JANUVIA®
MSD
sitagliptina, fosfato
Hipoglicemiante oral.
Apresentações.
JANUVIA® é apresentado em comprimidos revestidos de 25 mg e 50 mg acondicionados em caixas com 28 comprimidos e em comprimidos revestidos de 100 mg acondicionados em caixas com 14 e 28 comprimidos.
Uso Oral.
Uso Adulto.
Composição.
Ingrediente ativo: Cada comprimido revestido de JANUVIA® contém 32,13 mg, 64,25 mg e 128,5 mg de fosfato de sitagliptina, equivalente a 25 mg, 50 mg e 100 mg, respectivamente, de base livre. Ingredientes inativos: Cada comprimido revestido de JANUVIA® contém: celulose microcristalina, fosfato de cálcio dibásico anidro, croscarmelose sódica, estearato de magnésio e estearil fumarato de sódio. Além disso, o revestimento contém os seguintes ingredientes inativos: álcool polivinílico, polietilenoglicol (macrogol), talco, dióxido de titânio, óxido de ferro vermelho e óxido de ferro amarelo.
Indicações.
Monoterapia
JANUVIA® é indicado como adjuvante à dieta e à prática de exercícios para melhorar o controle glicêmico em pacientes com diabetes mellitus tipo 2.
Terapia Combinada
JANUVIA® também é indicado para pacientes com diabetes mellitus tipo 2 para melhorar o controle glicêmico em combinação com a metformina ou com um agonista de PPARc (por exemplo, tiazolidinediona) quando a dieta e os exercícios, além do agente único, não proporcionam controle glicêmico adequado.
Caract. farmacológicas.
Os comprimidos de JANUVIA® contêm sitagliptina, um inibidor potente e seletivo ativo por via oral da dipeptidil peptidase 4 (DPP-4), descrito quimicamente como: fosfato de 7-[(3R)-3-amino-1-oxo-4-(2,4,5-trifluorfenil)butil]-5,6,7,8-tetrahidro-3-(trifluormetil)-1,2,4-triazol[4,3-a] pirazina (1:1) monoidratado.
A fórmula empírica é C16H15F6N5O•H3PO4•H2O e o peso molecular é 523,32. A fórmula estrutural é: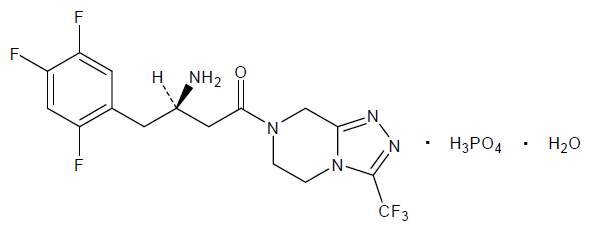
A sitagliptina é um pó cristalino, não higroscópico, branco a esbranquiçado. O composto é solúvel em água e N,N-dimetilformamida, discretamente solúvel em metanol, muito pouco solúvel em etanol, acetona e acetonitrila e insolúvel em isopropanol e acetato de isopropila.
Mecanismo de Ação
JANUVIA® pertence a uma classe de agentes hipoglicemiantes orais denominada inibidores da dipeptidil peptidase 4 (DPP-4), que melhoram o controle glicêmico em pacientes com diabetes tipo 2 por meio do aumento dos níveis de hormônios incretina ativos. Os hormônios incretina, inclusive o peptídeo-1 semelhante ao glucagon (GLP-1) e o peptídeo insulinotrópico dependente de glicose (GIP), são liberados pelo intestino ao longo do dia e seus níveis aumentam em resposta a uma refeição. As incretinas são parte de um sistema endógeno envolvido na regulação fisiológica da homeostase da glicose. Quando as concentrações sanguíneas de glicose estão normais ou elevadas, o GLP-1 e o GIP aumentam a síntese e a liberação de insulina das células pancreáticas beta por meio de vias sinalizadoras intracelulares que envolvem o AMP cíclico. O tratamento com GLP-1 ou com inibidores da DPP-4 em modelos animais de diabetes tipo 2 demonstrou melhorar a responsividade das células beta à glicose e estimular a biossíntese e a liberação de insulina. Com níveis de insulina mais altos, a captação tecidual de glicose é aumentada. Além disso, o GLP-1 diminui a secreção de glucagon pelas células pancreáticas alfa. A redução das concentrações de glucagon, associada a níveis mais altos de insulina, resulta em redução da produção hepática de glicose e conseqüente redução da glicemia. Quando as concentrações sanguíneas de glicose estão baixas, não são observadas estimulação da liberação de insulina e supressão da secreção de glucagon pelo GLP-1. O GLP-1 e o GIP não impedem a resposta normal do glucagon à hipoglicemia. A atividade do GLP-1 e do GIP é limitada pela enzima DPP-4, que hidrolisa rapidamente os hormônios incretina para produzir produtos inativos. A sitagliptina evita a hidrólise dos hormônios incretina pelo DPP-4, aumentando conseqüentemente as concentrações plasmáticas das formas ativas de GLP-1 e GIP. Ao aumentar os níveis de incretina ativa, a sitagliptina aumenta a liberação de insulina e diminui os níveis de glucagon de forma dependente da glicose. Em pacientes com diabetes tipo 2 com hiperglicemia, essas alterações nos níveis de insulina e de glucagon resultam em níveis mais baixos de hemoglobina A1c (HbA1c) e concentrações mais baixas da glicemia de jejum e pós-prandial. Embora a sitagliptina seja um inibidor potente e altamente seletivo da enzima DPP-4, ela não inibe as enzimas estreitamente relacionadas DPP-8 ou DPP-9. A inibição da DPP-8 ou da DPP-9, mas não da DPP-4, está associada a toxicidade nos modelos animais pré-clínicos e a alteração da função imunológica in vitro.
Farmacocinética
A farmacocinética da sitagliptina foi amplamente caracterizada em indivíduos saudáveis e em pacientes com diabetes tipo 2. Após a administração de uma dose de 100 mg por via oral a voluntários saudáveis, a sitagliptina foi rapidamente absorvida e as concentrações plasmáticas máximas (Tmáx mediano) ocorreram 1 a 4 horas após a dose. A área sob a curva (AUC) plasmática da sitagliptina aumentou de forma proporcional à dose. Após uma dose única de 100 mg por via oral a voluntários saudáveis, a AUC plasmática média da sitagliptina foi de 8,52 mcgM•h, a concentração máxima (Cmáx) foi de 950 nM e a meia-vida terminal aparente (t½) foi de 12,4 horas. No estado de equilíbrio, a AUC plasmática da sitagliptina aumentou aproximadamente 14% após doses de 100 mg em comparação com a primeira dose. Os coeficientes de variação intra-individuais e interindividuais para a AUC da sitagliptina foram pequenos (5,8% e 15,1%). A farmacocinética da sitagliptina foi, em geral, semelhante em voluntários saudáveis e em pacientes com diabetes tipo 2.
Absorção
A biodisponibilidade absoluta da sitagliptina é de aproximadamente 87%. Uma vez que a co-administração de uma refeição rica em gordura com JANUVIA® não exerceu efeito na farmacocinética, JANUVIA® pode ser administrado com ou sem alimentos.
Distribuição
Após uma dose única de 100 mg de sitagliptina por via intravenosa a voluntários saudáveis, o volume médio de distribuição no estado de equilíbrio é de aproximadamente 198 litros. A fração da sitagliptina que se liga reversivelmente às proteínas plasmáticas é pequena (38%).
Metabolismo
A sitagliptina é eliminada principalmente de forma inalterada na urina; o metabolismo é uma via de menor importância. Aproximadamente 79% da sitagliptina é excretada de forma inalterada na urina.
Após uma dose oral de [14C]sitagliptina, aproximadamente 16% da radioatividade foi excretada na forma de metabólitos de sitagliptina. Seis metabólitos foram detectados em níveis-traço e não se espera que contribuam para a atividade inibitória de DPP-4 plasmática da sitagliptina. Estudos in vitro indicaram que a principal enzima responsável pelo metabolismo limitado da sitagliptina foi a CIP3A4, com contribuição da CIP2C8.
Eliminação
Após a administração de uma dose de [14C]sitagliptina por via oral a voluntários saudáveis, aproximadamente 100% da radioatividade administrada foi eliminada nas fezes (13%) ou na urina (87%) uma semana após a administração. A t½ terminal aparente após uma dose de 100 mg de sitagliptina por via oral foi de aproximadamente 12,4 horas e a depuração renal foi de aproximadamente 350 mL/min.
A eliminação da sitagliptina ocorre principalmente por excreção renal e envolve secreção tubular ativa. A sitagliptina é um substrato para o transportador-3 aniônico orgânico humano (hOAT-3), que pode estar envolvido na eliminação renal da sitagliptina. A relevância clínica do hOAT-3 no transporte da sitagliptina não foi estabelecida. A sitagliptina também é um substrato da p-glicoproteína, que também pode estar envolvida na mediação da eliminação renal da sitagliptina. No entanto, a ciclosporina, um inibidor da p-glicoproteína, não reduziu a depuração renal da sitagliptina.
Populações Específicas
Insuficiência Renal: foi conduzido um estudo aberto, de dose única, para avaliar a farmacocinética de JANUVIA® (dose de 50 mg) em pacientes com graus variados de insuficiência renal crônica em comparação com voluntários saudáveis de controle. O estudo incluiu pacientes com insuficiência renal classificada (com base no clearance de creatinina) como leve (50 a < 80 mL/min), moderada (30 a < 50 mL/min) e grave ( < 30 mL/min), bem como pacientes com insuficiência renal terminal (IRT), em hemodiálise. O clearance de creatinina foi medido pelas determinações do clearance de creatinina urinária em 24 horas ou estimado a partir da creatinina sérica com base na fórmula de Cockcroft-Gault:
CrCl= [140 - idade (anos)] x peso (kg) [72 x creatinina sérica (mg/dL)] {x 0,85 para pacientes do sexo feminino}
Os pacientes com insuficiência renal leve não apresentaram aumento clinicamente significativo das concentrações plasmáticas da sitagliptina em comparação com os voluntários saudáveis de controle. Em comparação com os voluntários saudáveis de controle, observou-se aumento de aproximadamente 2 vezes na AUC plasmática da sitagliptina em pacientes com insuficiência renal moderada e aumento de aproximadamente 4 vezes em pacientes com insuficiência renal grave e naqueles com IRT em hemodiálise. A sitagliptina foi modestamente removida por hemodiálise (13,5% durante uma sessão de hemodiálise de 3 a 4 horas, iniciando-se 4 horas após a dose). Para atingir concentrações plasmáticas de sitagliptina semelhantes às observadas em pacientes com função renal normal, recomenda-se doses mais baixas para pacientes com insuficiência renal moderada e grave, bem como para pacientes com IRT e necessidade de hemodiálise (veja POSOLOGIA E ADMINISTRAÇÃO, Pacientes com Insuficiência Renal).
Insuficiência Hepática: após a administração de uma dose única de 100 mg de JANUVIA® a pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9), a AUC média e a Cmáx da sitagliptina aumentaram aproximadamente 21% e 13%, respectivamente, em comparação aos controles pareados saudáveis. Essas diferenças não são consideradas clinicamente significativas. Não é necessário ajuste posológico de JANUVIA® para pacientes com insuficiência hepática leve ou moderada.
Não existe experiência clínica em pacientes com insuficiência hepática grave (escore de Child-Pugh > 9). No entanto, como a sitagliptina é eliminada principalmente por via renal, não se espera que a insuficiência hepática grave afete a farmacocinética da sitagliptina.
Idosos: não é necessário ajuste posológico com base na idade. A idade não exerceu impacto clinicamente significativo na farmacocinética da sitagliptina com base em uma análise da farmacocinética populacional dos dados de estudos fases I e II. Os voluntários idosos (65 a 80 anos de idade) apresentaram concentrações plasmáticas da sitagliptina aproximadamente 19% mais altas em comparação com os voluntários mais jovens.
Pacientes Pediátricos: não foram conduzidos estudos com JANUVIA® em pacientes pediátricos.
Sexo: não é necessário ajuste posológico com base no sexo. O sexo não exerceu efeito clinicamente significativo na farmacocinética da sitagliptina com base em uma análise composta dos dados de farmacocinética de estudos fase I e em uma análise de farmacocinética populacional dos dados de estudos fases I e II.
Raça: não é necessário ajuste posológico com base na raça. A raça não exerceu efeito clinicamente significativo na farmacocinética da sitagliptina com base em uma análise composta dos dados de farmacocinética de estudos fase I e em uma análise de farmacocinética populacional dos dados de estudos fases I e II, que incluíram voluntários brancos, hispânicos, negros, asiáticos e de outros grupos raciais.
Índice de Massa Corporal (IMC): não é necessário ajuste posológico com base no IMC. O índice de massa corporal não exerceu efeito clinicamente significativo na farmacocinética da sitagliptina com base em uma análise composta dos dados de farmacocinética de estudos fase I e em uma análise de farmacocinética populacional dos dados de estudos fases I e II.
Diabetes Tipo 2: a farmacocinética da sitagliptina em pacientes com diabetes tipo 2 é, em geral, semelhante à de voluntários saudáveis.
Farmacodinâmica
Resultados de Eficácia
Em pacientes com diabetes tipo 2, a administração de doses únicas de JANUVIA® por via oral leva a inibição da atividade enzimática da DPP-4 por um período de 24 horas, o que resulta em aumento de 2 a 3 vezes nos níveis circulantes de GLP-1 e GIP ativos, aumento dos níveis plasmáticos de insulina e de peptídeo-C, redução das concentrações de glucagon, redução da glicemia de jejum e redução dos picos de glicose após uma sobrecarga oral de glicose ou refeição.
Em um estudo que envolveu pacientes com diabetes tipo 2 controlados inadequadamente com monoterapia com metformina, os níveis de glicose monitorados ao longo do dia foram significativamente mais baixos entre os pacientes que receberam 100 mg/dia de JANUVIA® em combinação com metformina em relação aos observados entre os pacientes que receberam placebo e metformina (veja Figura 1).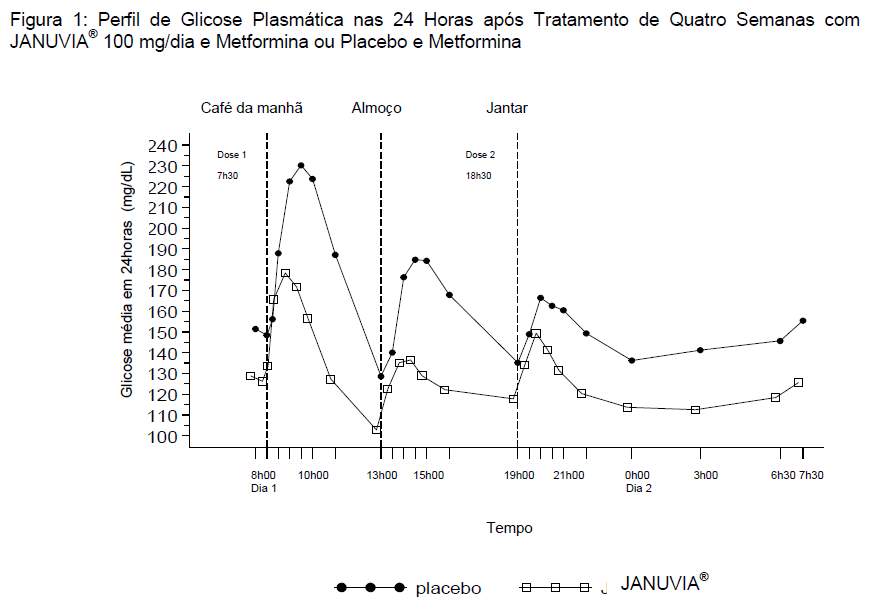
Nos estudos clínicos fase III, com duração de 18 e 24 semanas, o tratamento com 100 mg/dia em pacientes com diabetes tipo 2 melhorou significativamente a função da célula beta, conforme determinado por vários marcadores, inclusive HOMA- (Avaliação de um Modelo de Homeostase-), razão pró-insulina:insulina e medidas de responsividade da célula beta ao teste de tolerância à refeição amostradas freqüentemente.
Nos estudos fase II, a administração de 50 mg de JANUVIA® duas vezes ao dia não proporcionou eficácia glicêmica adicional em comparação com a dose de 100 mg uma vez ao dia.
Nos estudos que envolveram voluntários saudáveis, JANUVIA® não diminuiu a glicemia nem causou hipoglicemia, o que sugere que as ações insulinotrópicas e supressoras de glucagon do fármaco são dependentes da glicose.
Efeitos na Pressão Arterial
Em um estudo cruzado, randômico, controlado com placebo, conduzido em pacientes hipertensos que recebiam um ou mais anti-hipertensivos (inclusive inibidores da enzima conversora de angiotensina, antagonistas da angiotensina II, bloqueadores dos canais de cálcio, beta-bloqueadores e diuréticos), a co-administração de JANUVIA® foi geralmente bem tolerada. Nesses pacientes, JANUVIA® exerceu efeito redutor discreto na pressão arterial; em comparação com o placebo, o tratamento com 100 mg/dia de JANUVIA® reduziu a pressão arterial sistólica ambulatorial média de 24 horas em aproximadamente 2 mmHg. Não foram observadas reduções em voluntários normotensos.
Eletrofisiologia Cardíaca
Período basal
Em um estudo cruzado, randômico, controlado com placebo, 79 voluntários saudáveis receberam uma dose única de 100 mg e de 800 mg de JANUVIA® (8 vezes a dose recomendada) por via oral e placebo. A dose recomendada de 100 mg não exerceu efeito no intervalo QTc na concentração plasmática máxima ou em qualquer outro ponto de tempo durante o estudo. Após a dose de 800 mg,
o aumento máximo da alteração média do intervalo QTc corrigida pelo placebo em relação ao período basal três horas após a dose foi de 8,0 ms; este pequeno aumento não foi considerado clinicamente significativo. As concentrações plasmáticas máximas de 800 mg de sitagliptina foram aproximadamente 11 vezes mais altas do que as concentrações máximas após uma dose de 100 mg.
Os pacientes com diabetes tipo 2 que receberam 100 mg (N= 81) ou 200 mg de JANUVIA® (N= 63) diariamente não apresentaram alterações significativas no intervalo QTc com base nos dados de ECG obtidos no momento da concentração plasmática máxima esperada.
ESTUDOS CLÍNICOS
No total, 2.316 pacientes com diabetes tipo 2 foram distribuídos de modo randômico em quatro estudos clínicos fase III duplo-cegos, controlados com placebo, que avaliaram os efeitos da sitagliptina no controle glicêmico. No grupo estudado, foram comuns co-morbidades: 58% apresentavam hipertensão, 53%, dislipidemia, e mais de 50% eram obesos (IMC 30 kg/m2). A maioria dos pacientes (51,6% a 65,8%) preenchia os critérios diagnósticos do NCEP (National Cholesterol Education Program - Programa de Educação sobre o Colesterol dos EUA) para síndrome metabólica. Nesses estudos, a média de idade dos pacientes foi de 54,8 anos. Além disso, os pacientes dividiam-se em: 62% brancos, 18% hispânicos, 6% negros, 9% asiáticos e 4% de outros grupos raciais.
Foram conduzidos outros estudos clínicos duplo-cegos, controlados com placebo: um deles envolveu 151 pacientes japoneses com diabetes tipo 2 e outro, 91 pacientes com diabetes tipo 2 e insuficiência renal moderada a grave.
Em pacientes com diabetes tipo 2, o tratamento com JANUVIA® melhorou de forma clinicamente significativa a hemoglobina A1c (HbA1c), a glicemia de jejum (GJ) e a glicemia pós-prandial de 2 horas (GPP). JANUVIA® proporcionou melhora nas medidas de função das células beta (veja CARACTERÍSTICAS FARMACOLÓGICAS, Farmacodinâmica).
Estudos Clínicos de Monoterapia
Um total de 1.262 pacientes com diabetes tipo 2 participou de dois estudos duplo-cegos, controlados com placebo, com 18 e 24 semanas de duração, para avaliar a eficácia e a segurança da monoterapia com JANUVIA®. Os pacientes com controle glicêmico inadequado (HbA1c 7% a 10%) foram distribuídos de modo randômico para receber uma dose de 100 mg ou 200 mg de JANUVIA® ou placebo uma vez ao dia.
O tratamento com 100 mg/dia de JANUVIA® proporcionou melhoras significativas da HbA1c, da GJ e da GPP de 2 horas em comparação com o placebo (Tabelas 1 e 2). Esses estudos incluíram pacientes cujos níveis de HbA1c situavam-se em um amplo intervalo no período basal. A melhora da HbA1c não foi afetada por sexo, idade, raça, IMC no período basal, presença de síndrome metabólica ou índice-padrão de resistência à insulina (HOMA-IR). Os pacientes com diagnóstico de diabetes mais recente ( < 3 anos) ou com HbA1c no período basal mais elevada apresentaram maiores reduções de HbA1c. Em ambos os estudos, JANUVIA® proporcionou redução significativa da GJ em comparação com o placebo (-19,3 mg/dL no estudo de 18 semanas e -15,8 mg/dL no estudo de 24 semanas) em 3 semanas, o primeiro ponto de tempo no qual a GJ foi avaliada. No geral, a dose diária de 200 mg não proporcionou maior eficácia glicêmica do que a dose diária de 100 mg. O efeito de JANUVIA® nos desfechos lipídicos foi semelhante ao do placebo. O peso corporal não aumentou em relação ao período basal com a terapia com JANUVIA® em qualquer um dos estudos, em comparação a uma pequena redução observada entre os pacientes que receberam placebo (Tabela 2). A incidência observada de hipoglicemia no grupo que recebeu JANUVIA® foi semelhante à observada no grupo que recebeu placebo.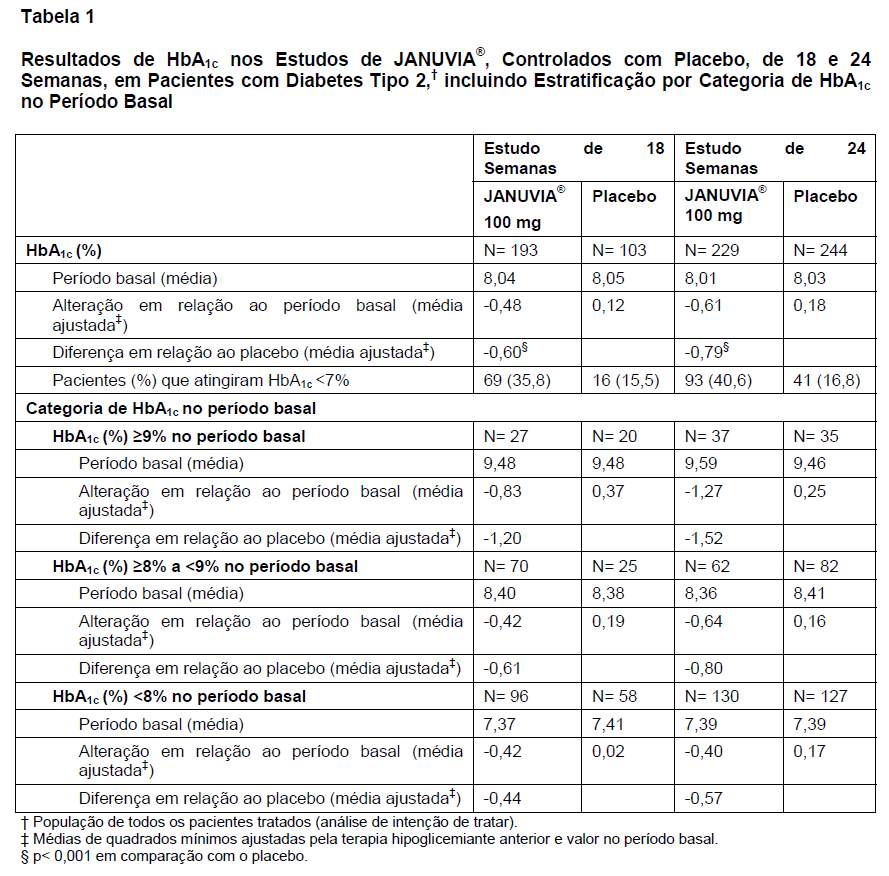
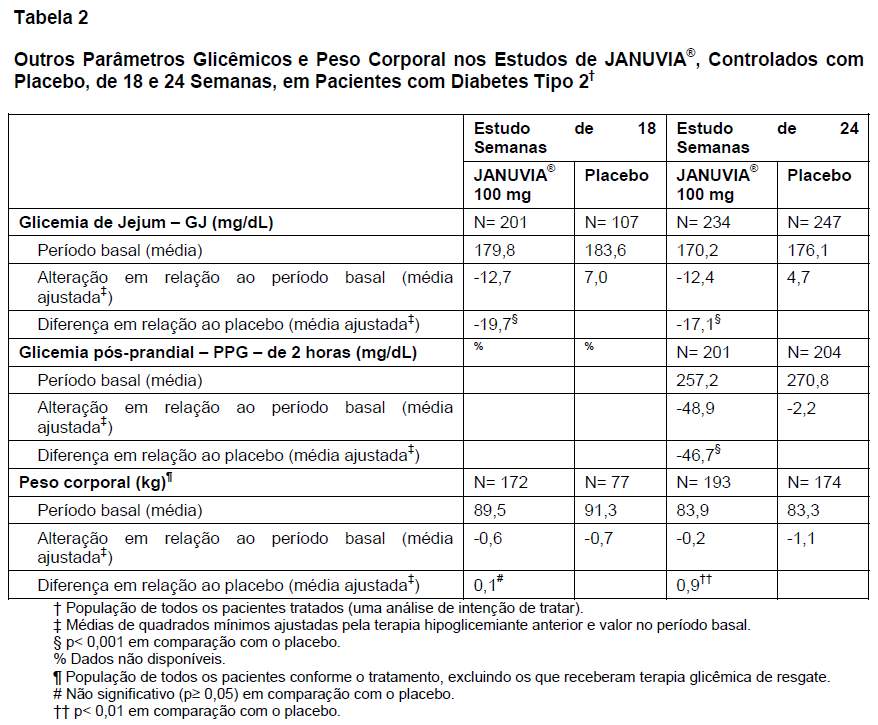
Outros Estudos de Monoterapia
Um estudo duplo-cego e controlado com placebo, que envolveu pacientes japoneses com diabetes tipo 2, avaliou a eficácia do tratamento com 100 mg de JANUVIA® uma vez ao dia em comparação com o placebo. Este estudo incluiu 151 pacientes (75 receberam JANUVIA® e 76 placebo) com média de idade de 55,3 anos e, no período basal, IMC de 25,2 kg/m2, HbA1c média de 7,6% e GJ média de 163 mg/DL. Após 12 semanas, JANUVIA® proporcionou redução de 1,05% da HbA1c em relação ao placebo (JANUVIA®, -0,65% de alteração em relação ao período basal; placebo, 0,41% [p < 0,001]). A GJ diminuiu 31,9 mg/dL em relação ao placebo (JANUVIA®, -22,5 mg/dL de alteração em relação ao período basal; placebo, 9,4 mg/dL [p < 0,001]).
Também foi conduzido um estudo multinacional, randômico, duplo-cego, controlado com placebo, para avaliar a segurança e a tolerabilidade de JANUVIA® em 91 pacientes com diabetes tipo 2 e insuficiência renal crônica (clearance de creatinina < 50 mL/min). Os pacientes com insuficiência renal moderada receberam 50 mg/dia de JANUVIA® e aqueles com insuficiência renal grave ou IRT em diálise receberam 25 mg/dia. Nesse estudo, a segurança e a tolerabilidade de JANUVIA® foram, em geral, semelhantes às do placebo. Além disso, as reduções da HbA1c e da GJ com JANUVIA® em comparação com o placebo foram, em geral, semelhantes às observadas em outros estudos de monoterapia (veja CARACTERÍSTICAS FARMACOLÓGICAS, Populações Específicas, Insuficiência Renal).
Terapia Combinada com Metformina
Um total de 701 pacientes com diabetes tipo 2 participou de um estudo de 24 semanas de duração, randômico, duplo-cego, controlado com placebo, que avaliou a eficácia de JANUVIA® em combinação com a metformina. Todos os pacientes iniciaram o tratamento com a metformina em monoterapia, cuja dose foi titulada para 1.500 mg/dia, no mínimo. Os pacientes foram distribuídos de modo randômico para receber a adição de 100 mg de JANUVIA® ou placebo, em dose única diária.
Em combinação com a metformina, JANUVIA® proporcionou melhoras significativas da HbA1c, da GJ, e da GPP de 2 horas em comparação com a associação de placebo e metformina (Tabela 3). A melhora da HbA1c não foi afetada pelos níveis de HbA1c no período basal, terapia hipoglicemiante anterior, sexo, idade, IMC no período basal, tempo transcorrido desde o diagnóstico de diabetes, presença de síndrome metabólica ou índice-padrão de resistência à insulina (HOMA-IR) ou secreção de insulina (HOMA-). O peso corporal diminuiu em relação ao período basal em ambos os grupos de tratamento.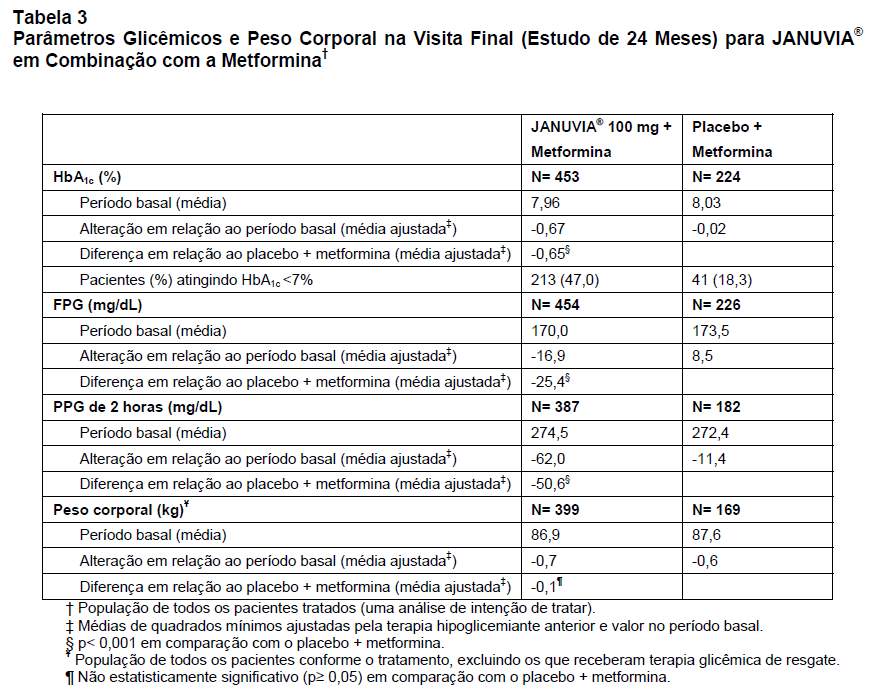
Terapia Combinada com Pioglitazona
Um total de 353 pacientes com diabetes tipo 2 participou de um estudo de 24 semanas de duração, randômico, duplo-cego e controlado com placebo, que avaliou a eficácia de JANUVIA® em combinação com a pioglitazona. No início do estudo, todos os pacientes receberam monoterapia com pioglitazona em uma dose de 30-45 mg por dia e, a seguir, foram distribuídos de modo randômico para receber a adição de 100 mg de JANUVIA® ou placebo, em dose única diária. Os desfechos glicêmicos avaliados incluíram HbA1c e glicemia de jejum.
Em combinação com a pioglitazona, JANUVIA® proporcionou melhoras significativas da HbA1c e da GJ em comparação com o placebo associado à pioglitazona (Tabela 4). A melhora da HbA1c não foi afetada pela HbA1c no período basal, terapia hipoglicemiante anterior, sexo, idade, raça, IMC no período basal, tempo transcorrido desde o diagnóstico de diabetes, presença de síndrome metabólica ou índice-padrão de resistência à insulina (HOMA-IR) ou secreção de insulina (HOMA-). O tratamento com JANUVIA® não aumentou significativamente o peso corporal em relação ao período basal em comparação com o placebo.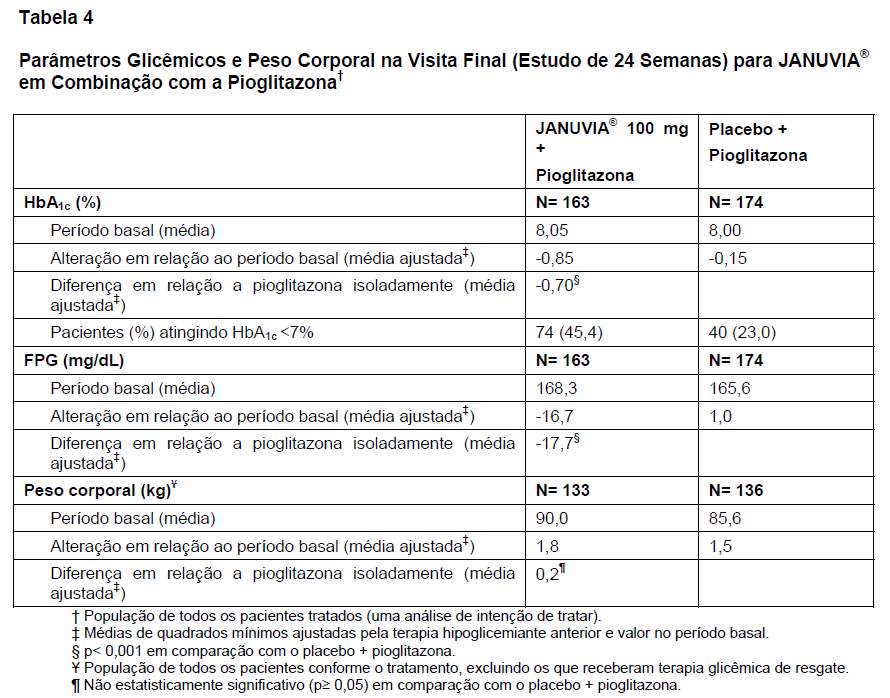
Contraindicações.
JANUVIA® é contra-indicado para pacientes com hipersensibilidade a qualquer um dos seus componentes.
Advertências.
Gerais
JANUVIA® não deve ser utilizado por pacientes com diabetes tipo 1 ou para o tratamento de cetoacidose diabética.
Hipoglicemia: nos estudos clínicos de JANUVIA® como monoterapia e JANUVIA® como parte da terapia combinada com a metformina ou a pioglitazona, as taxas de hipoglicemia relatadas com JANUVIA® foram semelhantes às observadas em pacientes que recebiam placebo. O uso de JANUVIA® em combinação com medicamentos que sabidamente causam hipoglicemia, como as sulfoniluréias ou a insulina, ainda não foi adequadamente estudado.
Insuficiência Renal: recomenda-se ajuste posológico para pacientes com insuficiência renal moderada ou grave e para pacientes com IRT que requeiram hemodiálise (veja POSOLOGIA E ADMINISTRAÇÃO, Pacientes com Insuficiência Renal).
Gravidez
Categoria de risco: B
A sitagliptina não foi teratogênica para ratos em doses orais de até 250 mg/kg ou para coelhos que receberam 125 mg/kg durante a organogênese (até 32 e 22 vezes, respectivamente, a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos). Em ratos, observou-se discreto aumento da incidência de malformações das costelas fetais (ausência, hipoplasia e costelas flutuantes) com doses orais de 1.000 mg/kg/dia (aproximadamente 100 vezes a exposição em humanos com base na dose diária recomendada de 100 mg/dia para humanos adultos). Na prole de ratos que receberam doses orais de 1.000 mg/kg/dia, foram observadas discretas reduções dos pesos corporais médios pré-desmame em ambos os sexos e ganhos de peso corporal pós-desmame em machos. No entanto, estudos de reprodução animal nem sempre são preditivos da resposta humana.
Não existem estudos adequados e bem controlados conduzidos em mulheres grávidas; portanto, não se conhece a segurança de JANUVIA® nessa população. O uso de JANUVIA®, assim como o de outros agentes hipoglicemiantes orais, não é recomendado na gravidez.
Lactação
A sitagliptina é secretada no leite de ratas lactantes. Não se sabe se a sitagliptina é secretada no leite humano; portanto, JANUVIA® não deve ser utilizado por uma mulher que esteja amamentando.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Uso Pediátrico
A segurança e a eficácia de JANUVIA® em pacientes pediátricos não foram estabelecidas.
Uso em Idosos
Nos estudos clínicos, a segurança e a eficácia de JANUVIA® em idosos (65 anos, N= 409) foram comparáveis às observadas em pacientes mais jovens ( < 65 anos). Não é necessário ajuste posológico com base na idade. A probabilidade de pacientes idosos apresentarem insuficiência renal é maior; assim como para outros pacientes, podem ser necessários ajustes posológicos para os idosos na presença de insuficiência renal significativa (veja POSOLOGIA E ADMINISTRAÇÃO, Pacientes com Insuficiência Renal).
Interações medicamentosas.
Nos estudos de interação medicamentosa, a sitagliptina não exerceu efeitos clinicamente significativos na farmacocinética dos seguintes fármacos: metformina, rosiglitazona, gliburida, sinvastatina, varfarina e anticoncepcionais orais. Com base nesses dados, a sitagliptina não inibe as isoenzimas do sistema do citocromo P450 (CIP) 3A4, 2C8 ou 2C9. Com base nos dados in vitro, também não é esperado que a sitagliptina iniba as isoenzimas 2D6, 1A2, 2C19 ou 2B6 ou induza a isoenzima 3A4 desse sistema enzimático.
Quando a digoxina foi co-administrada com o fosfato de sitaglipitina, houve discreto aumento na AUC (11%) e na média da Cmáx (18%) da digoxina; estes aumentos não parecem ser clinicamente significativos. Os pacientes em tratamento com digoxina devem ser monitorados de forma apropriada. Não é recomendado ajuste posológico da digoxina ou de JANUVIA® .
A AUC e a Cmáx da sitagliptina aumentaram aproximadamente 29% e 68%, respectivamente, em indivíduos que receberam a co-administração de uma dose única de 100 mg de JANUVIA® por via oral e uma dose única de 600 mg de ciclosporina por via oral, um potente inibidor investigativo da pglicoproteína. As alterações observadas na farmacocinética da sitagliptina não parecem ser clinicamente significativas. Não é recomendado ajuste posológico de JANUVIA® quando coadministrado com a ciclosporina ou outros inibidores da p-glicoproteína (por exemplo, cetoconazol).
Uma análise da farmacocinética populacional dos pacientes e voluntários saudáveis (N= 858) que utilizavam ampla variedade de medicamentos concomitantemente (83 medicamentos, aproximadamente metade dos quais eliminados por via renal) não mostrou efeitos clinicamente significativos desses medicamentos na farmacocinética da sitagliptina.
Efeito da Sitagliptina sobre Outros Fármacos
Nos estudos clínicos, a sitagliptina não alterou significativamente a farmacocinética da metformina, da gliburida, da sinvastatina, da rosiglitazona, da varfarina ou dos anticoncepcionais orais, fornecendo evidências in vivo de baixa propensão a causar interações medicamentosas com substratos do CIP3A4, CIP2C8, CIP2C9 e do transportador orgânico catiônico (TOC). Doses múltiplas da sitagliptina aumentaram discretamente as concentrações de digoxina; no entanto, esses aumentos não parecem ser clinicamente significativos e não são atribuídos a um mecanismo específico.
Metformina: a co-administração de doses múltiplas duas vezes ao dia de sitagliptina com a metformina, um substrato do TOC, não alterou significativamente a farmacocinética da metformina em pacientes com diabetes tipo 2; portanto, a sitagliptina não é um inibidor do transporte mediado por TOC.
Sulfoniluréia: a farmacocinética de uma dose única de gliburida, um substrato do CIP2C9, não foi significativamente alterada em voluntários que receberam doses múltiplas da sitagliptina. Não são esperadas interações clinicamente significativas com outras sulfoniluréias (por exemplo, glipizida, tolbutamida e glimepirida), as quais, a exemplo da gliburida, são eliminadas principalmente pelo CIP2C9.
Sinvastatina: a farmacocinética de uma dose única de sinvastatina, um substrato do CIP3A4, não foi alterada significativamente em voluntários que receberam doses múltiplas diárias da sitagliptina; portanto, a sitagliptina não é um inibidor do metabolismo mediado pelo CIP3A4.
Tiazolidinedionas: a farmacocinética de uma dose única de rosiglitazona não foi alterada significativamente em voluntários que receberam doses múltiplas diárias da sitagliptina; portanto, a sitagliptina não é um inibidor do metabolismo mediado pelo CIP2C8. Não são esperadas interações clinicamente significativas com a pioglitazona porque o metabolismo da pioglitazona é mediado predominantemente pelo CIP2C8 ou pelo CIP3A4.
Varfarina: doses múltiplas diárias de sitagliptina não alteraram significativamente a farmacocinética - conforme determinado pela medição dos enantiômeros S(-) ou R(+) varfarina - ou a farmacodinâmica
- conforme determinado pela medição da INR da protrombina - de uma dose única de varfarina. Uma vez que a S(-) varfarina é metabolizada principalmente pelo CIP2C9, esses dados também fundamentam a conclusão de que a sitagliptina não é um inibidor do CIP2C9.
Anticoncepcionais Orais: a co-administração com a sitagliptina não alterou significativamente a farmacocinética no estado de equilíbrio da noretindrona ou do etinilestradiol.
Digoxina: a sitagliptina exerce efeito mínimo na farmacocinética da digoxina. Após a administração de 0,25 mg de digoxina concomitantemente com 100 mg de JANUVIA® diariamente por 10 dias, a AUC plasmática da digoxina aumentou 11% e a Cmáx plasmática, 18%. Esses aumentos não parecem ser clinicamente significativos.
Os dados clínicos descritos abaixo sugerem que a sitagliptina não é passível de interações clinicamente significativas quando co-administrada com os seguintes medicamentos:
Metformina: a co-administração de doses múltiplas duas vezes ao dia de metformina com a sitagliptina não alterou significativamente a farmacocinética da sitagliptina em pacientes com diabetes tipo 2.
Ciclosporina: foi conduzido um estudo para determinar o efeito da ciclosporina, um potente inibidor da p-glicoproteína, sobre a farmacocinética da sitagliptina. A co-administração de uma dose única de 100 mg de JANUVIA® por via oral e de uma dose única de 600 mg de ciclosporina por via oral aumentou a AUC e a Cmáx da sitagliptina aproximadamente 29% e 68%, respectivamente. Essas modestas alterações na farmacocinética da sitagliptina não foram consideradas clinicamente significativas. A depuração renal da sitagliptina também não foi alterada significativamente. Portanto, não são esperadas interações significativas com outros inibidores da p-glicoproteína.
Farmacocinética Populacional: em uma análise da farmacocinética populacional dos estudos fases I e II, 83 medicamentos concomitantes, aproximadamente metade dos quais são predominantemente eliminados por via renal, foram selecionados quanto a potenciais efeitos sobre a farmacocinética da sitagliptina. As concentrações plasmáticas da sitagliptina não foram alteradas significativamente por nenhum dos medicamentos que foram avaliados. Esses resultados sugerem que a sitagliptina provavelmente não seja susceptível a interações com outros medicamentos.
TOXICOLOGIA ANIMAL
Toxicidade Aguda
A DL50 aproximada da sitagliptina administrada por via oral a ratos é > 3.000 mg/kg (dose máxima testada). Essa dose é equivalente a 200 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos. Em camundongos, a DL50 oral aproximada da sitagliptina é de 4.000 mg/kg. Essa dose é equivalente a > 385 vezes a exposição em humanos com base na dose diária recomendada de 100 mg/dia para humanos adultos.
Toxicidade Crônica
O potencial de toxicidade da sitagliptina foi avaliado em uma série de estudos de toxicidade em que foram utilizadas doses repetidas durante até 53 semanas em cães e até 27 semanas em ratos. Em cães que receberam a sitagliptina por via oral nas doses de 2, 10 e 50 mg/kg/dia, o nível no qual não se observou efeito foi de 10 mg/kg/dia (até 6 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos). Os sinais físicos relacionados ao tratamento observados no grupo que recebeu 50 mg/kg/dia incluíram respiração com a boca aberta, salivação, vômitos de espuma branca, ataxia, tremor, diminuição da atividade e/ou postura encurvada. Esses sinais foram transitórios, de grau leve, e sua incidência diminuiu durante o curso do estudo. Além disso, à histologia, observou-se degeneração muscular esquelética muito discreta a discreta nos estudos de toxicidade de 14 e 27 semanas com a dose de 50 mg/kg/dia, o que não foi observado no estudo de toxicidade de 53 semanas, indicando ausência de reprodutibilidade ou progressão dessa alteração com o aumento da duração do tratamento. A dose de 50 mg/kg/dia em cães resultou em valores de exposição sistêmica 26 vezes mais altos que os da exposição humana na dose diária recomendada de 100 mg/dia para humanos adultos. Em ratos, a sitagliptina administrada por via oral em doses de até 180 mg/kg/dia (até 23 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos) não causou toxicidade significativa. O único efeito relacionado ao fármaco observado foi salivação pós-dose, provavelmente relacionada à baixa palatabilidade do fármaco, com as doses de 60 mg/kg/dia e 180 mg/kg/dia.
Não se considera que as alterações relacionadas ao tratamento observadas em animais apresentem impacto clínico nas doses terapêuticas recomendadas para humanos.
Carcinogenicidade
Foi conduzido um estudo de carcinogenicidade de dois anos em ratos machos e fêmeas que receberam doses orais de sitagliptina de 50, 150 e 500 mg/kg/dia. Com a dose mais alta, houve aumento da incidência de adenomas e carcinomas hepáticos no grupo dos machos e de carcinomas hepáticos no grupo das fêmeas. Essa dose em ratos resulta em aproximadamente 58 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos. Esse nível de dose foi associado a hepatotoxicidade em ratos. O nível no qual não se observou efeito quanto à indução de neoplasia hepática foi de 150 mg/kg/dia, aproximadamente 19 vezes a exposição humana com base na dose recomendada de 100 mg/dia para humanos adultos. Como foi demonstrado que a hepatotoxicidade está relacionada com a indução de neoplasia hepática em ratos, essa incidência aumentada de tumores hepáticos em ratos foi provavelmente decorrente de toxicidade hepática crônica com a dose alta. A relevância clínica desses achados para humanos é desconhecida.
Foi conduzido um estudo de carcinogenicidade de dois anos em camundongos machos e fêmeas com doses orais de 50, 125, 250 e 500 mg/kg/dia. A sitagliptina não aumentou a incidência de tumor em qualquer órgão com doses de até 500 mg/kg/dia (aproximadamente 68 vezes a exposição em humanos com base na dose diária recomendada de 100 mg/dia para humanos adultos).
Mutagênese
A sitagliptina não foi mutagênica ou clastogênica em uma bateria de estudos de toxicologia genética, inclusive o ensaio bacteriano de Ames (teste de mutagênese microbiana), ensaio de aberração cromossômica em células de ovário de hâmster chinês (células CHO) (um ensaio de citogenética in vitro utilizando células CHO), um ensaio de eluição alcalina de DNA de hepatócito de ratos in vitro (um ensaio que mede a capacidade de um composto para induzir quebras no DNA monofilamentar) e um ensaio de micronúcleo in vivo.
Reprodução
Não foram observados efeitos adversos na fertilidade de ratos machos e fêmeas que receberam doses de até 1.000 mg/kg/dia (até aproximadamente 100 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos) de sitagliptina por via oral antes e durante o acasalamento.
Desenvolvimento
A sitagliptina não foi teratogênica em ratos em doses orais de até 250 mg/kg ou em coelhos que receberam 125 mg/kg durante a organogênese (até 32 e 22 vezes, respectivamente, a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos). Foi observado discreto aumento na incidência de malformações de costelas fetais (ausência, hipoplasia e costelas ondulantes) relacionada ao tratamento na prole de ratos com doses orais de 1.000 mg/kg/dia (aproximadamente 100 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos). O nível no qual não se observou efeito no desenvolvimento foi de 250 mg/kg/dia (32 vezes a exposição humana com base na dose diária recomendada de 100 mg/dia para humanos adultos). Na prole de ratos, foram observadas reduções relacionadas ao tratamento no peso corporal médio pré-desmame de ambos os sexos e ganho de peso corporal pós-desmame nos animais machos que receberam doses orais de 1.000 mg/kg de sitagliptina.
ACHADOS DE EXAMES LABORATORIAIS
Nos estudos clínicos, foi observa