JEMPERLI
GLAXOSMITHKLINE
dostarlimabe
Anticorpo monoclonal.
Apresentações.
Solução para diluição para infusão. Jemperli é apresentado em embalagem com 1 frasco-ampola de 10 ml contendo 500 mg de dostarlimabe (50 mg/ml).
USO INTRAVENOSO
USO ADULTO
Composição.
Cada 1 ml da solução para diluição para infusão contém: dostarlimabe 50 mg, excipientes* q.s.p. 1 ml
*Excipientes: ácido cítrico monoidratado, cloridrato de L-arginina, polissorbato 80, cloreto de sódio, citrato de sódio dihidratado, água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Jemperli é indicado como monoterapia para o tratamento de pacientes adultos com câncer endometrial recorrente ou avançado com deficiência de enzimas de reparo (dMMR) ou alta instabilidade de microssatélite (MSI-H), que progrediu durante ou após tratamento à base de platina.
2. RESULTADOS DE EFICÁCIA
Câncer endometrial com deficiência de enzimas de reparo (dMMR) ou alta instabilidade de microssatélite (MSI-H)
A eficácia e a segurança de dostarlimabe foram investigadas no estudo GARNET, um estudo de fase 1 de escalonamento de dose, multicêntrico, aberto, realizado em pacientes com câncer endometrial recorrente ou avançado que progrediram durante ou após tratamento à base de platina.
O estudo GARNET incluiu coortes de expansão em indivíduos com tumores sólidos recorrentes ou avançados que têm opções de tratamento disponíveis limitadas. A Coorte A1 recrutou pacientes com câncer endometrial com deficiência de enzimas de reparo (dMMR) ou alta instabilidade de microssatélite (MSI-H), que progrediu durante ou após tratamento à base de platina.
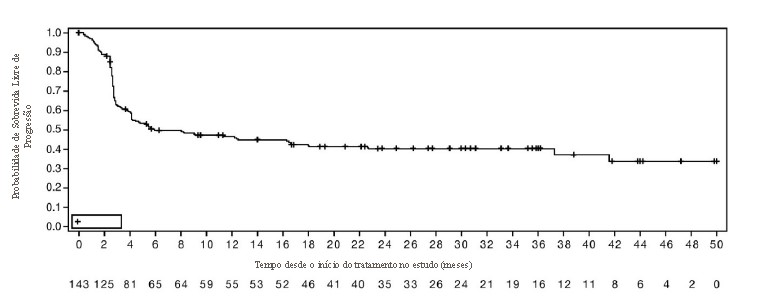
Os pacientes receberam 500 mg de Jemperli a cada 3 semanas por 4 ciclos, seguidos por 1000 mg a cada 6 semanas. O tratamento continuou até a toxicidade inaceitável ou até que a progressão da doença -sintomática, rapidamente progressiva -exigisse intervenção urgente ou que ocorresse um declínio no performance status (PS). Jemperli foi administrado por no máximo 220 semanas (51 meses) de tratamento e 24% dos indivíduos que receberam qualquer quantidade de dostarlimabe receberam tratamento > 102 semanas (2 anos). As principais medidas de resultado primário de eficácia foram taxa de resposta objetiva (ORR) e duração da resposta (DOR), avaliadas pela revisão cega de radiologistas centrais independentes (BICR), conforme o RECIST v1.1. Os desfechos secundários incluíram taxa de controle da doença (DCR) e sobrevida livre de progressão (PFS), ambos avaliados pela revisão BICR de acordo com RECIST v1.1; e sobrevida global (OS).
Todos os pacientes incluídos no conjunto de análises de eficácia primária e secundária tiveram um período mínimo de acompanhamento de 24 semanas a partir da primeira dose, independentemente de terem realizado um exame pós-tratamento.
Um total de 143 pacientes com câncer de endométrio dMMR/MSI-H foram avaliados quanto à eficácia no estudo GARNET. Entre esses 143 pacientes, as características basais foram: média de idade de 65 anos (52% com 65 anos ou mais); 77% branco, 3% asiático, 3% preto; e performance status (PS) do ECOG (Grupo de Oncologia Cooperativa Oriental) 0 (39%) ou PS 1 (61%). O número mediano de linhas anteriores de terapias foi um: 63% das pacientes tiveram uma linha anterior, 37% tiveram duas ou mais linhas anteriores. Quarenta e nove pacientes (34%) receberam tratamento apenas no cenário neoadjuvante ou adjuvante antes de participar do estudo.
A identificação da situação do tumor dMMR/MSI-H foi determinada prospectivamente com base em testes locais. Os ensaios de diagnóstico (IHQ [imuno-histoquímica], PCR [reação em cadeia da polimerase] ou NGS [sequenciamento de nova geração]) disponíveis foram usados para a detecção da expressão de dMMR / MSI-H em material tumoral. A maioria usou IHQ, por ser o ensaio mais comum disponível.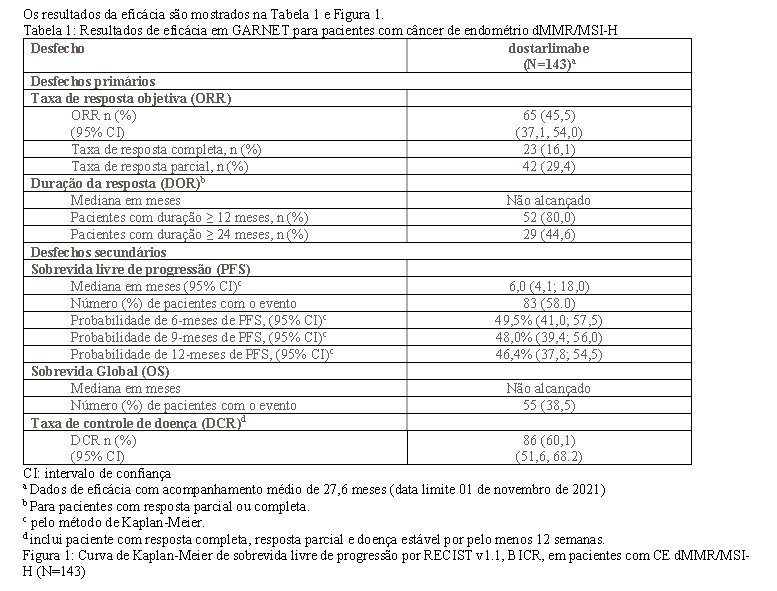
Pacientes idosos
Dos 515 pacientes tratados com dostarlimabe em monoterapia (população GARNET AI1 no momento do corte de dados 01 de Março de 2020), 51% tinham menos de 65 anos, 38% tinham 65-75 anos e 12% tinham 75 anos ou mais. Não se observou aumento nos riscos de segurança em sujeitos mais velhos em comparação com indivíduos mais jovens.
Nos 72 pacientes com CE dMMR/MSI-H (população IA1 no momento do corte de dados 01 de março de 2020) na análise de eficácia, a ORR por BICR (IC 95%) foi de 43,2% (27,1%, 60,5%) em pacientes com menos de 65 anos e 48,6% (31,4%, 66,0%) em pacientes com 65 anos ou mais.
Nos 105 pacientes com CE dMMR/MSI-H (população IA2 no momento do corte de dados em 01 de março de 2020) na análise de eficácia, a ORR por BICR (95% CI) foi de 45,3% (31,6%, 59,6%) em pacientes com menos de 65 anos e 44,2% (30,5%, 58,7%) em pacientes com 65 anos ou mais.
População pediátrica
A segurança e eficácia de dostarlimabe em crianças e adolescentes com menos de 18 anos de idade não foram estabelecidas.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: agente antineoplásico, anticorpo monoclonal e conjugado anticorpo medicamento.
Código ATC: L01FF07
Mecanismo de ação
Dostarlimabe é um anticorpo monoclonal humanizado (mAb) imunoglobulina G4 (IgG4) contra proteína de morte celular programada 1 (PD-1), derivado de uma linhagem celular estável do ovário de hamster chinês (CHO). A interação dos ligantes de PD-1, PD-L1 e PD-L2, ao receptor PD-1 encontrado nas células T inibe a proliferação de células T e a produção de citocinas. A regulação positiva dos ligantes PD-1 ocorre em alguns tumores e a sinalização por essa via pode contribuir para a inibição da vigilância imune ativa de tumores por células T. O dostarlimabe é um anticorpo monoclonal humanizado (mAb) do isótipo imunoglobulina G4 (IgG4) que se liga à PD-1, resultando em inibição da ligação com PD-L1 e PD-L2, liberando a inibição da resposta imune mediada por PD-1, incluindo a resposta imune antitumoral. Nos modelos de tumor singênico de camundongos, o bloqueio da atividade da PD-1 resultou em crescimento reduzido do tumor.
Efeitos farmacodinâmicos
Com base nas relações de eficácia e segurança na exposição, não há diferenças clinicamente significativas em eficácia e segurança ao dobrar a exposição ao dostarlimabe. A ocupação total do receptor, medida tanto pela ligação direta ao PD-1 quanto pelo ensaio funcional de produção de IL-2, foi mantida durante todo o intervalo de administração na dose terapêutica recomendada.
Farmacocinética
A farmacocinética de dostarlimabe foi caracterizada usando análise farmacocinética da população de 546 pacientes com vários tumores sólidos, incluindo 288 pacientes com câncer endometrial. A farmacocinética de dostarlimabe é proporcional à dose. O dostarlimabe mostra um acúmulo de aproximadamente 2 vezes (Cmín) quando administrado na dose terapêutica pretendida (500 mg administrados por via intravenosa a cada 3 semanas por 4 ciclos, seguidos por 1000 mg a cada 6 semanas), iniciando no ciclo 4 até o ciclo 12, consistente com a meia-vida terminal.
Absorção
Dostarlimabe é administrado por via intravenosa e, portanto, as estimativas de absorção não são aplicáveis.
Distribuição
O volume de distribuição médio geométrico do dostarlimabe no estado estacionário é de aproximadamente 5,26 L (CV% de 14,2%).
Metabolismo
O dostarlimabe é um mAb IgG4 terapêutico que espera-se ser catabolizado em pequenos peptídeos, aminoácidos e pequenos carboidratos pelo lisossomo através da endocitose por fase líquida ou mediada por receptor. Os produtos de degradação são eliminados por excreção renal ou devolvidos ao pool de nutrientes sem efeitos biológicos.
Eliminação
A depuração média geométrica é de 0,00682 L/h (CV% de 30,2%) no estado estacionário. A média geométrica da meia-vida terminal (t1/2) no estado estacionário é de 23,5 dias (CV% de 22,4%).
Linearidade/não linearidade
A exposição (concentração máxima [Cmáx] e a área sob a curva de concentração-tempo [AUC0-tau] e [AUC0-inf]), foi aproximadamente proporcional à dose.
Populações especiais de pacientes
Uma análise populacional da farmacocinética dos dados dos pacientes indica que não há efeitos clinicamente importantes de idade (faixa: 24 a 86 anos), sexo ou raça, etnia ou tipo de tumor na depuração do dostarlimabe. Esse modelo de farmacocinética populacional também indica que alterações na função renal (normal a moderada) e hepática (normal a comprometimento leve) não alteram a disposição do dostarlimabe.
Carcinogênese/mutagênese
Não foram realizados estudos para avaliar o potencial do dostarlimabe para carcinogenicidade ou genotoxicidade.
Toxicologia Reprodutiva
Não foram realizados estudos de reprodução animal com dostarlimabe. Acredita-se que a via PD-1/PD-L1 esteja envolvida na manutenção da tolerância ao feto durante a gravidez. Foi demonstrado que o bloqueio da sinalização do PD-L1 em modelos murinos de gravidez interrompe a tolerância ao feto e resulta em aumento na perda fetal.
Fertilidade
Não foram realizados estudos de fertilidade animal com dostarlimabe. Nos estudos toxicológicos de dose repetida de 1 e 3 meses em macacos, não houve efeitos notáveis nos órgãos reprodutores masculino e feminino; no entanto, muitos animais nesses estudos não estavam sexualmente maduros.
Toxicologia e/ou farmacologia animal
A segurança não clínica do dostarlimabe foi avaliada em estudos de toxicidade de dose repetida de 1 e 3 meses em macacos Cynomolgus, administrados com doses intravenosas de 10, 30 ou 100 mg/kg/semana. Nenhuma descoberta de significância toxicológica foi observada em ambos os estudos, exceto que um macaco macho recebendo 10 mg/kg/semana foi sacrificado devido a achados cutâneos crônicos e generalizados não resolvidos no estudo de três meses. O nível de efeito adverso não observado (NOAEL) foi ≥100 mg/kg no estudo de 1 mês, correspondendo a múltiplas exposições de 35 e 28 vezes a exposição em humanos em doses de 500 e 1000 mg, respectivamente. O NOAEL não foi determinado no estudo de três meses, pois a relação entre a eutanásia prematura do animal e dostarlimabe não pôde ser descartada.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes mencionados na seção COMPOSIÇÃO.
5. ADVERTÊNCIAS E PRECAUÇÕES Os dados descritos nesta seção refletem a exposição ao Jemperli como monoterapia em pacientes com tumores sólidos avançados ou recorrentes em um estudo aberto, de braço único e multicoorte (GARNET).
Reações adversas relacionadas ao sistema imune
Reações adversas relacionadas ao sistema imune, que podem ser graves ou fatais, podem ocorrer em pacientes tratados com anticorpos que bloqueiam a via da proteína-1 de morte celular programada/ligante-1 de morte programada (PD-1/PD L1), incluindo Jemperli. Enquanto reações adversas relacionadas ao sistema imunológico geralmente ocorrem durante o tratamento com anticorpos bloqueadores de PD-1/PD-L1, os sintomas também podem se manifestar após a descontinuação do tratamento. Reações adversas relacionadas ao sistema imune podem ocorrer em qualquer órgão ou tecido e podem afetar mais de um sistema do corpo simultaneamente. As reações adversas importantes relacionadas ao sistema imunológico listadas nesta seção não incluem todas as possíveis reações graves e fatais relacionadas ao sistema imunológico.
A identificação e o gerenciamento precoce das reações adversas relacionadas ao sistema imunológico são essenciais para garantir
o uso seguro dos anticorpos bloqueadores de PD-1/PD-L1. Monitorar sintomas e sinais de reações adversas relacionadas ao sistema imunológico. Avaliar a hematologia e bioquímica clínica, incluindo testes hepáticos, renais e testes da função tireoidiana, no período basal e periodicamente durante o tratamento. Para suspeitas de reações adversas relacionadas ao sistema imunológico, deve-se garantir uma avaliação adequada, incluindo consultas especializadas.
Com base na gravidade da reação adversa, Jemperli deve ser suspenso ou descontinuado permanentemente e administrar corticosteroides (1 a 2 mg/kg/dia de prednisona ou equivalente) ou outra terapia apropriada (veja abaixo o item Alterações de dose na seção 8. POSOLOGIA E MODO DE USAR). Após a melhora para Grau ≤1, a redução gradual de corticosteroides deve ser iniciada e continuada por 1 mês ou mais. Com base em dados limitados de estudos clínicos em pacientes cujas reações adversas não puderam ser controladas com o uso de corticosteroides, a administração de outros imunossupressores sistêmicos pode ser considerada. Instituir terapia de reposição hormonal para endocrinopatias, conforme necessário.
Jemperli deve ser permanentemente descontinuado para qualquer reação adversa relacionada ao sistema imunológico de Grau 3 que se repita e para qualquer toxicidade de reação adversa relacionada ao sistema imunológico de Grau 4, exceto para endocrinopatias controladas com reposição hormonal e a menos que especificado de outra forma na Tabela 3.
Pneumonite relacionada ao sistema imune:
Pneumonite foi relatada em pacientes recebendo Jemperli (consulte o item 9. REAÇÕES ADVERSAS). Os pacientes devem ser monitorados quanto a sinais e sintomas de pneumonite. A suspeita de pneumonite deve ser confirmada com imagens radiográficas e outras causas excluídas. Os pacientes devem ser tratados com modificações no tratamento com Jemperli e corticosteroides (consulte o item 8. POSOLOGIA E MODO DE USAR).
Colite relacionada ao sistema imune:
Jemperli pode causar colite relacionada ao sistema imunológico (consulte o item 9. REAÇÕES ADVERSAS). Monitore os pacientes quanto a sinais e sintomas de colite e gerencie com modificações no tratamento com Jemperli, agentes antidiarreicos e corticosteroides (consulte Posologia).
Hepatite relacionada ao sistema imune:
O Jemperli pode causar hepatite relacionada ao sistema imunológico. Monitore os pacientes periodicamente quanto a alterações na função hepática, conforme indicado, com base na avaliação clínica e gerencie com modificações no tratamento com Jemperli e corticosteroides (consulte o item 8. POSOLOGIA E MODO DE USAR).
Endocrinopatias relacionadas ao sistema imune:
Foram relatadas endocrinopatias relacionadas ao sistema imune, incluindo hipotireoidismo, hipertireoidismo, tireoidite, hipofisite, diabetes mellitus tipo 1, cetoacidose diabética e insuficiência adrenal, em pacientes que receberam Jemperli (consulte o item 9. REAÇÕES ADVERSAS).
Hipotireoidismo e hipertireoidismo: Ocorreram hipotireoidismo e hipertireoidismo relacionados ao sistema imune (incluindo tireoidite) em pacientes que receberam Jemperli, e o hipotireoidismo pode seguir o hipertireoidismo. Os pacientes devem ser monitorados quanto a testes anormais da função tireoidiana antes e periodicamente durante o tratamento e conforme indicado com base na avaliação clínica. Hipotireoidismo e hipertireoidismo relacionados ao sistema imune (incluindo tireoidite) devem ser tratados conforme recomendado no item 8. POSOLOGIA E MODO DE USAR.
Insuficiência adrenal: Ocorreu insuficiência adrenal relacionada à imunidade em pacientes que receberam Jemperli. Os pacientes devem ser monitorados quanto a sinais e sintomas clínicos de insuficiência adrenal. Para insuficiência adrenal sintomática, os pacientes devem ser tratados conforme recomendado no item 8. POSOLOGIA E MODO DE USAR.
Nefrite relacionada ao sistema imune
Jemperli pode causar nefrite relacionada ao sistema imunológico (consulte o item 9. REAÇÕES ADVERSAS). Monitore os pacientes quanto a alterações na função renal e gerencie com modificações no tratamento com Jemperli e corticosteroides (consulte o item 8. POSOLOGIA E MODO DE USAR).
Erupções cutâneas relacionadas ao sistema imune
Foram relatadas erupções cutâneas relacionadas ao sistema imune em pacientes que utilizaram Jemperli, incluindo pefingoide (consulte o item 9. REAÇÕES ADVERSAS). Os pacientes devem ser monitorados para sinais e sintomas de erupções cutâneas. Condições dermatológicas esfoliativas devem ser tratadas conforme recomendado (ver item 8. POSOLOGIA E MODO DE USAR). Eventos de síndrome de Stevens-Johnson ou necrólise epidérmica tóxica foram reportados em pacientes tratados com inibidores de PD-1.
Deve-se ter cautela ao considerar o uso de Jemperli em um paciente que já teve uma reação adversa cutânea grave ou com risco de morte em tratamento anterior com outros agentes imunoestimulantes anticâncer.
Artralgia relacionada ao sistema imune:
Foi relatada artralgia relacionada ao sistema imune em pacientes que receberam Jemperli. Os pacientes devem ser monitorados quanto a sinais e sintomas de artralgia. A suspeita de artralgia relacionada ao sistema imune deve ser confirmada e outras causas excluídas. Os pacientes deveriam ser tratados com modificações no tratamento com Jemperli e corticosteroides.
Outras reações adversas relacionadas ao sistema imune
Dado o mecanismo de ação de Jemperli, podem ocorrer outras reações adversas relacionadas ao sistema imunológico potenciais, incluindo acontecimentos potencialmente graves [por ex. miosite, miocardite, encefalite, neuropatia desmielinizante(incluindo síndrome de Guillain Barré), sarcoidose]. As reações adversas relacionadas ao sistema imunológico clinicamente significativas relatadas em menos de 1% dos pacientes tratados com Jemperli como monoterapia em ensaios clínicos incluem encefalite, anemia hemolítica autoimune, pancreatite, iridociclite, uveíte e cetoacidose diabética. Os pacientes devem ser monitorados quanto a sinais e sintomas de reações adversas relacionadas ao sistema imunológico e administrados conforme descrito no item 8. POSOLOGIA E MODO DE USAR.
Rejeição de transplante de órgão sólido foi relatada no cenário pós-comercialização em pacientes tratados com inibidores de PD1. O tratamento com Jemperli pode aumentar o risco de rejeição em receptores de transplante de órgãos sólidos. O benefício do tratamento com Jemperli versus o risco de possível rejeição de órgãos deve ser considerado nesses pacientes.
Complicações fatais e outras complicações graves podem ocorrer em pacientes que recebem transplante alogênico de célulastronco hematopoiéticas (TCTH) antes ou depois de serem tratados com um anticorpo bloqueador de PD-1/PD-L1. As complicações relacionadas ao transplante incluem doença hiperaguda do enxerto contra o hospedeiro (DECH), DECH aguda, DECH crônica, doença veno-oclusiva hepática após condicionamento de intensidade reduzida e síndrome febril que requer esteroide (sem uma causa infecciosa identificada). Essas complicações podem ocorrer apesar da terapia intermediária entre o bloqueio PD-1 / PD-L1 e o TCTH alogênico. Os pacientes devem ser acompanhados de perto em busca de evidências de complicações relacionadas ao transplante e intervenha imediatamente. Considerar o risco-benefício do tratamento com um anticorpo bloqueador de PD-1 / PD-L1 antes ou após um TCTH alogênico.
Pacientes excluídos dos estudos clínicos
Pacientes com o seguinte status foram excluídos do estudo GARNET: linha de base ECOG basal ≥ 2; metástases não controladas do sistema nervoso central ou meningite carcinomatosa; outras malignidades nos últimos 2 anos; imunodeficiência ou recebendo terapia imunossupressora em 7 dias; infecção ativa por HIV, hepatite B ou hepatite C; doença autoimune ativa que requer tratamento sistêmico nos últimos 2 anos excluindo terapia de reposição; história de doença pulmonar intersticial ou recebendo vacina com agente vivo dentro de 14 dias.
Reações relacionadas à infusão
Jemperli pode causar reações relacionadas à infusão, que podem ser graves (consulte o item 9. REAÇÕES ADVERSAS). Para reações graves à infusão (Grau 3) ou com risco de vida (Grau 4), interrompa a infusão e descontinue permanentemente o uso de Jemperli (consulte o item 8.POSOLOGIA E MODO DE USAR).
Gravidez e lactação
Fertilidade
Não foram realizados estudos de fertilidade com Jemperli.
Gravidez
Não existem dados disponíveis sobre o uso de Jemperli em mulheres grávidas. Não foram realizados estudos de reprodução em animais com Jemperli para avaliar seu efeito na reprodução e no desenvolvimento fetal. Com base em seu mecanismo de ação, Jemperli pode causar danos fetais quando administrado em uma mulher grávida. Modelos animais ligam a via de sinalização PD-1/PD-L1 à manutenção da gravidez através da indução da tolerância imunológica materna ao tecido fetal. Imunoglobulinas humanas IgG4 são conhecidas por atravessar a barreira placentária; portanto, Jemperli tem o potencial de ser transmitido da mãe para o feto em desenvolvimento. Aconselhe mulheres sobre o risco potencial para o feto.
Jemperli não é recomendado durante a gravidez. Aconselhe as mulheres em idade fértil a utilizar métodos contraceptivos altamente eficazes durante o tratamento com Jemperli e durante 4 meses após a última dose.
Categoria D. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Lactação
Não há informações sobre a presença de Jemperli no leite humano, ou seus efeitos na criança amamentada ou na produção do leite. Devido ao potencial de reações adversas graves em crianças amamentadas, aconselhe mulheres a não amamentarem durante
o tratamento e por 4 meses após a última dose de Jemperli.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Jemperli não tem influência ou tem influência insignificante na capacidade de dirigir e usar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos de interação medicamentosa com Jemperli. Os anticorpos monoclonais (mAbs), como o Jemperli, não são substratos para o citocromo P450 ou transportadores de medicamentos. Jemperli não é uma citocina e é improvável que seja um modulador de citocinas. Além disso, não é esperada interação farmacocinética (PK) medicamentosa do Jemperli com medicamentos de moléculas pequenas. Não há evidência de interação medicamentosa mediada pela depuração inespecífica da degradação de lisossomos para anticorpos.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de armazenamento
Os frascos devem ser armazenados sob refrigeração (entre 2 e 8°C). Não congelar. Mantenha na embalagem original até o momento do preparo para proteger da luz. Para condições de armazenamento após o preparo da solução para infusão, consulte o item 8. POSOLOGIA E MODO DE USAR. Por não conter conservantes, o produto não deve ser utilizado após o tempo de armazenamento recomendado. O prazo de validade do medicamento é de 24 meses a partir da data de fabricação.
Número do lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original. Após preparo, manter em temperatura ambiente (até 25°C) por não mais que 6 horas desde o momento da preparação
até o final da infusão OU manter sob refrigeração (entre 2 e 8°C) por não mais que 24 horas desde o momento da preparação até o final da infusão.
Aspectos físicos/características organolépticas
Jemperli é uma solução límpida a ligeiramente opalescente, incolor a amarelada, livre de partículas visíveis. Descarte o frasco-ampola se partículas visíveis forem observadas.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
A classificação do tumor dMMR/MSI-H deve ser determinada usando um teste com método validado como, por exemplo, imunohistoquímica, reação em cadeia da polimerase (PCR) e sequenciamento genético de nova geração (NGS).
Instruções de uso e manuseio
Medicamentos parenterais devem ser inspecionados visualmente quanto a partículas e descoloração antes da administração. Descarte o frasco se partículas visíveis forem observadas.
Jemperli é compatível com uma bolsa intravenosa (IV) feita de cloreto de polivinila (PVC) com ou sem di(2-etilhexil) ftalato (DEHP), etileno vinil acetato, polietileno (PE), polipropileno (PP) ou mistura de poliolefinas (PP+PE), e uma seringa de PP.
Como diluir a solução para infusão
Para a dose de 500 mg, retire 10 ml do frasco de Jemperli e transfira para uma bolsa intravenosa (IV) contendo solução para injeção de cloreto de sódio a 9 mg/ml (0,9%), ou solução para injeção de glicose a 50 mg/ml (5%). A concentração final da solução diluída deve estar entre 2 mg/ml a 10 mg/ml. Isso pode exigir a retirada de um volume de diluente da bolsa IV antes de adicionar um volume de Jemperli na bolsa IV.
-Por exemplo, se preparar uma dose de 500 mg em uma bolsa IV de 250 ml de diluente, para atingir uma concentração de 2 mg/ml, seria necessário retirar 10 ml de diluente da bolsa IV de 250 ml. Em seguida, 10 ml de Jemperli seriam retirados do frasco e transferidos para a bolsa IV.
Para a dose de 1000 mg, retire 10 ml de cada um dos dois frascos de Jemperli (retire 20 ml no total) e transfira para uma bolsa intravenosa contendo solução para injeção de cloreto de sódio a 9 mg/ml (0,9%) ou solução para injeção de glicose a 50 mg/ml (5%). A concentração final da solução diluída deve estar entre 2 mg/ml a 10 mg/ml. Isso pode exigir a retirada de um volume de diluente da bolsa IV antes de adicionar um volume de Jemperli na bolsa IV.
-Por exemplo, se preparar uma dose de 1.000 mg em uma bolsa IV de 500 ml de diluente, para atingir uma concentração de 2 mg/ml, seria necessário retirar 20 ml de diluente da bolsa IV de 500 ml. Em seguida, 10 ml de Jemperli seriam retirados de cada um dos dois frascos, totalizando 20 ml, e transferidos para a bolsa IV.
Misture a solução diluída por inversão suave. Não agite a bolsa de infusão final. Descarte qualquer parte não utilizada deixada no frasco.
A dose preparada pode ser armazenada:
-em temperatura ambiente até 25°C, por no máximo 6 horas, desde o momento da diluição até o final da infusão.
-sob refrigeração, de 2°C a 8°C, por não mais de 24 horas, desde o momento da diluição até o final da infusão. Se refrigerado, deixe a solução diluída atingir a temperatura ambiente antes da administração.
Como administrar a solução diluída
Jemperli deve ser administrado somente por infusão intravenosa, por um profissional de saúde, usando uma bomba de infusão intravenosa por 30 minutos. A tubulação deve ser feita de PVC, silicone curado com platina ou PP; acessórios de PVC ou policarbonato e agulhas de aço inoxidável. Um filtro de polietersulfona (PES) em linha de 0,2 ou 0,22 mícron deve ser usado durante a administração de Jemperli.
Jemperli não deve ser administrado como injeção intravenosa ou injeção em bolus. Não coadministre outros medicamentos através da mesma linha de infusão. Qualquer medicamento não utilizado ou residual deve ser descartado de acordo com as exigências locais.
POSOLOGIA
A dose recomendada de Jemperli como monoterapia é de 500 mg administrados como infusão intravenosa durante 30 minutos a cada 3 semanas em 4 ciclos, seguidos por 1000 mg a cada 6 semanas em todos os ciclos subsequentes.
O regime de administração é apresentado na Tabela 2.
A administração de Jemperli deve continuar de acordo com a dose e com o cronograma recomendado até a progressão da doença ou toxicidade inaceitável. Pacientes com progressão de doença radiológica não associada à deterioração clínica significativa, definida como nenhum sintoma novo ou agravado, nenhuma alteração no performance status (PS) por mais de duas semanas e nenhuma necessidade de terapia de resgate, podem continuar o tratamento.
Alterações de dose
A redução da dose não é recomendada. O atraso ou descontinuação da dose pode ser necessário com base na segurança e tolerabilidade individuais. As modificações recomendadas para gerenciar reações adversas estão fornecidas na Tabela 3.
Diretrizes detalhadas para o gerenciamento de reações adversas relacionadas ao sistema imunológico e reações relacionadas à infusão são descritas no item 5. ADVERTÊNCIAS E PRECAUÇÕES.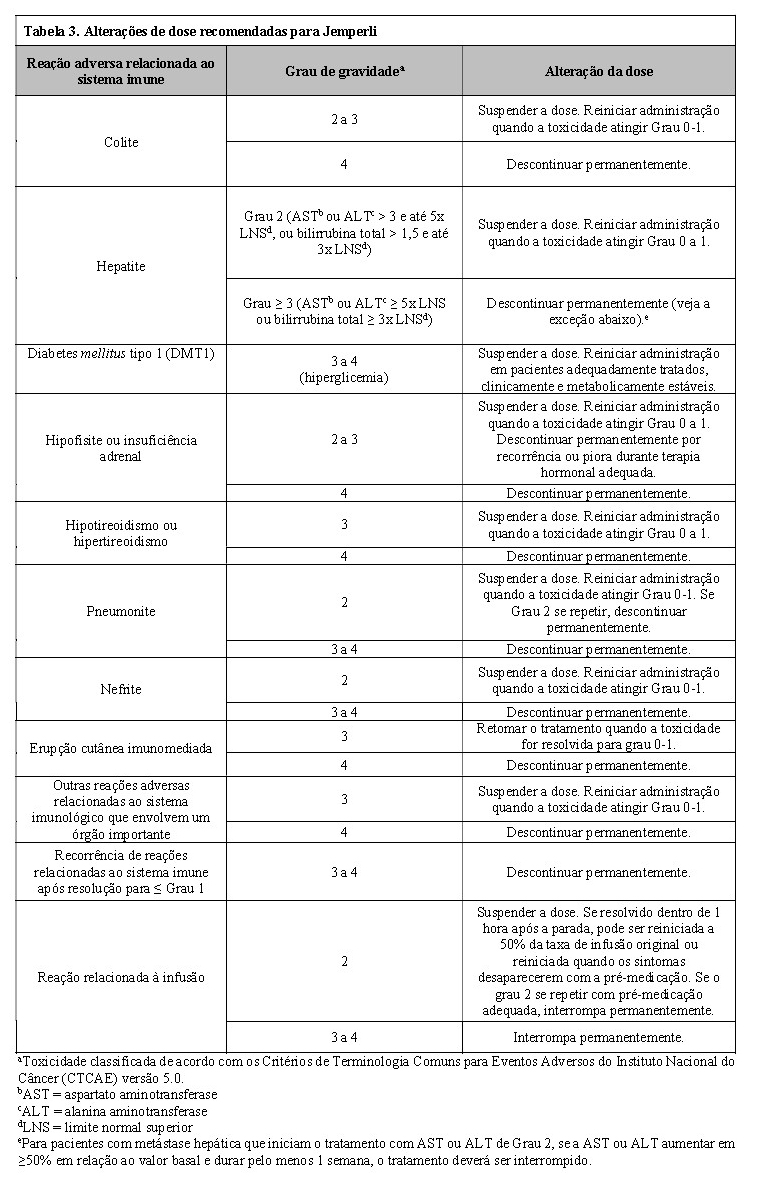
Crianças A segurança e eficácia de Jemperli em crianças e adolescentes com menos de 18 anos de idade não foram estabelecidas. Não há dados disponíveis.
Idosos
Nenhum ajuste da dose é recomendado em pacientes com idade igual ou superior a 65 anos. Os dados clínicos com Jemperli são limitados em pacientes com 75 anos ou mais.
Comprometimento renal
Nenhum ajuste de dose é recomendado em pacientes com insuficiência renal leve ou moderada. Os dados são limitados em pacientes com comprometimento renal grave ou doença renal terminal em diálise (consulte o item Farmacocinética na seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Comprometimento hepático
Nenhum ajuste de dose é recomendado em pacientes com comprometimento hepático leve. Os dados são limitados em pacientes com comprometimento hepático moderado ou grave (consulte o item Farmacocinética na seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
9. REAÇÕES ADVERSAS
Dados de estudos clínicos
As reações adversas observadas em pacientes com tumores sólidos recorrentes ou avançados que receberam monoterapia com Jemperli no estudo GARNET multicoorte aberto estão listados na Tabela 4. Reações adversas adicionais relacionadas ao sistema imunológico identificadas com base em dados agrupados gerados a partir de outros ensaios clínicos em pacientes com tumores sólidos recebendo Jemperli em combinação com vários tipos de terapias anticâncer também são mostradas na Tabela 4.
Essas reações são apresentadas por classe de sistema orgânico e por frequência. As frequências são definidas como: muito comum (≥ 1/10); comum (≥ 1/100 a < 1/10); incomum (≥ 1/1000 a < 1/100); raro (≥ 1/10000 a < 1/1000); muito raro ( < 1/10000) e desconhecido (não pode ser calculado a partir dos dados disponíveis).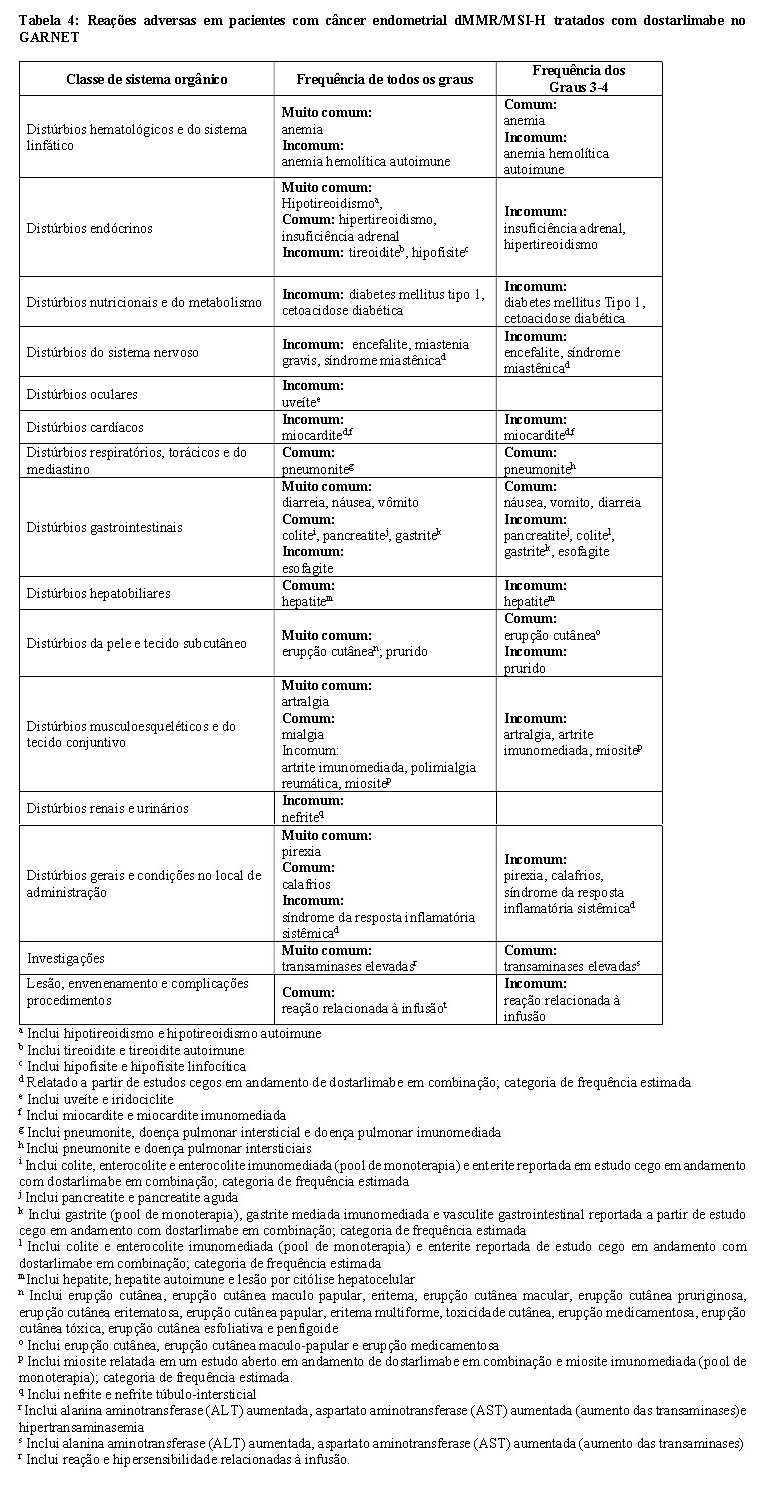
Imunogenicidade
Assim como todas as proteínas terapêuticas, existe potencial para imunogenicidade. A detecção da formação de anticorpos é altamente dependente da sensibilidade e especificidade do ensaio. Adicionalmente, a incidência observada da positividade de anticorpos (incluindo anticorpos neutralizantes) em um ensaio pode ser influenciada por vários fatores, incluindo metodologia de ensaio, manipulação de amostras, tempo de coleta de amostras, medicações concomitantes e doenças subjacentes. Por esses motivos, a comparação da incidência de anticorpos contra o dostarlimabe nos estudos descritos abaixo com a incidência de anticorpos em outros estudos ou com outros produtos pode ser enganosa.
No estudo GARNET, anticorpos contra o medicamento (ADA) foram testados em 384 pacientes que receberam Jemperli ea incidência de ADAs emergentes do tratamento com dostarlimabe foi de 2,5%. Anticorpos neutralizantes foram detectados em 1,3% dos pacientes. Nos pacientes que desenvolveram anticorpos antidostarlimabe, não houve evidência de farmacocinética, eficácia ou segurança alteradas de dostarlimabe.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Se houver suspeita de superdosagem, o paciente deve ser monitorado quanto a sinais ou sintomas de reações ou efeitos adversos, e medidas adequadas de tratamento devem ser instituídas imediatamente.
Em caso de intoxicação, ligue para 0800 722 6001 se você precisar de mais orientações.
Dizeres legais.
Reg. MS: 1.0107.0355
USO RESTRITO A HOSPITAIS
VENDA SOB PRESCRIÇÃO MÉDICA.