KEYTRUDATM, desde que mantidos sob cuidadosa observação; pode-se considerar a pré-medicação com antipirético e anti-histamínico.
Uso em população específica
Gravidez
Categoria D.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Não há dados sobre o uso de pembrolizumabe em mulheres grávidas. Estudos de reprodução animal não foram conduzidos com pembrolizumabe; entretanto, o bloqueio da sinalização do PD-L1 demonstrou interromper a tolerância ao feto e resultar no aumento de perda fetal em modelos murídeos de gravidez. Esses resultados indicam um risco potencial, baseado no seu mecanismo de ação, que a administração de KEYTRUDATM durante a gravidez possa causar danos fetais, incluindo taxas aumentadas de aborto ou de natimorto. A IgG4 (imunoglobulina) humana é conhecida por atravessar a barreira placentária, e o pembrolizumabe é um IgG4; assim, o pembrolizumabe tem o potencial de ser transmitido da mãe para o feto em desenvolvimento. KEYTRUDATM não é recomendado durante a gravidez a menos que o benefício clínico supere o risco potencial para o feto. Mulheres em idade fértil devem utilizar métodos anticoncepcionais efetivos durante o tratamento com KEYTRUDATM e por pelo menos 4 meses após a última dose de KEYTRUDATM.
Lactantes
Desconhece-se se KEYTRUDATM é secretado pelo leite humano. Como muitos fármacos são excretados no leite humano, deve-se decidir entre descontinuar a amamentação ou KEYTRUDATM, levando em conta o benefício da amamentação para a criança e o benefício do tratamento com KEYTRUDATM para a mulher.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
O pembrolizumabe pode apresentar uma pequena influência sobre a capacidade de dirigir e operar máquinas. Fadiga tem sido relatada após a administração de pembrolizumabe.
Atenção, diabéticos: contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
Estudos formais de interação farmacocinética de fármacos não foram conduzidos com KEYTRUDATM. Uma vez que o pembrolizumabe é eliminado da circulação pelo catabolismo, interações medicamentosas metabólicas não são esperadas. O uso de corticosteroides sistêmicos ou imunossupressores antes de iniciar o tratamento com KEYTRUDATM deve ser evitado por causa de suas potenciais interferências na atividade farmacodinâmica e na eficácia de KEYTRUDATM. Entretanto, corticosteroides sistêmicos ou outros imunossupressores podem ser usados depois do início de KEYTRUDATM para tratar reações adversas imunomediadas (veja 5. ADVERTÊNCIAS E PRECAUÇÕES).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar sob refrigeração (entre 2 e 8°C).
Proteger da luz. Não congelar. Não agitar.
Para condições de armazenamento após diluição do produto, veja 8. POSOLOGIA E MODO DE USAR.
O prazo de validade do medicamento é de 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
KEYTRUDATM apresenta-se na forma de solução límpida a levemente opalescente e incolor a levemente amarela. Descartar o medicamento caso note partículas visíveis nele.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Dose recomendada
A dose recomendada de KEYTRUDATM é de 2 mg/kg administrada por via intravenosa durante 30 minutos a cada três semanas. Os pacientes devem ser tratados com KEYTRUDATM até que haja progressão da doença ou toxicidade inaceitável. Respostas atípicas (por exemplo, um aumento inicial transitório no tamanho do tumor ou pequenas lesões novas, nos primeiros meses, seguidas por encolhimento do tumor) foram observadas. Pacientes clinicamente estáveis com evidência inicial de progressão da doença devem ser mantidos sob tratamento até a progressão da doença ser confirmada