KADCYLA
ROCHE
trastuzumabe
Antineoplásico.
Apresentações.
Kadcyla® 100 mg e 160 mg. Pó liofilizado para solução injetável. Cada embalagem contém um frasco-ampola de uso único com 100 mg ou 160 mg de pó liofilizado de trastuzumabe entansina para solução injetável para infusão via intravenosa após reconstituição e diluição.
INFUSÃO VIA INTRAVENOSA
USO ADULTO
Composição.
Kadcyla® 100 mg. Princípio ativo: cada frasco-ampola de uso único contém 100 mg de pó liofilizado de trastuzumabe entansina para solução injetável, destinado a veicular 5 mL (20 mg/mL). Kadcyla® 160 mg. Princípio ativo: cada frasco-ampola de uso único contém 160 mg de pó liofilizado de trastuzumabe entansina para solução injetável, destinado a veicular 8 mL (20 mg/mL). Excipientes: sacarose, ácido succínico, hidróxido de sódio e polissorbato 20.
Indicações.
Câncer de mama metastático
Kadcyla® é indicado em monoterapia para tratamento de pacientes com câncer de mama HER2-positivo metastático ou localmente avançado não ressecável, que tenham recebido tratamento prévio com trastuzumabe e um taxano.
Resultados de eficácia.
Câncer de mama metastático
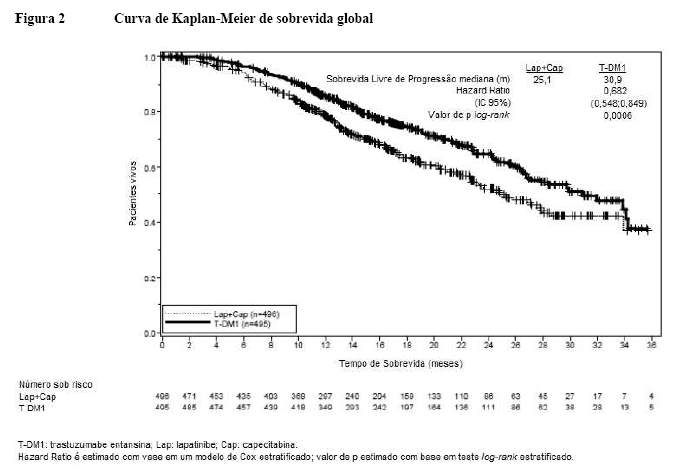
Um estudo clínico fase III, randomizado, multicêntrico, internacional, aberto (TDM4370g/BO21977) foi conduzido com pacientes com câncer de mama HER2-positivo metastático ou localmente avançado não ressecável que receberam terapia prévia à base de taxano e trastuzumabe, incluindo pacientes que receberam terapia prévia com trastuzumabe e um taxano como adjuvantes e que apresentaram recidiva dentro de seis meses após completarem a terapia adjuvante. Antes da inclusão, era necessário que amostras de tumor de mama fossem confirmadas como doença HER2-positiva, definida pelo escore de 3+ por IHQ (imunohistoquímica) ou amplificação genética por ISH (hibridização in situ). Características basais dos pacientes e do tumor foram bem equilibradas entre os grupos de tratamento. Para pacientes randomizados para Kadcyla®, a idade mediana foi de 53 anos, a maioria dos pacientes era mulheres (99,8%), a maioria era branca (72%) e 57% apresentavam doença positiva para receptor de estrógeno e/ou progesterona. O estudo comparou a segurança e a eficácia de Kadcyla® com a de lapatinibe mais capecitabina. No total, 991 pacientes foram randomizados para Kadcyla® ou lapatinibe mais capecitabina como vemos a seguir1:
• Braço Kadcyla®: 3,6 mg/kg via intravenosa (IV), em 30 a 90 minutos no Dia 1, de um ciclo de 21 dias,
• Braço controle (lapatinibe mais capecitabina): lapatinibe 1250 mg/dia via oral, uma vez por dia, de um ciclo de 21 dias, mais capecitabina 1000 mg/m2 via oral, duas vezes por dia, nos Dias 1 a 14 de um ciclo de 21 dias.
O tempo até progressão dos sintomas, definido por uma redução de 5 pontos no escore derivado do índice de evolução dos estudos - subescala mama (TOI-B) do questionário de Avaliação Funcional de Terapia de Câncer de Mama - Qualidade de Vida (FACT-B QoL) também foi avaliado durante o estudo clínico. Uma alteração de 5 pontos em TOI-B é considerada clinicamente significativa.1 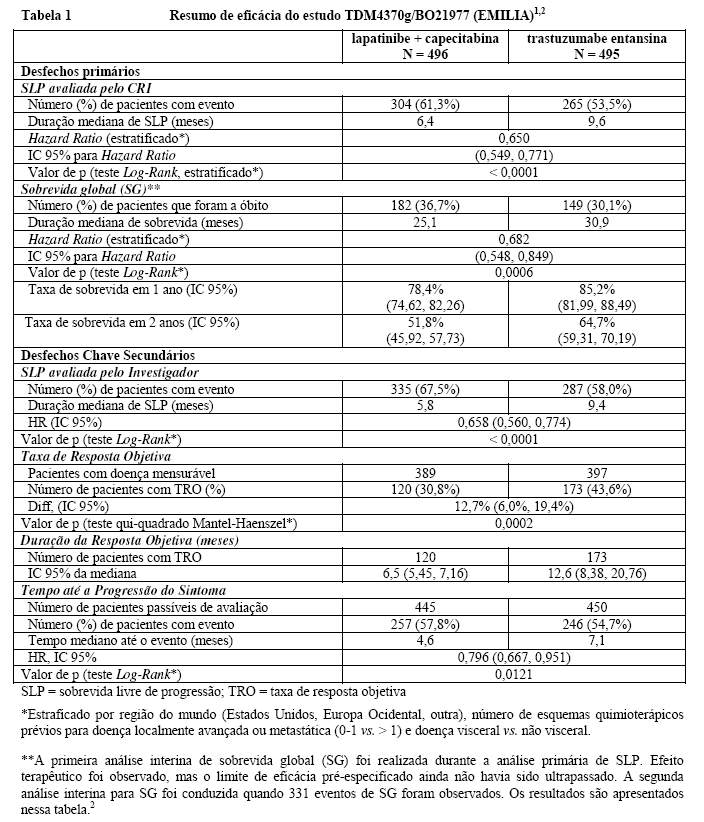
O benefício do tratamento observado no subgrupo de pacientes que não havia recebido, anteriormente, nenhuma terapia antineoplásica sistêmica no contexto metastático (n = 118), com hazard ratio para SLP de 0,51 (IC 95%: 0,30, 0,85) e para SG 0,661 (IC 95%: 0,32, 1,16). A mediana de SLP foi de 10,8 meses e de SG ainda não foi atingida para o grupo com Kadcyla®, quando comparadas com os 5,7 e 27,9 meses, respectivamente, para o grupo lapatinibe e capecitabina.1,2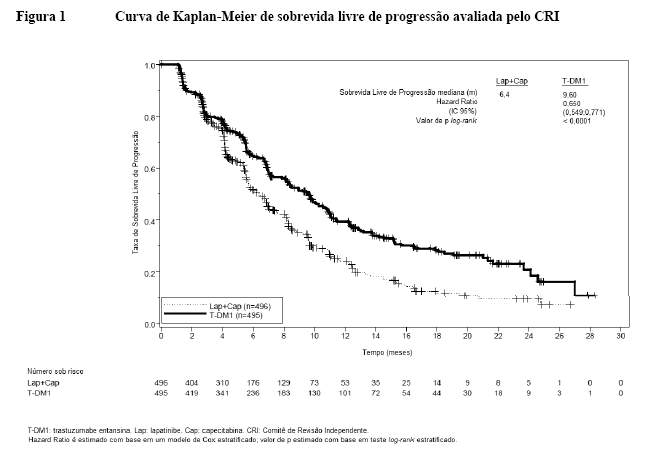
Um estudo randomizado, multicêntrico, aberto, fase II (TDM4450g/BO21976) avaliou os efeitos de Kadcyla® versus trastuzumabe mais docetaxel em pacientes com câncer de mama HER2-positivo metastático que não receberam quimioterapia prévia para doença metastática. Os pacientes foram randomizados para receber Kadcyla® 3,6 mg/kg IV a cada 3 semanas (n = 67) ou trastuzumabe 8 mg/kg IV em dose de ataque seguida por 6 mg/kg IV a cada 3 semanas mais docetaxel 75-100 mg/m2 IV a cada 3 semanas (n = 70).3
O desfecho primário foi SLP avaliada pelo investigador. A SLP mediana foi de 9,2 meses no braço trastuzumabe mais docetaxel e 14,2 meses no braço Kadcyla® (hazard ratio, 0,59; p = 0,035), com acompanhamento mediano de aproximadamente 14 meses nos dois braços. A TRO (taxa de resposta objetiva) foi de 58,0%, com trastuzumabe mais docetaxel e 64,2% com Kadcyla®. A duração mediana de resposta no braço controle foi de 9,5 meses e ainda não foi atingida com Kadcyla® .3
A queda dos escores FACT-B TOI foi menor no braço com Kadcyla® em comparação com o braço controle (tempo mediano até a progressão dos sintomas foi de 7,5 meses no braço com Kadcyla® e 3,5 meses no braço controle; hazard ratio, 0,58; p = 0,022).3
Um estudo fase II, aberto, de braço único (TDM4374g) avaliou os efeitos de Kadcyla® em pacientes com câncer de mama HER2-positivo localmente avançado não ressecável ou metastático. Todos os pacientes foram previamente tratados com terapias específicas para HER2 (trastuzumabe ou lapatinibe) e quimioterapia (antraciclina, taxano ou capecitabina) no contexto neoadjuvante, adjuvante, localmente avançado ou metastático. O número mediano de agentes antineoplásicos que os pacientes tinham recebido em qualquer contexto foi de 8,5 (intervalo de 5 a 19) e no contexto metastático foi de 7,0 (intervalo de 3 a 17), incluindo todos os agentes destinados ao tratamento do câncer de mama.4
Os pacientes (n = 110) receberam 3,6 mg/kg de Kadcyla® via intravenosa a cada 3 semanas até a progressão da doença ou toxicidade inaceitável.4
As análises chave de eficácia foram TRO baseada na revisão radiológica independente e duração de resposta objetiva. A TRO foi de 32,7% (IC 95%: 24,1, 42,1), n = 36 respondedores, de acordo com revisão do CRI e do investigador. A duração mediana de resposta de acordo com o CRI ainda não foi atingida (IC 95%, 4,6 meses até não estimável).4
Imunogenicidade1
Assim como em todas as proteínas para fins terapêuticos, existe o potencial para uma resposta imune ao trastuzumabe entansina. Dos 836 pacientes de seis estudos clínicos testados em diversos momentos para a resposta de anticorpos antiterapêuticos (ATAs) para Kadcyla®, quarenta e quatro pacientes (5,3%) apresentaram teste positivo para anticorpos anti-Kadcyla® em um ou mais momentos após a infusão, sendo que 28 desses tiveram amostras de referência negativas. A significância clínica de anticorpos contra o trastuzumabe entansina é ainda desconhecida.
Os resultados dos ensaios de imunogenicidade são altamente dependentes de diversos fatores, incluindo a sensibilidade, especificidade e metodologia dos ensaios, o manuseio das amostras, o tempo de coleta das amostras, o uso concomitante de medicamentos e a doença subjacente. Por essas razões, a comparação da incidência de anticorpos contra Kadcyla® com a de anticorpos contra outros produtos pode ser equivocada.
Referências Bibliográficas
1 Relatório de Estudo Clínico - TDM4370g/BO21977: Um Estudo Fase III, Randomizado, Aberto, Multicêntrico sobre Eficácia e Segurança de Trastuzumabe-MCC-DM1 vs. Capecitabina + Lapatinibe em Pacientes com Câncer de Mama HER2-Positivo Localmente Avançado ou Metastático que Tinham Recebido Terapia Prévia à Base de Trastuzumabe. Relatório de Pesquisa CSR TDM4370g/BO201977, Agosto 2012. (CDS v1.0).
2 Verma S., Miles D., Gianni L., Krop I. E., Welslau M., Baselga J., Pegram M., Oh D.Y., Diéras V., Guardino, E., Fang L., Lu M. W., Olsen S., and Blackwell K.. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N Engl J Med, v.367, n.19, p.1783 - 1791, 2012.
3 Relatório de Estudo Clínico - TDM4450g: Um Estudo Fase II, Randomizado, Multicêntrico sobre Eficácia e Segurança de Trastuzumabe-MCC-DM1 vs.Trastuzumabe (Herceptin®) e Docetaxel (Taxotere®) em Pacients com Câncer de Mama Metastático HER2-Positivo que Não Tinham Recebido Quimioterapia Prévia Para Doença Metastática. Relatório de Pesquisa CSR TDM4450g/BO21976, Julho 2012. (CDS v1.0).
4 Relatório de Estudo Clínico - TDM4374g: Um Estudo Fase II, Braço único, Aberto de Trastuzumabe-MCC-DM1 Administrado Por Via Intravenosa Em Pacientes com Câncer de Mama Metastático HER2-Positivo. Relatório de Pesquisa CSR TDM4374g, 20 Maio 2010 (CDS v1.0).
Caract. farmacológicas.
Propriedades Farmacodinâmicas
Mecanismo de Ação
Kadcyla®, trastuzumabe entansina, é um conjugado de anticorpo-medicamento que tem HER2 como alvo e contém a IgG1 anti-HER2 humanizada, trastuzumabe, ligada de forma covalente com a droga inibitória de microtúbulo DM1 (um derivado de maitansina) através do ligante tioéter estável MCC (4-[N-maleimidometil] ciclohexano-1-carboxilato). Entansina diz respeito ao complexo MCC-DM1. Em média, 3,5 moléculas de DM1 são conjugadas a cada molécula de trastuzumabe.
A conjugação de DM1 a trastuzumabe confere seletividade do agente citotóxico para células tumorais que superexpressam HER2, aumentando assim a veiculação intracelular de DM1 diretamente às células malignas. Com a ligação ao HER2, trastuzumabe entansina sofre internalização mediada por receptor e subsequente degradação lisossomal, resultando na liberação de catabólitos citotóxicos contendo DM1 (principalmente lisina-MCC-DM1).
Kadcyla® tem os mecanismos de ação de trastuzumabe e DM1.
• Trastuzumabe entansina, como o trastuzumabe, se liga ao subdomínio IV do domínio extracelular HER2 (DEC), bem como a receptores Fcc e complemento C1q. Além disso, Kadcyla®, como trastuzumabe, inibe a dispersão do DEC de HER2, inibe a sinalização através da via da fosfatidilinositol 3-quinase (PI3-K) e faz a mediação de citotoxicidade celular através de anticorpos (ADCC) em células de câncer de mama humano que superexpressam HER2.
• DM1, o componente citotóxico de Kadcyla®, liga-se à tubulina. Pela inibição da polimerização da tubulina, tanto DM1 quanto Kadcyla® fazem com que as células parem na fase G2/M do ciclo celular, finalmente levando à apoptose da célula. Os resultados de ensaios de citotoxicidade in vitro mostram que DM1 é de 20 a 200 vezes mais potente do que os taxanos e alcaloides da vinca.
• O ligante MCC é projetado para limitar a liberação sistêmica e aumentar e direcionar a veiculação de DM1, como demonstrado pela detecção de níveis muito baixos de DM1 livre no plasma.
Propriedades Farmacocinéticas
Kadcyla® é administrado por via intravenosa. Não foram feitos estudos com outras vias de administração.
Distribuição
Kadcyla®, quando administrado por via intravenosa a cada 3 semanas, apresentou farmacocinética linear entre doses, variando de 2,4 a 4,8 mg/kg. Pacientes que receberam doses menores ou iguais a 1,2 mg/kg apresentaram uma depuração plasmática mais rápida.
Pacientes no estudo TDM4370g/BO21977 que receberam 3,6 mg/kg de Kadcyla® via intravenosa a cada 3 semanas apresentaram concentração sérica máxima (Cmax) de trastuzumabe entansina de 83,4 (± 16,5) mg/mL. Com base em análise de farmacocinética populacional, depois da administração intravenosa, o volume central de distribuição de trastuzumabe entansina foi de 3,13 L e se aproximou do volume plasmático.
Metabolismo
Espera-se que trastuzumabe entansina sofra catabolismo através de proteólise em lisossomos celulares, sem nenhum envolvimento significativo de isoenzimas do citocromo P450. Catabólitos, incluindo Lys-MCC-DM1, MCC-DM1 e DM1, são detectados em baixos níveis em plasma humano. No Estudo TDM4370g/BO21977, a média dos níveis máximos de DM1 no Ciclo 1 depois da administração de Kadcyla® foi constantemente baixa e foi em média 4,61 ± 1,61 ng/mL.
Estudos de metabolismo in vitro em microssomos hepáticos humanos sugerem que DM1, um componente de trastuzumabe entansina, seja metabolizado principalmente por CYP3A4 e, em menor extensão, por CYP3A5.
Eliminação
Com base na análise farmacocinética (PK) populacional, depois da administração intravenosa de Kadcyla® em pacientes com câncer de mama HER2-positivo metastático, a eliminação de Kadcyla® foi de 0,68 L/dia e a meia-vida de eliminação (t1/2) foi de aproximadamente 4 dias. Nenhum acúmulo de Kadcyla® foi observado depois de administração repetida de infusão intravenosa a cada 3 semanas.
Com base em análise de PK populacional (n = 671), peso corpóreo, albumina, soma de maior diâmetro de lesões alvo por Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST), domínio extracelular de dispersão HER2 (DEC), concentrações basais de trastuzumabe e AST foram identificados como covariáveis estatisticamente significativas para parâmetros farmacocinéticos de trastuzumabe entansina. No entanto, a magnitude do efeito dessas covariáveis sobre exposição a trastuzumabe entansina sugere que, com exceção do peso corpóreo, é improvável que essas covariáveis tenham algum efeito clinicamente significativo sobre a exposição a Kadcyla® . Portanto, o peso corpóreo, baseado na dose de 3,6 mg/kg a cada 3 semanas sem a correção para as outras covariáveis, é considerado apropriado. Em estudos não clínicos, catabólitos de trastuzumabe entansina, incluindo DM1, Lys-MCC-DM1 e MCCDM1, são principalmente excretados na bile com eliminação mínima na urina.
Farmacocinética em populações especiais
A análise farmacocinética populacional de Kadcyla® mostrou que a etnia não parece influenciar a farmacocinética de Kadcyla®. Como a maioria dos pacientes nos estudos clínicos de Kadcyla® era de mulheres, o efeito do sexo sobre a farmacocinética de Kadcyla® não foi avaliado formalmente.
Uso em idosos
A análise farmacocinética populacional de Kadcyla® mostrou que a idade não afetou a farmacocinética de Kadcyla® . Nenhuma diferença significativa foi observada na farmacocinética de Kadcyla® entre pacientes < 65 anos (n = 577), pacientes entre 65 e 75 anos (n = 78) e pacientes > 75 anos (n = 16).
Insuficiência renal
A análise farmacocinética populacional de Kadcyla® mostrou que a depuração plasmática de creatinina não afeta a farmacocinética de Kadcyla®. A farmacocinética de Kadcyla® em pacientes com insuficiência renal leve [depuração plasmática de creatinina (Clcr) 60 - 89 mL/min, n = 254] ou moderada (CLcr 30 a 59 mL/min, n = 53) foi similar à de pacientes com função renal normal (CLcr ≥ 90 mL/min, n = 361). Dados farmacocinéticos em pacientes com insuficiência renal grave (CLcr 15 a 29 mL/min) são limitados (n = 1); portanto, nenhuma recomendação de dose pode ser feita.
Nenhum estudo farmacocinético formal foi conduzido em pacientes com insuficiência hepática.
Segurança pré-clínica
Carcinogenicidade
O trastuzumabe entansina não foi testado para carcinogenicidade.
Mutagenicidade
Não se observou nenhuma evidência de atividade mutagênica em um ensaio de mutação bacteriana reversa in vitro de DM1. Em um ensaio de micronúcleo in vivo de trastuzumabe entansina em macacos cynomolgus, não foi observada nenhuma evidência de dano cromossômico para células de medula óssea. No entanto, em ensaio de micronúcleo em medula óssea de rato, DM1 foi positivo para formação de micronúcleos depois de uma dose única baixa na faixa de concentração de DM1 medida em seres humanos recebendo trastuzumabe entansina, confirmando que trastuzumabe entansina é um aneugênio e/ou clastogênio.
Comprometimento da fertilidade
Estudos dedicados a fertilidade não foram conduzidos com trastuzumabe entansina. No entanto, com base nos resultados de estudos de toxicidade geral em animais, podem ser esperados efeitos adversos sobre a fertilidade.
Teratogenicidade
Estudos dedicados ao desenvolvimento embrio-fetal não foram conduzidos em animais com trastuzumabe entansina. Toxicidade de trastuzumabe para o desenvolvimento foi identificada no contexto clínico, embora não fosse prevista no programa não clínico. Além disso, a toxicidade de maitansina para o desenvolvimento foi identificada em estudos não clínicos, o que sugere que DM1, o componente maitansinoide citotóxico de trastuzumabe entansina, inibidor de microtúbulos, será também teratogênico e potencialmente embriotóxico.
Contraindicações.
Kadcyla® é contraindicado a pacientes com hipersensibilidade a trastuzumabe entansina ou a qualquer um dos excipientes contidos no medicamento.
Advertências e precauções.
Gerais
Pacientes tratados com Kadcyla® precisam ter tumores confirmados como HER2-positivos de acordo com avaliação da superexpressão da proteína HER2 ou amplificação de gene.
Toxicidade pulmonar
Casos de pneumopatia intersticial (PPI), incluindo pneumonite, alguns levando à síndrome do desconforto respiratório agudo ou evolução fatal, foram reportados em estudos clínicos com Kadcyla® (vide item "Reações Adversas"). Sinais e sintomas incluem dispneia, tosse, fadiga e infiltrados pulmonares.
Recomenda-se que o tratamento com Kadcyla® seja definitivamente descontinuado em pacientes com diagnóstico de PPI ou pneumonite.
Pacientes com dispneia ao repouso devido a complicações de doença maligna avançada e comorbidades podem apresentar um risco maior de eventos pulmonares.
Hepatotoxicidade
Hepatotoxicidade, predominantemente na forma de aumentos assintomáticos das concentrações de transaminases séricas (elevações de transaminases Graus 1-4), foi observada durante tratamento com Kadcyla® em estudos clínicos (vide item "Reações Adversas"). As elevações de transaminases foram geralmente transitórias com um pico de elevação no Dia 8 após o tratamento e subsequente recuperação ao Grau 1 ou menor antes do próximo ciclo. Observou-se também, um efeito cumulativo de Kadcyla® sobre as transaminases. Na maioria dos casos, pacientes com níveis elevados de transaminases apresentaram melhora para Grau 1 ou normal dentro de 30 dias a partir da última dose de Kadcyla® .
A função hepática deve ser monitorada antes da introdução do tratamento e a cada dose de Kadcyla®. Reduções de dose ou descontinuação para transaminases séricas e bilirrubina total aumentadas são especificadas no item "Posologia e Modo de Usar - Modificações de dose".
Kadcyla® não foi estudado em pacientes com transaminases séricas > 2,5 x LSN (limite superior da normalidade) ou bilirrubina total > 1,5 x LSN antes da introdução do tratamento. O tratamento com Kadcyla® em pacientes com transaminases séricas > 3 x LSN e bilirrubina total concomitante > 2 x LSN deve ser definitivamente descontinuado.
Foram identificados casos de HNR do fígado a partir de biópsias hepáticas em pacientes tratados com Kadcyla®. HNR é uma condição hepática rara, caracterizada por transformação benigna disseminada de parênquima hepático em pequenos nódulos regenerativos. HNR pode levar à hipertensão portal não cirrótica. O diagnóstico de HNR pode ser confirmado apenas por histopatologia. HNR deve ser considerada em todos os pacientes com sintomas clínicos de hipertensão portal e/ou cirrose do tipo padrão observada na tomografia computadorizada (TC) do fígado, mas com transaminases normais e sem outras manifestações de cirrose. Com um diagnóstico de HNR, Kadcyla® precisa ser descontinuado definitivamente.
Disfunção ventricular esquerda
Pacientes tratados com Kadcyla® apresentam risco aumentado para desenvolvimento de disfunção ventricular esquerda. Fração de ejeção de ventrículo esquerdo (FEVE) < 40% foi observada em pacientes tratados com Kadcyla® e, portanto, insuficiência cardíaca congestiva (ICC) sintomática é um potencial risco. Testes padrão de função cardíaca [ecocardiograma ou angiografia radioisotópica e cintilografia sincronizada das câmaras cardíacas (MUGA)] devem ser realizados antes do início e em intervalos regulares (p.ex., a cada três meses) durante o tratamento com Kadcyla®.O tratamento com Kadcyla® não foi estudado em pacientes com FEVE < 50% antes do início do tratamento. Orientações específicas relativas às modificações de dose e descontinuação são fornecidas no item "Posologia e Modo de Usar - Modificações de dose".
Reações relacionadas à infusão
O tratamento com Kadcyla® não foi estudado em pacientes que tinham descontinuado definitivamente trastuzumabe por causa de reações relacionadas à infusão (RRI); o tratamento com Kadcyla® não é recomendado para esses pacientes.
Reações relacionadas à infusão, caracterizadas por um ou mais dos seguintes sintomas - rubor, calafrios, febre, dispneia, hipotensão, sibilos, broncoespasmo e taquicardia - foram reportadas em estudos clínicos de Kadcyla®. Em geral, esses sintomas não foram graves (vide item "Reações Adversas"). Na maioria dos pacientes, essas reações foram resolvidas durante várias horas ou um dia depois de encerrada a infusão. O tratamento com Kadcyla® deve ser interrompido em pacientes com RRI graves. O tratamento com Kadcyla® deve ser definitivamente descontinuado em caso de reação relacionada à infusão potencialmente fatal (vide item "Posologia e Modo de Usar - Modificações de dose").
Reações de hipersensibilidade
Os pacientes devem ser observados cuidadosamente para verificar o aparecimento de reações de hipersensibilidade, especialmente durante a primeira infusão. Hipersensibilidade, incluindo reações tipo anafilactoides graves, foi observada em estudos clínicos durante tratamento com Kadcyla®. As medicações para tratar essas reações, bem como equipamento de emergência, devem estar disponíveis para uso imediato.
Trombocitopenia
Trombocitopenia, ou número reduzido de plaquetas, foi reportada em pacientes em estudos clínicos com Kadcyla®.A maioria desses pacientes apresentou eventos Grau 1 ou 2 (≥ 50.000/mm3), sendo que o ponto mais baixo ocorre em torno do Dia 8 e geralmente regridem para grau 0 ou 1 (≥ 75.000/mm3) por ocasião da dose seguinte programada. Em estudos clínicos, a incidência e gravidade da trombocitopenia foram maiores em pacientes asiáticos.
Casos de eventos de hemorragia com desfecho fatal têm sido observados. Casos graves de eventos hemorrágicos, incluindo hemorragia do sistema nervoso central, têm sido relatados em estudos clínicos com Kadcyla®; estes eventos foram independentes da etnia. Em alguns dos casos observados, os pacientes também estavam recebendo terapia anticoagulação.
Pacientes com trombocitopenia ( < 100.000/mm3) e pacientes recebendo tratamento anticoagulante devem ser monitorados cuidadosamente durante o tratamento com Kadcyla® . Recomenda-se que o número de plaquetas seja monitorado antes de cada aplicação de Kadcyla® . Kadcyla® não foi estudado em pacientes com número de plaquetas < 100.000/mm3 antes do início do tratamento. No caso de número de plaquetas diminuído para Grau 3 ou maior ( < 50.000/mm3), não administre Kadcyla® até que o número de plaquetas se recupere para Grau 1 (≥ 75.000/mm3). Por favor, vide item "Posologia e Modo de Usar - Modificações de dose".
Neurotoxicidade
Neuropatia periférica, principalmente Grau 1 e predominantemente sensorial, foi reportada em estudos clínicos com Kadcyla®.
O tratamento com Kadcyla® deve ser temporariamente descontinuado em pacientes que apresentarem neuropatia periférica Graus 3 ou 4 até que os sintomas sejam resolvidos ou regridam para ≤ Grau 2. Os pacientes devem ser monitorados clinicamente de forma contínua para sinais/sintomas de neurotoxicidade.
Extravasamento
Em estudos clínicos com Kadcyla®, foram observadas reações secundárias a extravasamento. Essas reações foram geralmente leves e constituídas de eritema, sensibilidade, irritação cutânea, dor ou edema no local de infusão. Essas reações foram observadas mais frequentemente dentro de 24 horas de infusão. O tratamento específico para extravasamento de Kadcyla® não é conhecido até o momento. O local de infusão deve ser monitorado cuidadosamente, verificando possível infiltração subcutânea durante a administração da droga.
Capacidade para dirigir e operar máquinas
Estudos sobre os efeitos na capacidade de dirigir e operar máquinas não foram realizados.
Exames laboratoriais
Vide item "Advertências e Precauções -Hepatotoxicidade e Trombocitopenia".
Uso em populações especiais
Gravidez e lactação
Categoria de risco na gravidez: D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Não existem estudos clínicos com Kadcyla® em gestantes. Nenhum estudo reprodutivo e de toxicologia para desenvolvimento foi conduzido com Kadcyla®.
O trastuzumabe, um componente de Kadcyla®, pode provocar dano ou óbito fetal quando administrado à gestante. No contexto pós-comercialização, casos de oligoidrâmnio, alguns associados com hipoplasia pulmonar fatal, foram reportados em gestantes recebendo trastuzumabe. Estudos em animais de laboratório com maitansina, uma entidade química estreitamente relacionada à mesma classe maitansinoide de DM1, sugerem que se pode esperar que DM1, o componente citotóxico de Kadcyla®, inibidor de microtúbulos, seja teratogênico e potencialmente embriotóxico.
A administração de Kadcyla® a gestantes não é recomendada. Mulheres que engravidarem precisam entrar em contato com seu médico e devem ser orientadas sobre a possibilidade de dano fetal. Se uma gestante for tratada com Kadcyla® , recomenda-se monitoramento e cuidados por uma equipe multidisciplinar.
As pacientes com possibilidade de engravidar devem ser orientadas sobre o uso efetivo de contracepção durante tratamento com Kadcyla® e por, pelo menos, seis meses depois da conclusão do tratamento.
Não se sabe se Kadcyla® é excretado no leite humano. Como muitas drogas são excretadas no leite humano e como existe potencial para reações adversas graves por Kadcyla® nos lactentes em aleitamento materno, as mulheres precisam descontinuar a amamentação antes de iniciar o tratamento com Kadcyla®. As mulheres podem iniciar o aleitamento seis meses depois de concluído o tratamento.
Uso pediátrico
A segurança e a eficácia de Kadcyla® não foram estabelecidas em crianças abaixo dos 18 anos de idade.
Existem dados insuficientes para estabelecer a segurança e a eficácia de Kadcyla® em pacientes com 75 anos de idade ou mais.
Insuficiência renal
Vide item "Características Farmacológicas - Farmacocinética em populações especiais".
Insuficiência hepática
A segurança e a eficácia de Kadcyla® em pacientes com insuficiência hepática não foram estabelecidas.
Atenção: Este medicamento contém açúcar, portanto, deve ser usado com cautela em portadores de diabetes.
Até o momento, não há informações de que Kadcyla® (trastuzumabe entansina) possa causar doping.
Interações medicamentosas.
Não foram conduzidos estudos formais em humanos sobre interação medicamentosa com Kadcyla® . Estudos sobre metabolismo in vitro em microssomos hepáticos humanos sugerem que DM1, um componente do trastuzumabe entansina, é metabolizado principalmente pelo CYP3A4 e, em menor extensão, por CYP3A5. DM1 não induz nem inibe
o metabolismo mediado por P450 in vitro. Deve-se tomar cuidado quando Kadcyla® é coadministrado com inibidores potentes de CYP3A.
Recomenda-se sempre consultar a bula dos medicamentos utilizados concomitantemente pelo paciente, devido às potenciais interações medicamentosas.
Cuidados de armazenamento.
Armazenamento
Frascos-ampola
Armazenar os frascos-ampola sob refrigeração (temperatura entre 2 e 8 °C).
Prazo de validade do pó em frasco-ampola fechado
Este medicamento possui prazo de validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Prazo de validade da solução reconstituída
Os frascos-ampola do produto reconstituído com água estéril para injetáveis devem ser utilizados imediatamente depois da reconstituição. Se não forem utilizados imediatamente, os frascos-ampola reconstituídos podem ser armazenados durante até 24 horas em temperatura entre 2 e 8 °C, e devem ser descartados depois deste período.
Não congelar a solução reconstituída.
Após aberto e reconstituído com água estéril para injetáveis, válido por até 24 horas, se mantido sob refrigeração em temperatura entre 2 e 8 °C; não pode ser congelado.
Prazo de validade da solução para infusão contendo o produto reconstituído
A solução reconstituída de trastuzumabe entansina, diluída em bolsas de cloreto de polivinila (PVC) ou de poliolefina sem látex e sem PVC, contendo solução de cloreto de sódio 0,9% ou cloreto de sódio 0,45%, pode ser armazenada em temperatura entre 2 e 8 °C, durante até 24 horas antes do uso. Material particulado pode ser observado durante o armazenamento, se diluído em solução de cloreto de sódio 0,9%; portanto, é necessário um filtro em linha de polietersulfona (PES) de 0,22 mm para administração (vide item "Posologia e Modo de Usar - Instruções especiais para uso e manipulação").
Após reconstituição e diluição em bolsa com solução de cloreto de sódio 0,9% ou 0,45%, válido por até 24 horas, se mantido em temperatura entre 2 e 8 °C não pode ser congelado.
Características físicas e organolépticas Kadcyla® apresenta-se sob forma de pó branco a quase branco. A cor da solução reconstituída varia de incolor a marrom clara e deve estar livre de partículas visíveis, límpida a discretamente opalescente.
Antes de usar, observe o aspecto do medicamento.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
Para evitar erros na medicação, é importante verificar os rótulos do frasco-ampola para garantir que a droga que está sendo preparada e administrada é Kadcyla® (trastuzumabe entansina) e não trastuzumabe.
A terapia com Kadcyla® deve ser administrada exclusivamente sob a supervisão de um profissional de saúde com experiência no tratamento de pacientes com câncer.
Os pacientes tratados com Kadcyla® devem apresentar tumores HER2-positivos, definidos como um escore de 3+ pela imunohistoquímica (IHQ) ou uma razão ≥ 2,0 por hibridização in situ (ISH) em um teste validado.
Para aumentar a rastreabilidade dos medicamentos biológicos, o nome comercial do produto administrado deve ser claramente registrado (ou declarado) no prontuário médico do paciente.
A substituição de Kadcyla® por qualquer outro medicamento biológico exige o consentimento do médico prescritor.
Kadcyla® precisa ser reconstituído e diluído por um profissional de saúde e administrado na forma de infusão intravenosa (vide item "Posologia e Modo de Usar -Instruções especiais para uso e manipulação"). Não administre como injeção intravenosa direta ou em bolus.
Instruções especiais para uso e manipulação
Deve ser usada técnica asséptica adequada. Procedimentos adequados para preparação de drogas quimioterápicas devem ser empregados.
O produto reconstituído não contém conservante e se destina exclusivamente a uso único. Despreze qualquer porção não utilizada.
• Utilizando uma seringa estéril, injete lentamente 5 mL de água estéril para injetáveis no frasco-ampola de 100 mg ou 8 mL de água estéril para injetáveis no frasco-ampola de 160 mg de trastuzumabe entansina. A concentração final da solução reconstituída será de, aproximadamente, 20 mg/mL.
• Faça movimentos circulares suaves com o frasco-ampola até que esteja completamente dissolvido. NÃO AGITE<33>
• Armazene trastuzumabe entansina reconstituído em temperatura entre 2 e 8 °C; descarte o trastuzumabe entansina não utilizado após 24 horas.
A solução reconstituída deve ser inspecionada visualmente na busca de material particulado e alterações de cor antes da administração. A solução reconstituída deve estar livre de partículas visíveis, límpida a discretamente opalescente. A cor da solução reconstituída deve ser incolor a marrom clara. Não utilize o medicamento se a solução reconstituída contiver partículas visíveis ou estiver turva ou com alteração de cor.
Instruções para diluição
Volume (mL) = Peso corpóreo (kg) x dose (mg/kg)/20 mg/mL (concentração de solução reconstituída)
A quantidade apropriada da solução deve ser retirada do frasco-ampola e adicionada a uma bolsa de infusão contendo 250 mL de cloreto de sódio 0,45% ou cloreto de sódio 0,9%. Não deve ser usada solução de dextrose (5%). O cloreto de sódio 0,45% pode ser usado sem um filtro em linha de polietersulfona (PES) de 0,22 mm. Se for usado cloreto de sódio 0,9% para infusão, é necessário um filtro em linha de polietersulfona (PES) de 0,22 mm. Assim que a infusão for preparada, ela deve ser aplicada imediatamente. Se não for utilizada imediatamente, a infusão pode ser armazenada durante até 24 horas em um refrigerador em temperatura entre 2 e 8 oC. A infusão não pode ser congelada nem agitada durante o armazenamento.
Incompatibilidades
A solução de dextrose (5%) não deve ser usada, porque provoca agregação de proteínas. O trastuzumabe entansina não deve ser misturado nem diluído com outras drogas.
Esquema de dosagem
A dose recomendada de Kadcyla® é de 3,6 mg/kg, administrada em infusão intravenosa a cada 3 semanas (ciclo de 21 dias) até a progressão da doença ou ocorrência de toxicidade inaceitável.
A dose inicial deve ser administrada na forma de infusão intravenosa durante 90 minutos. Os pacientes devem ser observados durante a infusão e por, pelo menos, 90 minutos depois da dose inicial para verificar o eventual aparecimento de febre, calafrios ou outras reações relacionadas à infusão. O local de infusão deve ser monitorado cuidadosamente para verificar possível infiltração subcutânea durante a administração da droga (vide item "Advertências e Precauções -Extravasamento").
Se as primeiras infusões forem bem toleradas, as doses subsequentes de Kadcyla® podem ser administradas em infusões de 30 minutos e os pacientes devem ser observados durante as infusões e por, pelo menos, 30 minutos depois delas.
A velocidade de infusão de Kadcyla® deve ser diminuída ou interrompida se o paciente desenvolver sintomas relacionados à infusão (vide item "Advertências e Precauções"). Descontinue Kadcyla® na presença de reações à infusão potencialmente fatais.
A dose máxima de Kadcyla® que pode ser administrada é 3,6 mg/kg a cada 3 semanas. O medicamento não deve ser administrado em doses maiores que esse valor.
Dose atrasada ou perdida
Se uma dose programada for perdida, ela deve ser administrada o mais brevemente possível; não aguarde até o próximo ciclo planejado. O esquema de administração deve ser ajustado para manter um intervalo de 3 semanas entre as doses. A infusão pode ser administrada na velocidade que o paciente tolerou a infusão mais recente.
Modificações de dose
O manejo de eventos adversos sintomáticos pode exigir interrupção temporária, redução de dose ou descontinuação de tratamento com Kadcyla®, conforme as orientações fornecidas nas Tabelas 2-6.
A dose de Kadcyla® não deve ser reescalonada depois de ter sido feita uma redução de dose.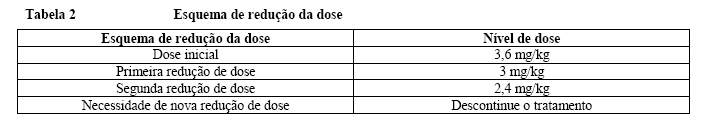




Instruções posológicas especiais
Idosos
Não é necessário nenhum ajuste de dose de Kadcyla® para pacientes com idade > 65 anos (vide item "Advertências e Precauções - Uso em idosos").
Crianças
A segurança e a eficácia de Kadcyla® não foram estabelecidas em pacientes pediátricos.
Insuficiência renal
Não é necessário nenhum ajuste da dose inicial de Kadcyla® em pacientes com insuficiência renal leve ou moderada (vide item "Características Farmacológicas -Farmacocinética em populações especiais"). A potencial necessidade para ajuste de dose em pacientes com insuficiência renal grave não pode ser determinada porque os dados são insuficientes.
Insuficiência hepática
A segurança e a eficácia de Kadcyla® não foram estudadas em pacientes com insuficiência hepática.
Reações adversas.
Estudos Clínicos
A segurança de Kadcyla® foi avaliada em mais de 880 pacientes em estudos clínicos. A Tabela 7 resume as reações adversas à droga (RADs) que foram reportadas em associação com o uso de Kadcyla® em estudos clínicos.
Nesta seção, as seguintes categorias de frequência foram usadas: muito comuns ( > 1/10), comuns ( > 1/100 a < 1/10), incomuns ( > 1/1.000 a < 1/100).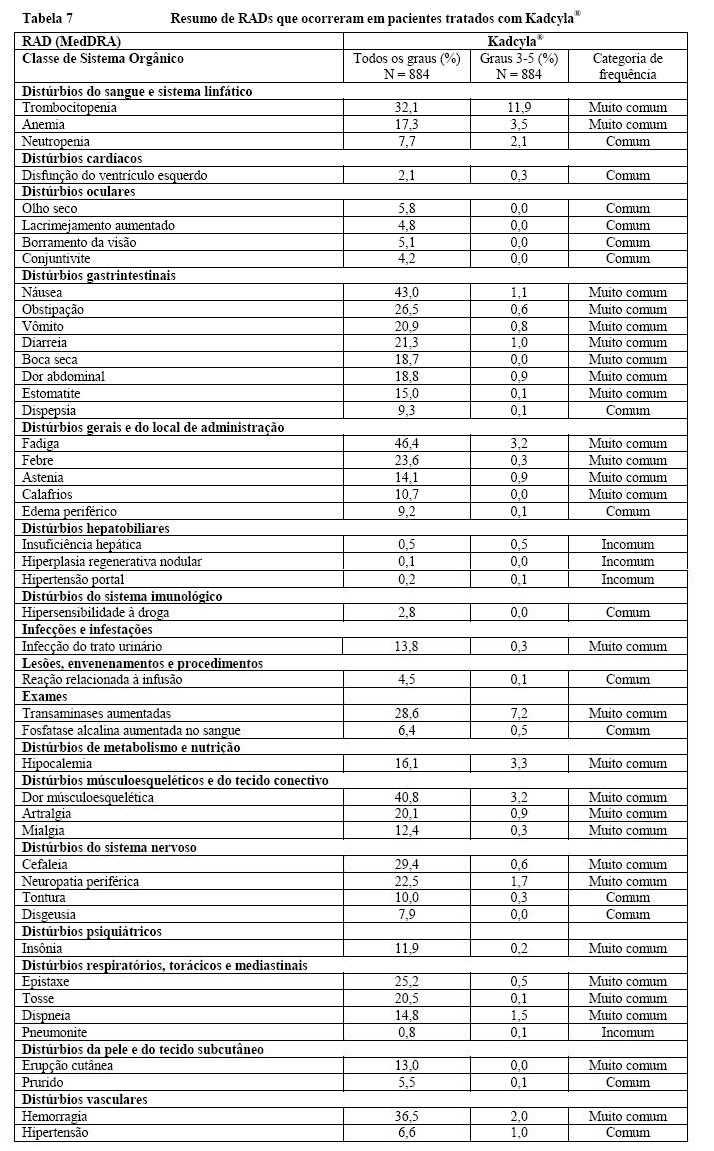
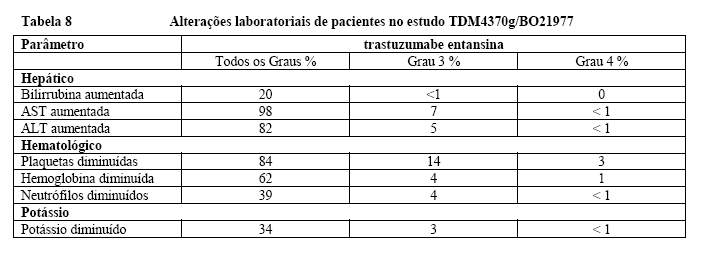
Alterações laboratoriais
A tabela a segui