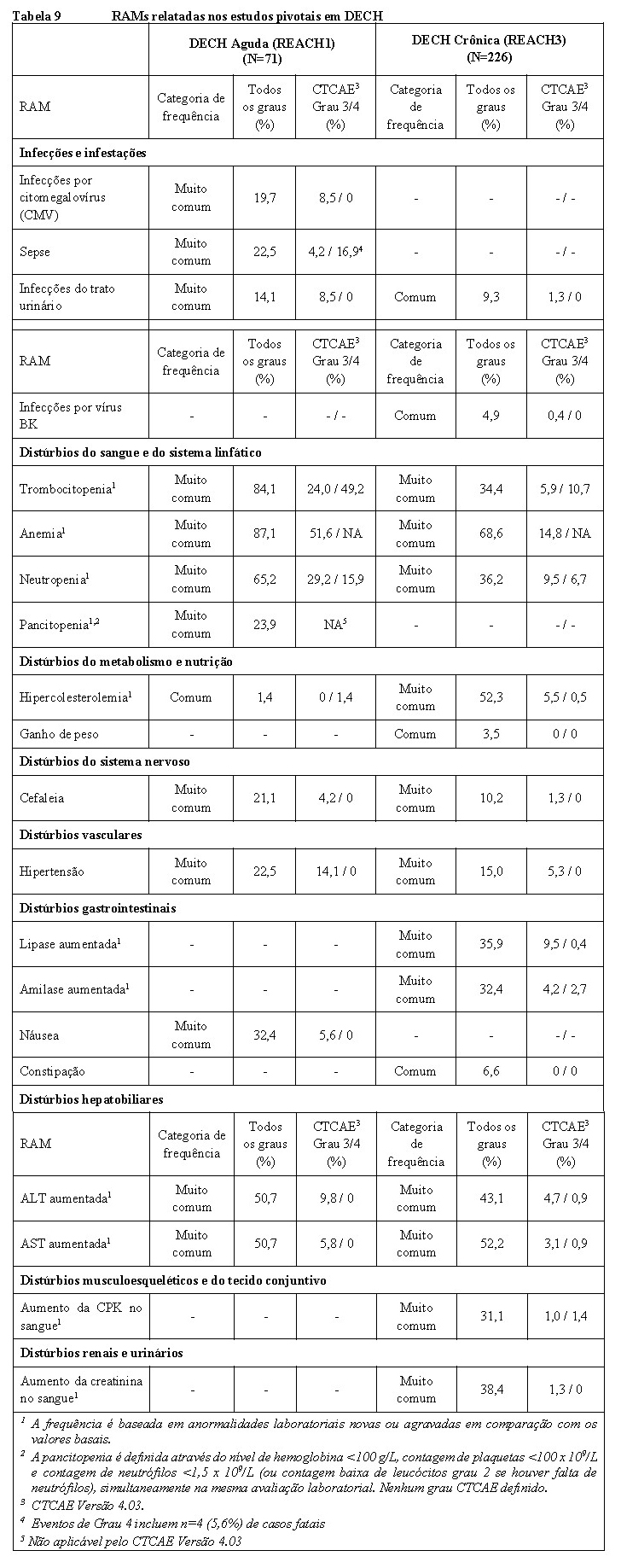
JAKAVI
NOVARTIS
ruxolitinibe
Inibidor da proteína quinase.
Apresentações.
Jakavi® 5 mg, 10 mg, 15 mg ou 20 mg - embalagens contendo 60 comprimidos.
VIA ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 12 ANOS (vide indicações)
Composição.
Cada comprimido de Jakavi® 5 mg contém 6,60 mg de fosfato de ruxolitinibe (equivalente a 5 mg de ruxolitinibe).
Cada comprimido de Jakavi® 10 mg contém 13,20 mg de fosfato de ruxolitinibe (equivalente a 10 mg de ruxolitinibe).
Cada comprimido de Jakavi® 15 mg contém 19,80 mg de fosfato de ruxolitinibe (equivalente a 15 mg de ruxolitinibe).
Cada comprimido de Jakavi® 20 mg contém 26,40 mg de fosfato de ruxolitinibe (equivalente a 20 mg de ruxolitinibe).
Excipientes: lactose monoidratada, celulose microcristalina, amidoglicolato de sódio, hiprolose, povidona, dióxido de silício, estearato de magnésio.
Informações técnicas.
1. INDICAÇÕES
Mielofibrose (MF)
Jakavi® é indicado para o tratamento de pacientes com mielofibrose de risco intermediário ou alto, incluindo mielofibrose primária, mielofibrose pós-policitemia vera ou mielofibrose pós trombocitemia essencial.
Policitemia vera (PV)
Jakavi® é indicado para o tratamento de pacientes com policitemia vera que são intolerantes ou resistentes à hidroxiureia ou à terapia citorredutora de primeira linha.
Doença do enxerto contra hospedeiro (DECH) aguda
Jakavi® é indicado para o tratamento de pacientes com doença do enxerto contra hospedeiro aguda com 12 anos ou mais que apresentam resposta inadequada aos corticosteroides.
Doença do enxerto contra hospedeiro (DECH) crônica
Jakavi® é indicado para o tratamento de pacientes com doença do enxerto contra hospedeiro crônica com 12 anos ou mais que apresentam resposta inadequada aos corticosteroides ou outras terapias sistêmicas.
2. RESULTADOS DE EFICÁCIA
Mielofibrose
Dois estudos randomizados de Fase 3 (COMFORT-I e COMFORT-II) 2,1
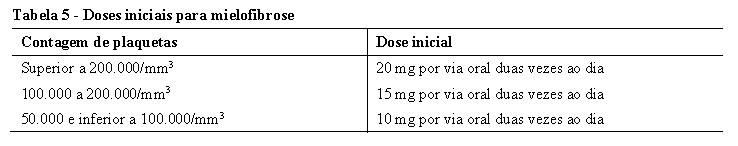
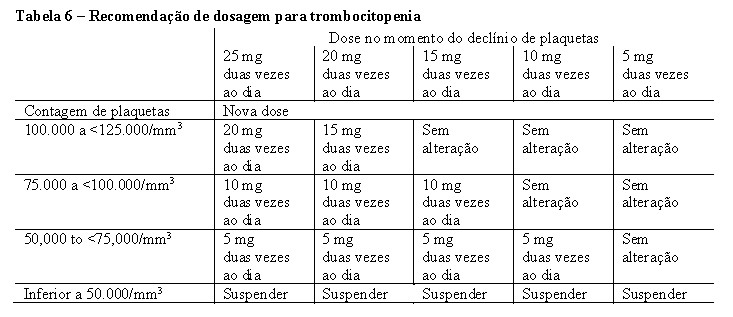
foram conduzidos em pacientes com Mielofibrose (MF) (Mielofibrose Primária (MFP), Mielofibrose Pós-Policitemia Vera (MF-PPV) ou Mielofibrose Pós-Trombocitemia Essencial (MF-PTE)). Nos dois estudos, os pacientes apresentaram esplenomegalia palpável pelo menos 5 cm abaixo da margem costal e categoria de risco intermediário 2 (2 fatores prognósticos) ou alto risco (3 ou mais fatores prognósticos) com base nos Critérios de Consenso do Grupo de Trabalho Internacional (IWG). Os fatores prognósticos que compreendem os critérios do IWG consistem em idade > 65 anos, presença de sintomas constitucionais (perda de peso, febre, sudorese noturna), anemia (hemoglobina < 10 g/dL), leucocitose (história de contagem de leucócitos > 25 x 109/L) e blastos circulantes ≥ 1%. A dose inicial de Jakavi® teve como base a contagem de plaquetas. Pacientes com uma contagem de plaquetas entre 100.000 e 200.000/mm3 iniciaram Jakavi® 15 mg duas vezes ao dia e pacientes com uma contagem de plaquetas > 200.000/mm3 iniciaram Jakavi® 20 mg duas vezes ao dia. De 301 pacientes, 111 (36,9%) tiveram uma base de contagem de plaquetas entre 100.000 e 200.000/mm3, e 190 (63,1%) tiveram uma base de contagem de plaquetas > 200,000/mm3. Pacientes com contagens de plaquetas ≤100,000/mm3 não foram elegíveis para os estudos COMFORT. A dose inicial máxima segura (MSSD) de 10 mg duas vezes ao dia para pacientes com uma contagem basal de plaquetas entre ≥50.000 e < 100.000/mm3 foi confirmada pelo EXPAND, um estudo de determinação de dose, fase Ib, aberto em pacientes com MFP, MF-PPV ou MF-PTE, onde as doses foram, então, individualizadas com base na tolerabilidade e na eficácia, com doses máximas de 20 mg duas vezes ao dia para pacientes com contagens de plaquetas entre 100.000 a ≤ 125.000/mm3, de 10 mg duas vezes ao dia para pacientes com contagens de plaquetas entre 75.000 a ≤ 100.000/mm3, e de 5 mg duas vezes ao dia para pacientes com contagens de plaquetas entre 50.000 a ≤ 75.000/mm3.
COMFORT-I 2 foi um estudo duplo-cego, randomizado, controlado por placebo em 309 pacientes refratários ou que não eram candidatos para a terapia disponível. Os pacientes receberam doses de Jakavi® ou placebo correspondente. O objetivo primário de eficácia foi a proporção de indivíduos que atingiram redução ≥ 35% no volume do baço desde o basal na Semana 24, conforme medição por ressonância magnética (RM) ou tomografia computadorizada (TC).
Os objetivos secundários incluíram a duração da manutenção da redução ≥ 35% desde o basal no volume do baço, proporção de pacientes que tiveram uma redução ≥ 50% na pontuação total de sintomas desde o basal até a Semana 24, conforme medição do diário do Formulário de Avaliação dos Sintomas de Mielofibrose Modificado (FASMM) v2.0, alteração na pontuação total de sintomas desde o basal até a Semana 24, conforme medição do diário do FASMM v2.0 e sobrevida global.
COMFORT-II 1 foi um estudo randomizado e aberto em 219 pacientes. Os pacientes foram randomizados em uma proporção de 2:1 para Jakavi® versus melhor terapia disponível (MTD). A MTD foi escolhida pelo investigador caso a caso. No braço de MTD, 47% dos pacientes receberam hidroxiureia e 16% dos pacientes receberam glicocorticoides. O objetivo primário de eficácia foi a proporção de pacientes que atingiu redução ≥ 35% no volume do baço desde o basal na Semana 48, conforme medição por RM ou TC.
Um objetivo secundário no COMFORT-II foi a proporção de pacientes que atingiram redução ≥ 35% no volume do baço medida por RM ou TC desde o basal até a Semana 24. A duração da manutenção de redução ≥ 35% desde o basal nos pacientes respondedores também foi um objetivo secundário.
No COMFORT-I, os dados demográficos do basal dos pacientes e as características da doença foram semelhantes entre os braços de tratamento. A idade mediana foi de 68 anos, com 61% dos pacientes com mais de 65 anos de idade e 54% sendo homens. Cinquenta por cento (50%) dos pacientes apresentaram MFP, 31% apresentaram MF-PPV e 18% apresentaram MF-PTE. Vinte e um (21%) dos pacientes tiveram transfusões de sangue em até 8 semanas a partir da inclusão no estudo. A contagem mediana de plaquetas foi de 251.000/mm3. Setenta e seis por cento dos pacientes apresentaram a mutação codificando a substituição V617F presente na proteína JAK. Os pacientes tiveram um comprimento de baço mediano palpável de 16 cm. No basal, 37,4% dos pacientes no braço Jakavi® apresentaram anemia Grau 1, 31,6% Grau 2 e 4,5% Grau 3, enquanto que no braço de placebo, 35,8% apresentaram Grau 1, 35,1% Grau 2, 4,6% Grau 3 e 0,7% Grau 4. Trombocitopenia Grau 1 foi encontrada em 12,9% dos pacientes no braço de Jakavi® e 13,2% no braço de placebo 2.
No COMFORT-II, os dados demográficos do basal dos pacientes e as características da doença foram semelhantes entre os braços de tratamento. A idade mediana foi de 66 anos, com 52% dos pacientes com mais de 65 anos de idade e 57% sendo homens. Cinquenta e três por cento (53%) dos pacientes apresentaram MFP, 31% apresentaram MF-PPV e 16% apresentaram MF-PTE. Dezenove por cento (19%) dos pacientes foram considerados dependentes de transfusão no basal. Os pacientes apresentaram um comprimento mediano de baço palpável de 15 cm.
No basal, 34,2% dos pacientes no braço de Jakavi® apresentaram anemia de Grau 1, 28,8% Grau 2, e 7,5% Grau 3, enquanto que no braço de BAT 37% apresentaram Grau 1, 27,4% Grau 2, 13,7% Grau 3, e 1,4% Grau 4. Trombocitopenia de Grau 1 foi encontrada em 8,2% dos pacientes no braço de Jakavi® e 9,6% no braço da MTD 1. As análises de eficácia do objetivo primário no COMFORT-I e COMFORT-II são apresentadas na Tabela 1. Uma proporção significativamente maior de pacientes nos braços de Jakavi® atingiram redução ≥35% no volume do baço desde o basal nos dois estudos, em comparação ao placebo no COMFORT-I e MTD no COMFORT-II.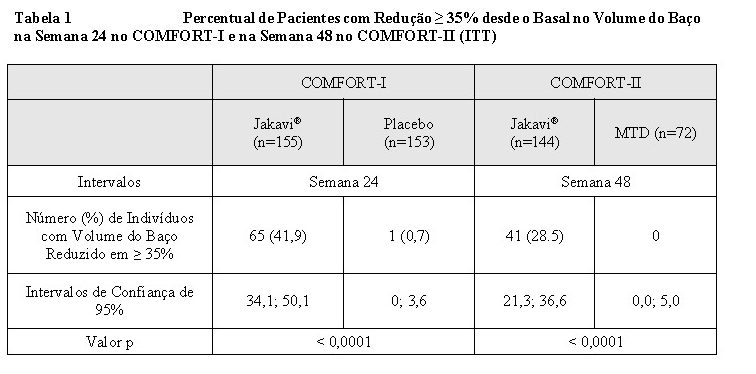
No estudo COMFORT-I, 41,9% dos pacientes no braço de Jakavi® atingiram redução ≥ 35% no volume do baço desde o basal em comparação a 0,7% no braço de placebo na Semana 24. Uma proporção semelhante de pacientes no grupo de Jakavi® atingiram redução ≥ 50% no comprimento do baço palpável.
No estudo COMFORT-II, 28,5% dos pacientes no braço de Jakavi® atingiram redução ≥ 35% no volume do baço desde o basal em comparação a nenhum (0%) no braço da MTD na Semana 48. Um objetivo secundário foi a proporção de pacientes que atingiram redução ≥ 35% no volume do baço na Semana 24. Uma proporção significativamente maior de pacientes no grupo de Jakavi®, 46 pacientes (31,9%) atingiram redução ≥ 35% no volume do baço desde o basal em comparação a nenhum (0%) paciente no grupo da melhor terapia disponível (valor p < 0,0001).
Uma proporção significativamente maior de pacientes no braço de Jakavi® atingiram redução ≥ 35% desde o basal no volume do baço independente da presença ou ausência da mutação JAK2V617F ou subtipo da doença (MFP, MF-PPV, MF-PTE).
A figura 1 apresenta um gráfico em cascata da alteração percentual desde o basal no volume do baço na Semana 24 no COMFORT-I. Entre os 139 pacientes no braço de Jakavi® que apresentaram as duas avaliações do volume do baço no basal e na Semana 24, todos (exceto dois pacientes tiveram algum nível de redução no volume do baço na Semana 24, com redução mediana de 33%. Entre os 106 pacientes no braço de placebo que apresentaram avaliações do volume do baço no basal e na Semana 24, houve um aumento mediano de 8,5%.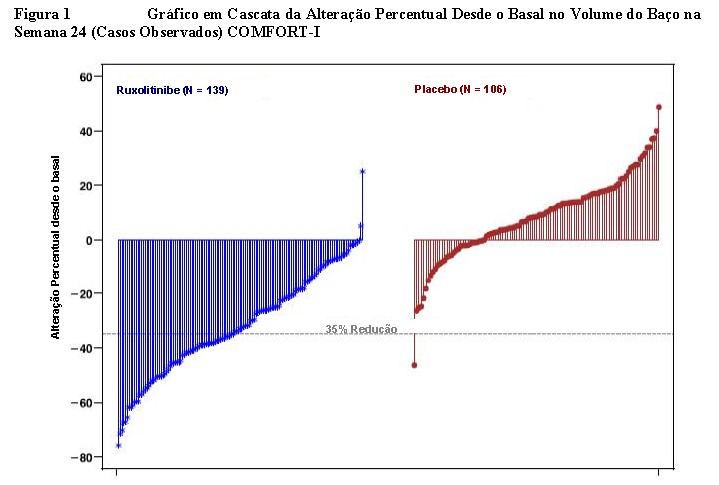
A figura 2 apresenta um gráfico em cascata da alteração percentual desde o basal no volume do baço na Semana 48 no estudo COMFORT-II. Entre os 98 pacientes no braço de Jakavi® que apresentaram as duas avaliações do volume do baço no basal e na Semana 48, a redução mediana no volume do baço na Semana 48 foi de 28%. Entre os 34 pacientes no braço da MTD que apresentaram avaliações do volume do baço no basal e na Semana 48, houve um aumento mediano de 8,5%.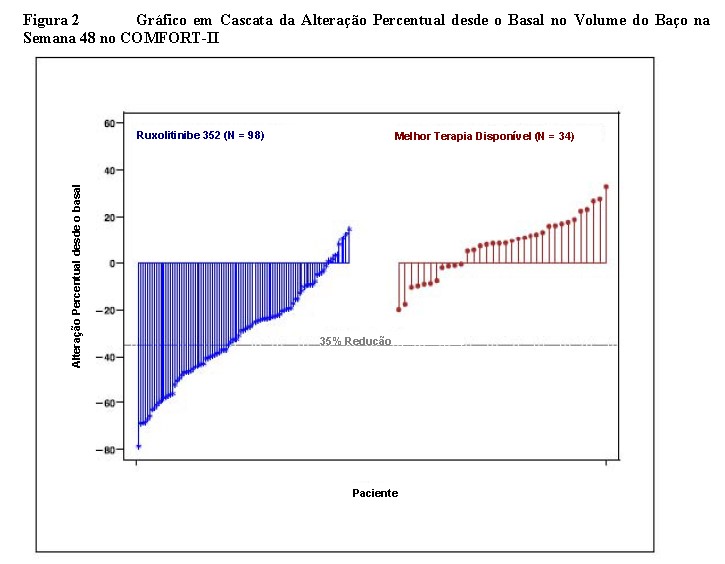
A probabilidade de duração da 1ª redução ≥ 35% do volume do baço até um aumento de 25% desde o nadir e perda de resposta no COMFORT-I e COMFORT-II apresentado na Tabela 2.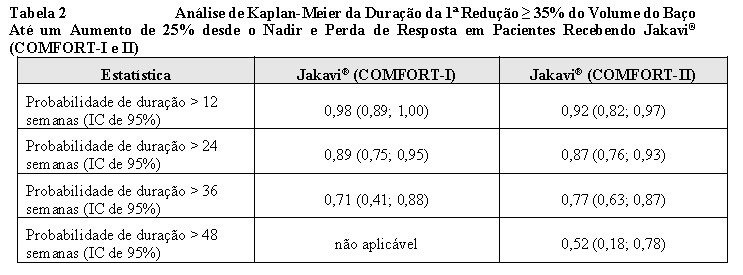
Entre os 80 pacientes que apresentaram redução ≥ 35% em qualquer momento no COMFORT-I e os 69 pacientes no COMFORT-II, a probabilidade de um paciente manter uma resposta com Jakavi® por pelo menos 24 semanas foi de 89% e 87% no COMFORT-I e COMFORT-II, respectivamente, e a probabilidade de manutenção da resposta por pelo menos 48 semanas foi de 52% no COMFORT-II.
Jakavi® melhora os sintomas relacionados à MF e a qualidade de vida (QOL) em pacientes com MFP, MF-PPV e MF-PTE. No COMFORT-I, os sintomas de MF foram capturados utilizando-se o diário do Formulário de Avaliação dos Sintomas de Mielofibrose Modificado (FASMM) v2.0 como um diário eletrônico, o qual os pacientes preenchiam todos os dias. A alteração desde o basal na pontuação total na Semana 24 foi um objetivo secundário neste estudo. Uma proporção significativamente maior de pacientes no braço de Jakavi® atingiram melhora ≥ 50% desde o basal na pontuação total dos sintomas na semana 24 comparado ao braço placebo (45,9% e 5,3%, respectivamente, p < 0,0001 usando o teste do Qui-quadrado).
Uma melhora na qualidade de vida global foi medida pela European Organization for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire (QLQ)-C30 em ambos COMFORT-I e COMFORT-II. O estudo COMFORT-I comparou Jakavi® com placebo por 24 semanas e o estudo COMFORT-II comparou Jakavi® com a MTD por 48 semanas. No basal, para os dois estudos, as pontuações da subescala individual de EORTC QLQ-C30 para os braços de Jakavi® e comparador foram similares. Na Semana 24, no COMFORT-I, o braço de Jakavi® demonstrou uma melhora significativa da saúde global / QOL do EORTC QLQ-C30 comparado ao braço de placebo (alteração média de +12,3 e -3,4 para Jakavi® e placebo, respectivamente, p < 0,0001). Na semana 24 e na semana 48, o grupo de Jakavi® no COMFORT-II apresentou uma tendência em direção a uma melhora maior da saúde global/ QOL comparada à MTD, um objetivo exploratório, consistente com os achados do COMFORT-II.
No estudo COMFORT-I, após acompanhamento médio de 34,3 meses, a taxa de mortes em pacientes randomizados no braço Jakavi® foi de 27,1% (42 de 155 pacientes) versus 35,1% (54 de 154) dos pacientes randomizados com placebo. Houve uma redução de 31,3% no risco de morte no braço Jakavi® quando comparado ao placebo (HR 0,687; IC 95% 0,459-1,029; p = 0,0668)3.
No estudo COMFORT-I, após acompanhamento médio de 61,7 meses, a taxa de mortes em pacientes randomizados no braço Jakavi® foi de 44,5% (69 de 155 pacientes) versus 53,2% (82 de 154) dos pacientes randomizados com placebo. Houve uma redução de 31% no risco de morte no braço Jakavi® quando comparado ao placebo (HR 0,69; IC 95% 0,50-0,96; p = 0,025)
No estudo COMFORT-II, após acompanhamento médio de 34,7 meses, a taxa de mortes em pacientes randomizados com Jakavi® foi de 19,9% (29 de 146 pacientes) versus 30,1% (22 de 73 pacientes) em pacientes randomizados com melhor terapia disponível (MTD). Houve uma redução no risco de morte de 52% no braço Jakavi® comparado ao braço MTD (HR 0,48; IC 95% 0,28-0,85; p = 0,009)3. Na análise final, após acompanhamento médio de 55,9 meses, a taxa de mortes em pacientes randomizados com ruxolitinibe foi de 40,4% (59 de 146 pacientes) versus 47,9% (35 de 73 pacientes) em pacientes randomizados com melhor terapia disponível (MTD). A redução no risco de morte é consistente com a encontrada no estudo COMFORT-I de 33% no braço ruxolitinibe comparado ao braço MTD (HR 0,67; IC 95% 0,44-1,02; p = 0,062).
Policitemia vera [7-9]
Um estudo de fase 2, multicêntrico, aberto, randomizado, não controlado e com regime de dose variável foi conduzido para estabelecer a dose de 10 mg de ruxolitinibe duas vezes ao dia como uma dose ativa, segura e bem tolerada em pacientes com PV avançada refratários à hidroxiureia ou para quem o tratamento com hidroxiureia estava contraindicado. O estudo consistiu de grupos 1 e 2, que recrutaram 34 pacientes com PV.8
Um estudo (RESPONSE) randomizado, aberto, ativo-controlado, de fase 3,7,9 foi conduzido com 222 pacientes com PV que eram resistentes ou intolerantes à hidroxiureia. Um total de 110 pacientes foram randomizados para o braço de Jakavi® e 112 pacientes para o braço MTD. A dose inicial de Jakavi® foi de 10 mg duas vezes ao dia. As doses foram então ajustadas individualmente aos pacientes com base na tolerabilidade e eficácia, com a dose máxima de 25 mg duas vezes ao dia. MTD foi selecionada pelo investigador, paciente por paciente. Os tratamentos incluídos em MTD foram: hidroxiureia (59,5%), interferona/interferona peguilada (1,7%), anagrelida (7,2%), pipobromana (1,8%) e observação (15,3%).
Dados demográficos do basal e as características da doença foram comparáveis entre os dois braços de tratamento. A idade média foi de 60 anos (faixa de 33 a 90 anos). Pacientes no braço de Jakavi® apresentaram diagnóstico PV por uma média de 8,2 anos e tinham recebido previamente hidroxiureia por uma média de aproximadamente de 3 anos. A maioria dos pacientes ( > 80%) tinha recebido pelo menos duas flebotomias nas últimas 24 semanas antes da triagem.
O desfecho primário composto foi a proporção de pacientes que atingiram tanto a ausência de elegibilidade de flebotomia (controle HCT) e ≥ 35% de redução do volume do baço em relação ao basal na semana 32.
A elegibilidade de flebotomia foi definida como HCT > 45% confirmado que é, pelo menos, 3 pontos percentuais maior do que o HCT obtido no basal ou um HCT > 48% confirmado, o que for menor. Desfechos secundários principais incluíram a proporção de pacientes que atingiram o desfecho primário e que permaneceram livres de progressão na semana 48, e a proporção de pacientes que atingiram remissão hematológica completa na semana 32.
O estudo cumpriu o seu objetivo principal e uma maior proporção de pacientes no braço de Jakavi® alcançaram o desfecho primário composto e cada um dos seus componentes individuais. Significativamente, mais pacientes com Jakavi® (23%) em comparação com MTD (0,9%) obtiveram uma resposta primária (p < 0,0001). O controle do HTC foi conseguido em 60% dos pacientes no braço Jakavi® em comparação com 18,75% no braço MTD, e redução de ≥ 35% do volume do baço foi obtido em 40% dos pacientes no braço Jakavi® em comparação com 0,9% no braço MTD (figura 3).
Ambos os desfechos secundários foram atingidos: a proporção de pacientes que atingiram uma remissão hematológica completa foi de 23,6% com Jakavi® em comparação a 8,0% com MTD (p = 0,0013), e a proporção de pacientes que atingiram uma resposta primária duradoura na semana 48 foi de 20% com Jakavi® e de 0,9% com BAT (p < 0,0001).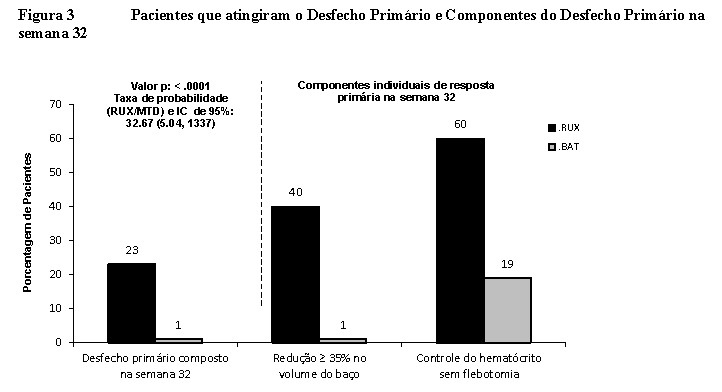
Os sintomas foram avaliados usando a pontuação MPN-Symptoms Assessment Form (SAF) escore total de sintomas (TSS) do diário eletrônico do paciente que consiste de 14 questões. Na semana 32, 49% e 64% dos pacientes tratados com Jakavi® conseguiram uma redução ≥ 50% no TSS-14 e TSS-5, respectivamente, em comparação com apenas 5% e 11% dos pacientes em MTD.
A percepção de benefício do tratamento foi medida pelo questionário Impressão Global de Mudança do Paciente (PGIC). Um total de 66% dos pacientes tratados com Jakavi® em comparação com 19% em MTD, relataram uma melhora tão cedo quanto 4 semanas após o início do tratamento. A melhora na percepção de benefício do tratamento também foi maior em pacientes tratados com Jakavi® na semana 32 (78% versus 33%).
Análises adicionais do estudo RESPONSE para verificar a durabilidade da resposta, foram conduzidas na semana 80 e na semana 256 seguindo a randomização. Dos 25 pacientes que alcançaram a resposta primária na semana 32, 3 pacientes progrediram até a semana 80 e 6 pacientes até a semana 256. A probabilidade de ter mantido uma resposta da semana 32 até a semana 80 e semana 256 foi de 92% e 74%, respectivamente (vide tabela 3).
Um segundo estudo randomizado, aberto, controlado-ativo de fase IIIb (RESPONSE 2)9, foi conduzido em 149 pacientes com PV que foram resistentes ou intolerantes à hidroxiureia, mas sem esplenomegalia palpável. Setenta e quatro pacientes foram randomizados para o braço Jakavi® e 75 pacientes para o braço MTD. A dose inicial e ajustes da dose de Jakavi® e a MTD selecionada pelo investigador foram semelhantes ao estudo RESPONSE. Os dados demográficos basais e as características da doença foram comparadas entre os dois braços de tratamento e foram semelhantes a população de pacientes do estudo RESPONSE. O desfecho primário foi a proporção de pacientes que atingiram o controle de HCT (ausência de elegibilidade de flebotomia) na semana 28. O desfecho chave secundário foi a proporção de pacientes que atingiram a remissão hematológica completa na semana 28.
O estudo RESPONSE-2 cumpriu o seu objetivo primário com uma maior proporção de pacientes no braço Jakavi® (62,2%) comparado ao braço MTD (18,7%), atingindo seu desfecho primário (p < 0,0001). O desfecho chave secundário também foi cumprido com, significativamente, mais pacientes atingindo uma remissão hematológica completa no braço Jakavi® (23,0%) comparado ao braço MTD (5,3%; p=0,0019). Na semana 28, a proporção de pacientes que atingiram uma redução de ≥ 50% na carga de sintomas como mensurado pela pontuação total de sintomas MPN-SAF foi de 45,3% no braço Jakavi® e 22,7% no braço MTD.
Doença do enxerto contra hospedeiro
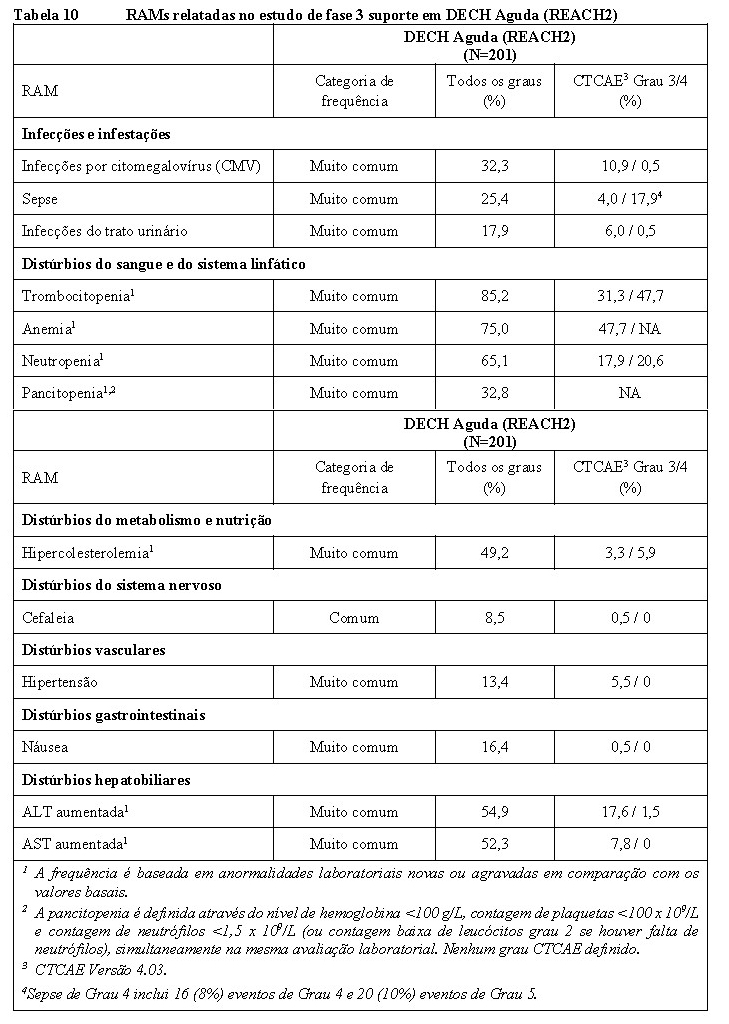
A eficácia clínica de Jakavi® em pacientes com 12 anos de idade ou mais com DECH aguda foi demonstrada com base em um estudo pivotal de Fase II (REACH1) e um estudo de Fase III suporte (REACH2).
Em pacientes com 12 anos de idade ou mais com DECH crônica, a eficácia clínica de Jakavi® foi demonstrada com base em um estudo de Fase III (REACH3).
A dose inicial de Jakavi® foi de 10 mg duas vezes ao dia.
Doença de enxerto contra hospedeiro aguda
No estudo principal REACH 1, que incluiu 71 participantes, o desfecho primário foi a taxa de resposta global (TRG) no Dia 28, definida como a proporção de pacientes com uma resposta completa (RC), resposta parcial muito boa (RPMB) ou uma resposta parcial (RP) (de acordo com as modificações do CIBMTR (Center for International Blood and Marrow Transplant Research) para o índice de resposta IBMTR (International Bone Marrow Transplant Registry).
O desfecho secundário principal foi a duração da resposta (DDR) em seis meses, definida como o tempo desde a primeira resposta até a progressão de DECH ou morte, avaliada quando todos os participantes que ainda estão em estudo completam a visita do Dia 180.
O estudo REACH 1 atingiu o limite predeterminado para um resultado positivo do estudo (limite inferior do IC de 95% para TRG do Dia 28 ≥ 40%). Quarenta participantes (56,3% [IC de 95%: 44,0, 68,1]) demonstraram uma resposta no Dia 28, incluindo 19 participantes (26,8%) que alcançaram uma RC, 6 participantes que alcançaram uma RPMB (8,5%) e 15 participantes que alcançaram uma RP (21,1%).
Um outro desfecho secundário foi Sobrevida Livre de Falha (SLF), um tempo composto para desfecho de evento definido como o tempo desde o início da medicação até i) recidiva ou recorrência da doença subjacente, ii) mortalidade sem recidiva, ou iii) adição ou início de outra terapia sistêmica.
O estudo REACH2, multicêntrico, randomizado, aberto, de fase 3, com um total de 309 pacientes, também atingiu seu desfecho principal. A TRG no Dia 28 de tratamento, definida como a proporção de pacientes que alcançaram uma RC ou RP de acordo com Harris (2016), foi maior no braço Jakavi® (62,3%) em comparação com o braço MTD (39,4%). Houve uma diferença estatisticamente significativa entre os braços de tratamento (teste estratificado de Cochrane-Mantel-Haenszel p < 0,001, unilateral, Razão de Chances (OR): 2,64; IC de 95%: 1,65-4,22). Houve também uma proporção maior de respondedores completos no braço Jakavi® -34,40% em comparação com o braço MTD (19,4%).
O estudo atingiu seu principal desfecho secundário. A TRG durável no Dia 56 foi de 39,6% (IC de 95%: 31,8-47,8) no braço Jakavi® e 21,9% (IC de 95%: 15,7, 29,3) no braço MTD. Houve uma diferença estatisticamente significativa entre os dois braços de tratamento (OR: 2,38; IC de 95%: 1,43-3,94; p < 0,001).
A proporção de pacientes com RC foi de 26,6% no braço Jakavi® vs. 16,1% no braço MTD. No geral, 49 pacientes originalmente randomizados para o braço MTD cruzaram para o braço Jakavi®.
Para o desfecho secundário SLF, houve menos eventos no braço Jakavi® (91; 59,1%) do que no braço MTD (121; 78,1%). Entre os pacientes randomizados, a taxa de incidência estimada de um evento SLF em um mês foi menor no braço Jakavi® (18,47%; IC de 95%: 12,74-25,04) do que no braço MTD (49,13%; IC de 95%: 40,94-56,80). Dados adicionais de acompanhamento permanecem favoráveis ao Jakavi®.
A mediana de SLF com Jakavi® foi significativamente maior do que no braço MTD (4,86 meses vs. 1,02 meses; HR: 0,49, IC de 95%: 0,37-0,63; p < 0,0001).
Doença do enxerto contra hospedeiro crônica
No estudo REACH3, multicêntrico, aberto, de fase 3, um total de 329 pacientes com DECH crônica moderada ou grave refratária a corticosteroides foram randomizados 1:1 para Jakavi® (165 pacientes) ou MTD (164 pacientes). Os pacientes foram estratificados pela gravidade da DECH crônica no momento da randomização. A refratariedade aos corticosteroides foi determinada quando os pacientes não apresentaram resposta ou tiveram progressão da doença após a administração de prednisona na dose mínima de 1mg/kg/dia por pelo menos 7 dias, ou persistência da doença por 4 semanas na dose de 0,5 ou 1mg/kg/dia ou quando falharam na redução de corticosteroide em dose < 0,25mg/kg/dia por duas vezes.
A MTD foi selecionada pelo investigador de acordo com a necessidade de cada paciente e incluiu fotoferese extracorpórea (ECP), metotrexato em baixa dose (MTX), micofenolato mofetil (MMF), inibidores de mTOR (everolimo ou sirolimo), infliximabe, rituximabe, pentostatina, imatinibe ou ibrutinibe.
Além de Jakavi® ou MTD, os pacientes poderiam ter recebido tratamento de suporte para transplante alogênico de células-tronco alogênico padrão, incluindo medicamentos anti-infecciosos e suporte de transfusão, bem como profilaxia padrão para DECH crônica e medicamentos de tratamento iniciados antes da randomização, incluindo corticosteroides sistêmicos e CNIs (ciclosporina ou tacrolimo). As terapias com corticosteroides tópicos ou inalatórios puderam ser continuadas de acordo com as diretrizes institucionais
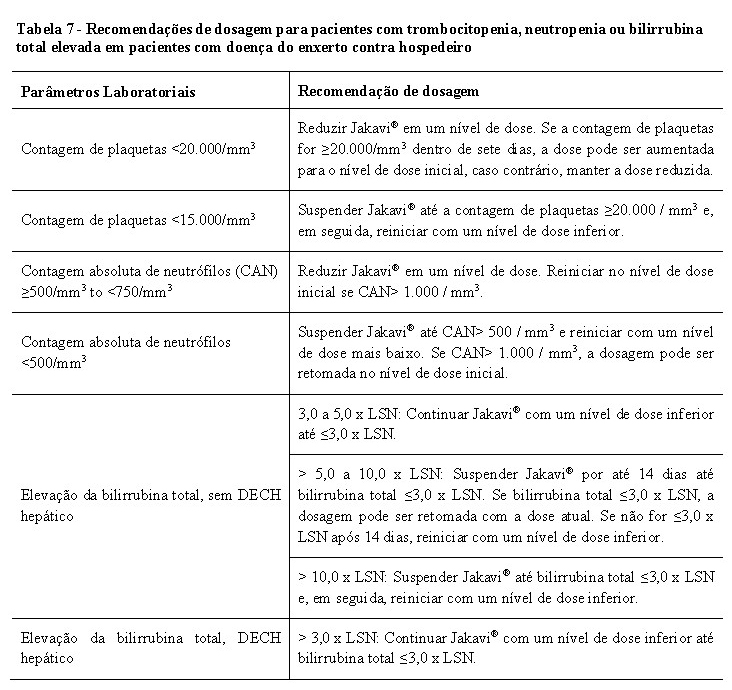
Os pacientes randomizados para o braço MTD foram autorizados a passar para o braço Jakavi® após a visita no Dia 1 do Ciclo 7 (semana 24). A redução gradual de Jakavi® foi permitida após a visita do Dia 1 do Ciclo 7.
Os dados demográficos basais e as características da doença foram balanceados entre os dois braços de tratamento. A idade mediana foi de 49 anos (faixa de 12 a 76 anos). O estudo incluiu 3,6% de adolescentes, 61,1% de homens e 75,4% de pacientes brancos. A maioria dos pacientes participantes possuía doença maligna subjacente.
A gravidade no diagnóstico de DECH crônica refratária a corticosteroides foi balanceada entre os dois braços de tratamento, com 41% e 45% moderado e 59% e 55% grave, nos braços Jakavi® e MTD, respectivamente.
A resposta insuficiente dos pacientes aos corticosteroides no braço Jakavi® e MTD foi caracterizada por i) uma falta de resposta ou progressão da doença após o tratamento com corticosteroides por pelo menos 7 dias com 1mg/ kg/dia de equivalentes de prednisona (37,6% e 44,5%, respectivamente), ii) persistência da doença após 4 semanas com 0,5 mg/kg/dia (35,2% e 25,6%), ou iii) dependência de corticosteroides (27,3% e 29,9%, respectivamente).
Entre todos os pacientes, 73% e 45% tiveram envolvimento de pele e pulmão no braço Jakavi®, comparado a 69% e 41% no braço MTD.
As terapias sistêmicas de DECH crônica mais frequentemente utilizadas foram: apenas corticosteroides (43% no braço Jakavi® e 49% no braço MTD) e corticosteroides + CNIs (41% pacientes no braço Jakavi® e 42% no braço MTD).
O desfecho primário foi a taxa de resposta global (TRG) no Dia 1 do Ciclo 7, definida como a proporção de pacientes em cada braço com uma resposta completa (RC) ou uma resposta parcial (RP) sem a necessidade de terapias sistêmicas adicionais para uma progressão mais precoce, resposta mista ou não-resposta com base na avaliação do investigador de acordo com os critérios do NIH (National Institutes of Health).
Os principais desfechos secundários foram sobrevida livre de falha (SLF) e proporção de pacientes com melhora do escore de sintomas de Lee modificado (mLSS) no Dia 1 do Ciclo 7. A SLF, um desfecho composto de tempo para ocorrência de evento, incorporou o primeiro dos seguintes eventos: i) recidiva ou recorrência da doença subjacente ou morte devido à doença subjacente, ii) mortalidade sem recidiva ou iii) adição ou início de outra terapia sistêmica para DECH crônica.
O estudo REACH3 atingiu seu objetivo primário. A TRG na semana 24 foi maior no braço Jakavi® (49,7%) comparado ao braço MTD (25,6%). Houve uma diferença estatisticamente significativa entre os braços de tratamento (teste estratificado de Cochrane-Mantel-Haenszel p < 0,0001, unilateral, OR: 2,99; IC de 95%: 1,86-4,80). Os resultados são apresentados na Tabela 4.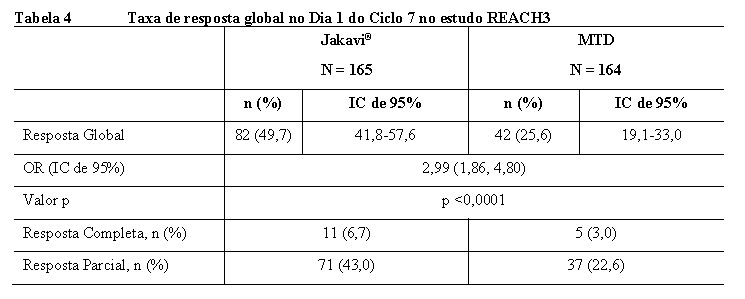
Ambos os principais desfechos secundários também foram alcançados. A SLF demonstrou uma superioridade estatisticamente significativa de Jakavi® versus MTD (HR: 0,37; IC de 95%: 0,268-0,51) com uma redução de risco de 63% (ver Figura 4). A probabilidade de SLF em 6 meses (IC de 95%) foi de 74,9% (67,5%-80,9%) e 44,5% (36,5%-52,1%) para os braços Jakavi® e MTD, respectivamente. A maioria dos eventos de SLF foram adição ou início de outra terapia sistêmica para DECHc'. A probabilidade de ocorrência destes eventos em 6 meses foi de 13,5% e 48,5% para os braços Jakavi® e MTD, respectivamente.
A taxa de resposta de acordo com a melhora de ≥7 pontos do escore total de sintomas (TSS) a partir do basal do mLSS mostrou uma diferença estatisticamente significativa (p = 0,0011) entre os braços Jakavi® (24,2%) e MTD (11%).
Outro desfecho secundário foi a melhor resposta global (MRG) definida como a proporção de pacientes que alcançaram TRG (RC + RP) em qualquer ponto de tempo até o Dia 1 do Ciclo 7. A MRG até o Dia 1 do Ciclo 7 foi maior no braço Jakavi® (76,4%) do que no braço MTD (60,4%).
A probabilidade estimada de manutenção de MRG em 12 meses foi maior no braço Jakavi® comparado ao braço MTD (68,5% [IC de 95%: 58,9-76,3] vs 40,3% [IC de 95%: 30,3-50,2]).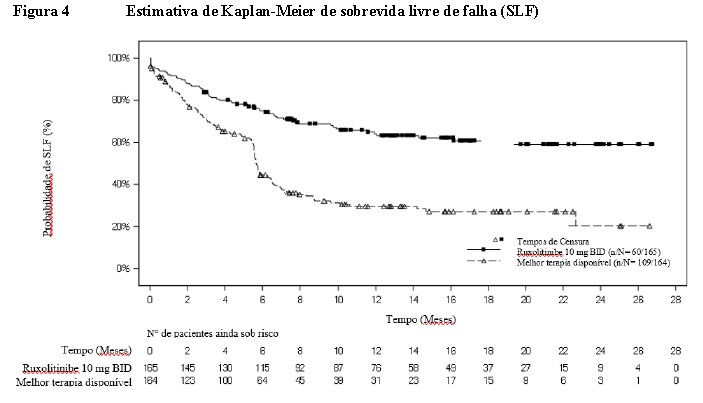
Referências bibliográficas
1. CINC424A2352 (INCB 18424-352): A Randomized Study of the JAK Inhibitor INCB018424 Tablets Compared to Best Available Therapy in Subjects with Primary Myelofibrosis (PMF), Post-Polycythemia Vera-Myelofibrosis (PPV-MF) or Post-Essential Thrombocythemia Myelofibrosis (PET-MF). [1] (dados em arquivo)
2. INCB 18424-351: A Randomized, Double-blind, Placebo-controlled Study of the JAK Inhibitor INCB018424 Tablets Administered Orally to Subjects with Primary Myelofibrosis (PMF), Post Polycythemia-Vera Myelofibrosis (PPV-MF) or Post Essential Thrombocythemia-Myelofibrosis (PET-MF). [2] (dados em arquivo)
3. 2.5 Clinical Overview. Treatment of adult patients with primary myelofibrosis (PMF), post-polycythemia vera-myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF). Novartis, 17-Oct-2013. [33] (dados em arquivo)
4. 2.5 Clinical Overview - Ruxolitinib (INC424/INCB18424). May 2011. [29] (dados em arquivo)
5. Mesa R. et al. Effect of Ruxolitinib Therapy on Myelofibrosis-Related Symptoms and Other Patient-Reported Outcomes in COMFORT-I: A Randomized, Double-Blind, Placebo-Controlled Trial. JCO, 2013 (10): 1285-92).
6. Mesa R et al. Comparison of placebo and BAT for the treatment of MF in the phase 3 COMFORT studies. Haematologica, 2014, 99 92) 292-8.
7. Vannucchi AM, Kiladjian JJ, Griesshammer M, Masszi T, Durrant S, Passamonti F, Harrison CN, Pane F, Zachee P, Mesa R, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015 Jan 29; 372(5):426-35.
8. Verstovsek, S et al. "A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea." Cancer 2014; 120:513-520.
9. CINC 424B2401 Interim Clinical Study Report Week 28 - Randomized, open label, multicenter phase IIIb study evaluating the efficacy and safety of ruxolitinib versus best available therapy in patients with polycythemia vera who are hydroxyurea resistant or intolerant (RESPONSE 2). Novartis. Apr-2016 (dados em arquivo).
10. Jagasia MH et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group Report. Biol Blood Marrow Transplant. 2015 Mar; 21(3): 389-401.e1.
11. Clinical Study Report INCB 18424-271 (REACH1). Dec 2019 (dados em arquivo)
12. Clinical Study Report CINC424C2301 (REACH2). Novartis. 2020 (dados em arquivo)
13. Clinical Study Report CINC424D2301 (REACH3). Novartis. 2020 (dados em arquivo)
14. Harris AC, Young R, Devine S, et al (2016). International, multicenter standardization of acute graft-versus-host disease clinical data collection: a report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transplant; 22:4-10
15. 2.5 Clinical Overview - Myelofibrosis in patients with low baseline platelet count (≥50×109/L and < 100×109/L) - CINC424A2201 (EXPAND). Novartis. 14-Oct-2020 (dados em arquivo).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico
Agentes antineoplásicos, Inibidor de proteína-quinase. Código ATC: L01XE18.
Propriedades farmacodinâmicas
O ruxolitinibe inibe a fosforilação de STAT3 induzida por citocina no sangue total de indivíduos sadios e pacientes com MF e PV. O ruxolitinibe resultou na inibição máxima da fosforilação de STAT3 2 horas após a dosagem. A fosforilação retornou praticamente para o valor basal em 8 horas, tanto em indivíduos sadios quanto em pacientes com mielofibrose, não indicando nenhum acúmulo de metabólitos originais ou ativos.
Elevações do basal nos marcadores inflamatórios associados a sintomas constitucionais como TNFa, IL-6, e CRP em pacientes com MF haviam diminuído após o tratamento com Jakavi®. Pacientes com MF não se tornaram refratários aos efeitos farmacodinâmicos do tratamento com Jakavi® com o passar do tempo. Da mesma forma, pacientes com PV também apresentaram elevações em relação ao basal dos marcadores inflamatórios e estes marcadores foram reduzidos após o tratamento com Jakavi®.
Em um estudo de QT completo em indivíduos sadios, não havia nenhuma indicação quanto ao efeito prolongador do QT/QTc do ruxolitinibe em doses únicas e até uma dose supraterapêutica de 200 mg, indicando que o ruxolitinibe não tem nenhum efeito na repolarização cardíaca.
Mecanismo de ação
O ruxolitinibe é um inibidor seletivo das Janus Quinases Associadas (JAKs) JAK1 e JAK2 (valores de IC50 de 3,3 nM e 2,8 nM para as enzimas JAK1 e JAK2, respectivamente). Elas medem a sinalização de uma série de citocinas e fatores de crescimento que são importantes para a hematopoiese e função imune. A sinalização de JAK envolve o recrutamento de transdutores de sinais e ativadores da transcrição (STATs) para receptores da citocina, ativação e localização subsequente de STATs para o núcleo, levando à modulação da expressão do gene. A desregulação da via JAK-STAT tem sido associada a vários cânceres e aumento da proliferação e sobrevida de células malignas.
A MF e a PV são neoplasias mieloproliferativas (NMP) conhecidas por estarem associadas à sinalização desregulada da JAK1 e JAK2. Acredita-se que a base para a desregulação inclua níveis altos de citocinas circulantes que ativam a via JAK-STAT, mutações de ganho de função, tais como JAK2V617F e silenciamento dos mecanismos regulatórios negativos. Pacientes com MF exibem sinalização da JAK desregulada, independente do status mutacional da JAK2V617F. Mutações ativadas da JAK2 (V617F ou exon 12) são encontradas em mais de 95% dos pacientes com PV.
O ruxolitinibe inibe a sinalização de JAK-STAT e a proliferação celular de modelos celulares dependentes de citocina de malignidades hematológicas, bem como de células Ba/F3 para aumento independente de citocina pela expressão da proteína JAK2V617F mutada, com IC50 variando de 80 a 320 nM. Em um modelo murino de NMP positiva para JAK2V617F, administração oral de ruxolitinibe evitou a esplenomegalia, reduziu preferencialmente as células mutantes JAK2V617F no baço, reduziu as citocinas inflamatórias circulantes (ex.: TNF-a, IL-6) e resultou em prolongamento significativo na sobrevida em camundongos nas doses que não causaram efeitos mielosupressores.
As vias de sinalização JAK-STAT desempenham um papel na regulação do desenvolvimento, proliferação e ativação de vários tipos de células imunes importantes para a patogênese da DECH. Em um modelo de rato com DECH aguda, a administração oral de ruxolitinibe foi associada à diminuição da expressão de citocinas inflamatórias em homogenatos do cólon e à redução da infiltração de células imunes no cólon.
Propriedades farmacocinéticas (PK)
- Absorção
O ruxolitinibe é uma molécula de classe 1 de acordo com o Sistema de Classificação Biofarmacêutico, com alta permeabilidade, alta solubilidade e rápidas características de dissolução. Em estudos clínicos, o ruxolitinibe é rapidamente absorvido após a administração oral, com uma concentração plasmática máxima (Cmáx) atingida aproximadamente 1 hora após a dose. Com base no estudo de equilíbrio de massa em humanos, a absorção oral do ruxolitinibe foi 95% ou mais. A Cmáx e a exposição total (AUC) médias de ruxolitinibe aumentaram proporcionalmente em uma variação de dose única de 5 a 200 mg. Não houve nenhuma alteração clinicamente relevante na PK do ruxolitinibe com a administração de refeição com alto teor de gordura. A Cmáx média foi moderadamente reduzida (24%) enquanto a AUC média foi praticamente inalterada (aumento de 4%) com a dosagem com uma refeição de alto teor de gordura.
- Distribuição
O volume médio de distribuição no estado estacionário é de 72 litros em pacientes com MF com uma variabilidade interindividual de 29,4% e 75 litros em pacientes com PV com uma variabilidade interindividual de 22,6%. Em concentrações clinicamente relevantes de ruxolitinibe, a ligação às proteínas plasmáticas in vitro é de aproximadamente 97%, principalmente à albumina. Em um estudo autorradiográfico de corpo total em ratos, demonstrou-se que o ruxolitinibe não penetra a barreira hematoencefálica.
- Biotransformação/metabolismo
Estudos in vitro indicam que a CYP3A4 e a CYP2C9 são as principais enzimas responsáveis pelo metabolismo do ruxolitinibe. O composto original é a entidade predominante em humanos, representando aproximadamente 60% do material relacionado ao medicamento em circulação. Dois metabólitos principais e ativos foram identificados no plasma de indivíduos sadios, representando 25% e 11% da AUC original. Esses metabólitos possuem de metade a um quinto da atividade farmacológica original relacionada ao JAK. A soma de todos os metabólitos ativos contribui para 18% da farmacodinâmica geral do ruxolitinibe. Nas concentrações clinicamente relevantes, o ruxolitinibe não inibe as CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4 e não é um indutor potente da CYP1A2, CYP2B6 ou CYP3A4 com base nos estudos in vitro.
- Eliminação
Após uma dose única oral de ruxolitinibe [14C] em indivíduos adultos sadios, a eliminação foi predominantemente via metabolismo, com 74% da radioatividade excretada na urina e 22% excretada via fezes. O medicamento inalterado constituiu menos de 1% da radioatividade total excretada. A meia-vida média de eliminação de ruxolitinibe é de aproximadamente 3 horas.
- Linearidade/ não linearidade
A proporcionalidade da dose foi demonstrada em estudos de dose ún