INTELENCETM
JANSSEN-CILAG
etravirina
Antiviral.
Apresentações.
Comprimidos de 100 mg em frasco com 120 comprimidos.
USO ADULTO
USO ORAL
Composição.
Cada comprimido contém 100 mg de etravirina.
Excipientes: celulose microcristalina, croscarmelose sódica, dióxido de silício coloidal, estearato de magnésio, hipromelose, lactose monoidratada.
Indicações.
Intelence™, em associação com inibidores de protease potencializados (IP boosted) e outros medicamentos anti-retrovirais, é indicado para o tratamento da infecção pelo vírus da imunodeficiência humana do tipo 1 (HIV-1) em pacientes adultos com experiência no tratamento anti-retroviral, com evidência de replicação viral e que apresentemresistência à inibidores da transcriptase reversa não-análogo de nucleosídeo (ITRNN) e resistência a inibidor de protease (IP).
Essa indicação baseia-se nas análises de 48 semanas de 2 estudos de Fase III randomizados, duplo-cegos e controlados por placebo em pacientes previamente expostos a tratamento e com resistência a ITRNN (presente na seleção e/ou documentada) e inibidor da protease (IP), onde o Intelence™ administrado com um esquema de base (EB) foi estatisticamente superior ao placebo com um EB na proporção de pacientes que atingiram carga viral indetectável confirmada (carga viral < 50 cópias/mL) e aumento da contagem de células CD4 em relação à linha de base.
O histórico do tratamento e, quando disponível, o teste de resistência, deve orientar o uso de Intelence™. Intelence™ não é recomendado para uso em combinações contendo apenas ITRNs em pacientes que apresentaram falha virológica com um regime contendo um inibidor da transcriptase reversa não-análogo de nucleosídeo (ITRNN) e inibidor da transcriptase reversa análogo de nucleosídeo/nucleotídeo (ITRN).
Resultados de eficácia.
Experiência clínica
Pacientes previamente expostos a tratamento
As evidências de eficácia de Intelence™ baseiam-se nas análises dos dados de 48 semanas de 2 estudos em andamento de Fase III, randomizados, duplo-cegos e controlados por placebo (DUET-1 e DUET-2). Esses estudos tiveram desenhos idênticos e foi observada eficácia semelhante de Intelence™ em cada estudo. Os resultados a seguir são os dados agrupados dos dois estudos.
Os pacientes infectados pelo HIV-1 previamente expostos a tratamento que apresentavam no plasma carga viral > 5000 cópias/mL e tinham uma ou mais mutações associadas à resistência a ITRNN no screening ou em uma genotipagem prévia (ou seja, resistência prévia) foram admitidos. Esses pacientes também apresentavam 3 ou mais das seguintes mutações primárias de IP: D30N, V32I, L33F, M46I/L, I47A/V, G48V, I50L/V, V82A/F/L/S/T, I84V, N88S ou L90M no screening e recebiam um esquema anti-retroviral estável por pelo menos 8 semanas. A randomização foi estratificada pelo possível uso da enfuvirtida (ENF) no esquema de base (EB), uso prévio de darunavir/ritonavir e pela carga viral no screening. Essa análise incluiu 612 pacientes no DUET-1 e 591 no DUET-2 que haviam completado 48 semanas de tratamento ou descontinuado previamente.
Na semana 48, a taxa de resposta virológica foi avaliada em pacientes que receberam Intelence™ (200 mg 2x/dia) adicionada ao esquema de base versus pacientes que receberam placebo adicionado ao esquema de base. O esquema de base consistiu de darunavir/ritonavir 600/100 mg 2x/dia e, no mínimo, 2 outros agentes anti-retrovirais selecionados pelo investigator (ITRN com ou sem a ENF). 45,6% dos pacientes no grupo Intelence™ e 46,9% dos pacientes no grupo placebo utilizaram a ENF na terapia anti-retroviral de base. 25,5% dos pacientes no grupo Intelence™ utilizaram a ENF pela primeira vez (de novo) em comparação a 26,5% dos pacientes no grupo placebo. 20,0% dos pacientes no grupo Intelence™ reutilizaram a ENF em comparação a 20,4% dos pacientes no grupo placebo. A resposta virológica foi definida como a obtenção de carga viral indetectável confirmada (carga viral < 50 cópias/mL).
A tabela a seguir mostra os resultados de eficácia em 48 semanas dos pacientes no grupo Intelence™ e dos pacientes no grupo placebo dos estudos agrupados DUET-1 e DUET-2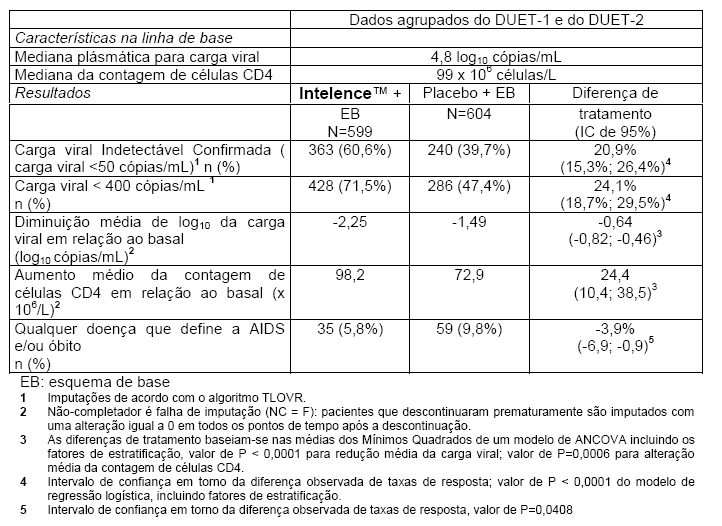
Como houve um efeito de interação significativo entre o tratamento e a ENF, a análise primária foi realizada para 2 estratos de ENF (pacientes em reuso ou não usando a ENF versus pacientes usando a ENF de novo). Os resultados na análise agrupada do DUET-1 e do DUET-2 na semana 48 demonstraram que o grupo Intelence™ foi superior ao grupo placebo independentemente de se a ENF foi usada pela primeira vez ou não. Na população de pacientes em reuso ou não usando a ENF, a proporção de pacientes com carga viral < 50 cópias/mL foi de 57,0% no grupo Intelence™ e 33,0% no grupo placebo (uma diferença de 24,0%, p < 0,0001). No grupo de pacientes que utilizaram a ENF pela primeira vez, 71,2% dos pacientes no grupo Intelence™ atingiram carga viral < 50 cópias/mL em comparação a 58,5% no grupo placebo (uma diferença de 12,7%, p = 0,0199).
Na semana 48, significativamente poucos pacientes no grupo Intelence™ (35 pacientes, 5,8%) atingiram um desfecho clínico (doenças definidoras de AIDS ou morte) se comparado ao grupo placebo (59 pacientes, 9,8%) (p = 0,0408).
Resultados relatados pelo paciente
Nos estudos agrupados DUET, os pacientes no grupo Intelence™ demonstraram em 48 semanas uma melhora estatisticamente significativa em relação à linha de base na subescala de Bem-Estar Físico do questionário FAHI (avaliação funcional da infecção pelo vírus da imunodeficiência humana) relatado pelo paciente. Essa melhora foi estatisticamente maior nos pacientes do grupo Intelence™ comparado à pacientes no grupo placebo. Para a subescala de Bem-Estar Funcional e Global, nenhuma diferença estatística foi encontrada.
Análise de resultado virológico e genótipo ou fenótipo basais
No DUET-1 e no DUET-2, a presença na linha de base de 3 ou mais das seguintes mutações: V90I, A98G, L100I, K101E, K101P, V106I, V179D, V179F, Y181C, Y181I, Y181V, G190A e G190S (RAMs - resistência associada a mutações de Intelence™) foi associada a uma diminuição da resposta virológica ao Intelence™ (vide a tabela a seguir). Essas mutações individuais ocorreram na presença de outras RAMs de ITRNN. A V179F nunca esteve presente sem a Y181C.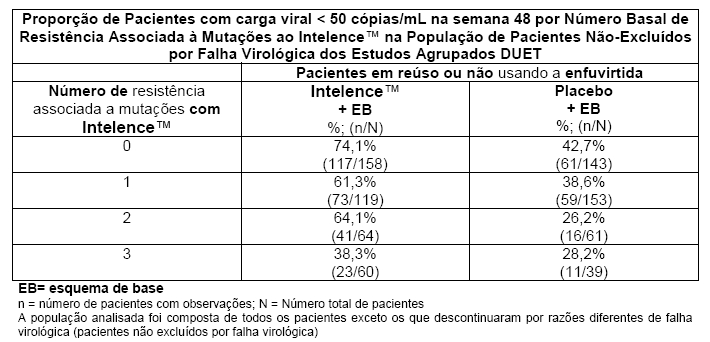
A K103N, que foi a mutação do ITRNN mais prevalente no DUET-1 e no DUET-2 na linha de base, não foi identificada como uma mutação associada à resistência ao Intelence™. A presença dessa mutação não afetou a resposta no grupo Intelence™.
Demonstrou-se que o fenótipo da etravirina na linha de base (alteração da sensibilidade em relação à referência) é um fator preditivo da resposta virológica. As taxas de resposta avaliadas de acordo com o fenótipo da etravirina na linha de base são mostradas na tabela a seguir. Esses grupos de fenótipo na linha de base baseiam-se em algumas populações de pacientes do DUET-1 e do DUET-2 e não se destinam a representar breakpoints de sensibilidade clínica definitivos para o Intelence™. Os dados são fornecidos para dar aos médicos informações sobre a probabilidade de sucesso virológico com base na sensibilidade à etravirina pré-tratamento em pacientes previamente expostos a tratamento.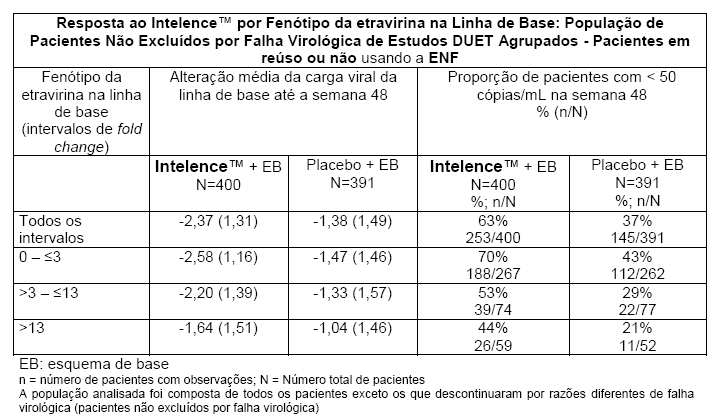
Para a carga viral indetectável ( < 50 cópias/mL), a redução absoluta do risco (RRA) é de aproximadamente 17,87, o que corresponde, portanto, ao número necessário para tratar (NNT) de aproximadamente 6.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
Mecanismo de ação
A etravirina é um ITRNN do vírus da imunodeficiência humana do tipo 1 (HIV-1). A etravirina liga-se diretamente à transcriptase reversa (TR) e bloqueia as atividades da DNA polimerase RNA-dependente e DNA-dependente através da quebra do sítio catalítico da enzima. A etravirina pode ligar-se à transcriptase reversa a pelo menos 2 modos conformacionais distintos. Em um determinado modo de ligação, a flexibilidade torsional da etravirina permite o acesso à numerosas variantes conformacionais enquanto o seu desenho compacto permite o reposicionamento e reorientação (tradução e rotação) dentro do pocket. A etravirina não inibe as DNA polimerases a, b e c humanas.
Atividade antiviral in vitro
A etravirina apresenta atividade contra cepas de laboratório e de isolados clínicos do HIV-1 do tipo selvagem em linhagens de células T agudamente infectadas, células mononucleares do sangue periférico humano e monócitos/macrófagos humanos com valores medianos de CE50 variando de 0,9 a 5,5 nM (ou seja, 0,4 a 2,4 ng/mL).
A etravirina demonstra atividade antiviral in vitro contra um painel de vírus HIV-1 do grupo M (subtipos A, B, C, D, E, F, G) e grupo O com CE50 variando de 0,7 a 21,7 nM. Esses valores de CE50 ficam bem abaixo do intervalo de concentração de toxicidade celular de 50% de 15 a > 100 mM.
O valor de CE50 da etravirina para o HIV-1 aumenta com um fator mediano de 5,8 na presença de soro humano.
Não se observa antagonismo entre a etravirina e qualquer dos anti-retrovirais estudados. A etravirina demonstra atividade antiviral aditiva em combinação com os IPs amprenavir, atazanavir, darunavir, indinavir, lopinavir, nelfinavir, ritonavir, tipranavir e saquinavir; os ITRNs zalcitabina, didanosina, estavudina, abacavir e tenofovir; os ITRNNs efavirenz, delavirdina e nevirapina; o inibidor de fusão enfuvirtida; inibidor de integrase raltegravir e o antagonista de CCR5 maraviroque. A etravirina demonstra atividade antiviral aditiva a sinérgica em combinação aos ITRNs entricitabina, lamivudina e zidovudina.
Resistência
Em um grupo de 65 cepas de HIV-1 com substituição da posiçãode um único aminoácido da transcriptase reversa associadas à resistência ao ITRNN, incluindo a K103N e a Y181C que são as mais frequentemente observadas, a etravirina demonstra atividade antiviral potente contra 56 dessas cepas. As substituições de aminoácidos, que resultam na maior resistência à etravirina em cultura de células, são a Y181I (fold change de 13 - em valor de CE50) e a Y181V (fold change de 17 - em valor de CE50). A atividade antiviral da etravirina em cultura de células contra 24 cepas de HIV-1 com múltiplas substituições de aminoácidos associada a resistência a ITRNs e/ou IPs é comparável à observada contra o HIV-1 do tipo selvagem.
A seleção in vitro de cepas resistentes à etravirina originárias do HIV-1 do tipo selvagem de diferentes origens e subtipos, bem como do HIV-1 resistentes a ITRNN, foi feita com altos e baixos inóculos virais. No inóculo viral alto, o aparecimento de cepas resistentes do HIV-1 do tipo selvagem foi retardado ou prevenido nas concentrações de 40 nM ou 200 nM. O mesmo foi observado com cepas resistentes que apresentam as mutações únicas K103N e Y181C associadas à resistência a ITRNN. Independentemente do desenho experimental e da cepa do HIV-1 original, o desenvolvimento de resistência contra a etravirina necessita caracteristicamente de múltiplas mutações na transcriptase reversa, entre as quais as seguintes foram observadas mais frequentemente: L100I, E138K, E138G, V179I, Y181C e M230I.
Nos estudos de fase III, DUET-1 e DUET-2, as mutações que se desenvolveram com maior frequência nos pacientes com falha virológica ao esquema contendo Intelence™ foram V179F, V179I e Y181C, que geralmente apareceram no contexto de várias outras resistências associadas à mutações (RAMs) a ITRNN. Em todos os estudos conduzidos com Intelence™ em pacientes infectados pelo HIV-1, as seguintes mutações apareceram com maior frequência: L100I, E138G, V179F, V179I, Y181C e H221Y.
Resistência cruzada
Foi observada limitada resistência cruzada entre a etravirina e o efavirenz in vitro em 3 das 65 cepas mutantes de HIV-1 contendo uma mutação associada a resistência a ITRNN. Para as outras cepas, as posições dos aminoácidos associadas a diminuição da sensibilidade à etravirina e ao efavirenz foram diferentes. A etravirina continua com valor de CE50 < 10 nM contra 83% dos 6.171 isolados clínicos resistentes a delavirdina, efavirenz e/ou nevirapina. Não se recomenda o tratamento de pacientes com delavirdina, efavirenz ou nevirapina após falha virológica de um regime contendo etravirina.
Propriedades Farmacocinéticas
As propriedades farmacocinéticas da etravirina foram avaliadas em adultos saudáveis e em pacientes adultos infectados pelo HIV-1 previamente expostos a tratamento. A exposição à etravirina foi um pouco menor nos pacientes infectados pelo HIV-1 do que nos indivíduos saudáveis.
Absorção
Não existe disponível uma formulação intravenosa da etravirina, portanto, a biodisponibilidade absoluta de Intelence™ é desconhecida. Após a administração oral com alimentos, a concentração plasmática máxima da etravirina é geralmente alcançada em 4 horas. Em indivíduos saudáveis, a absorção da etravirina não é afetada pela administração concomitante por via oral da ranitidina ou do omeprazol, que são medicamentos conhecidos por aumentarem o pH gástrico.
Efeito dos alimentos sobre a absorção
A exposição à etravirina é semelhante quando administrada após uma refeição calórica normal padrão (561 kcal) ou uma refeição de alto teor calórico com alto teor de gordura (1.160 kcal). Em comparação à administração após uma refeição calória normal padrão, as exposições diminuíram quando a etravirina foi tomada antes de uma refeição calória normal padrão (17%), após um croissant (20%) ou em jejum (51%). Portanto, para atingir a exposição ideal, ao Intelence™ deve ser tomado após uma refeição.
Distribuição
A etravirina apresenta taxa de ligação a proteínas plasmáticas de 99,9%, principalmente à albumina (99,6%) e à glicoproteína ácida a1 (97,66%-99,02%) in vitro. A distribuição da etravirina em outros locais além do plasma (p. ex., fluido cerebroespinal, secreções do trato genital) ainda não foi avaliada em humanos.
Metabolismo
Os experimentos in vitro com microssomas hepáticos humanos (MHHs) indicam que a etravirina sofre principalmente metabolismo oxidativo pelo sistema hepático do citocromo P450 (CYP) 3A e, em menor extensão, pela família da CYP2C seguido da glicuronidação.
Eliminação
Após a administração de uma dose de 14C-etravirina radiomarcada, 93,7% e 1,2% da dose administrada da 14C-etravirina puderam ser recuperadas nas fezes e na urina, respectivamente. A etravirina inalterada representou nas fezes 81,2% a 86,4% da dose administrada. A etravirina inalterada não foi detectada na urina. A meia-vida de eliminação terminal da etravirina foi de aproximadamente 30-40 horas.
Populações especiais
Crianças e adolescentes: a farmacocinética da etravirina em pacientes pediátricos ainda está em estudo. Não há dados suficientes neste momento para recomendar uma dose (veja o item Posologia).
Idosos: a análise de farmacocinética populacional em pacientes infectados pelo HIV demonstrou que a farmacocinética da etravirina não é consideravelmente diferente na faixa etária avaliada (18 a 77 anos) (veja os itens Posologia e Advertências).
Sexo: não foram observadas diferenças farmacocinéticas significativas entre homens e mulheres. Um número limitado de mulheres foi incluído nos estudos.
Raça: a análise de farmacocinética populacional da etravirina em pacientes infectados pelo HIV indicou que a raça não teve efeito aparente sobre a exposição à etravirina.
Insuficiência hepática: a etravirina é metabolizada e eliminada principalmente pelo fígado. Em um estudo que comparou 8 pacientes com insuficiência hepática leve (pontuação A de Child-Pugh) a 8 controles pareados e 8 pacientes com insuficiência hepática moderada (pontuação B de Child-Pugh) a 8 controles pareados, a disposição farmacocinética de doses múltiplas da etravirina não sofreu alteração em pacientes com insuficiência hepática leve a moderada. Não é necessário ajustar a dose nos pacientes com insuficiência hepática leve ou moderada. A farmacocinética de Intelence™ não foi estudada em pacientes com insuficiência hepática grave (pontuação C de Child-Pugh) (veja os itens Posologia e Advertências).
Co-infecção pelos vírus da hepatite B e/ou hepatite C: a análise de farmacocinética populacional dos Estudos DUET-1 e DUET-2 mostrou depuração reduzida de Intelence™ nos pacientes infectados pelo HIV-1 com co-infecção pelos vírus da hepatite B e/ou C. Com base no perfil de segurança (veja o item Reações Adversas), não é necessário ajustar a dose em pacientes co-infectados pelos vírus da hepatite B e/ou C.
Insuficiência renal: a farmacocinética da etravirina ainda não foi estudada em pacientes com insuficiência renal. Os resultados de um estudo de equilíbrio de massa com 14C-etravirina radioativa mostraram que < 1,2% da dose administrada da etravirina é excretada na urina. O fármaco inalterado não foi detectado na urina, de forma que é de se esperar que o impacto da insuficiência renal sobre a eliminação da etravirina seja mínimo. Por apresentar alta taxa de ligação a proteínas plasmáticas, é improvável que a etravirina seja significativamente removida por hemodiálise ou diálise peritoneal (veja os itens Posologia e Advertências)
Dados pré-clínicos de segurança
Foram conduzidos estudos de toxicologia em animais com a etravirina em camundongos, ratos, coelhos e cães. Em camundongos, os principais órgãos-alvo identificados foram o fígado e o sistema de coagulação. A cardiomiopatia hemorrágica só foi observada em camundongos machos e foi considerada secundária à coagulapatia grave mediada pela via da vitamina K. Isso não é considerado relevante em humanos. Em ratos, os principais órgãos-alvo identificados foram o fígado, a tireóide e o sistema de coagulação. A exposição em camundongos foi equivalente à exposição em humanos, ao passo que em ratos foi abaixo da exposição clínica na dose recomendada. Em cães, foram observadas alterações no fígado e na vesícula biliar nas exposições aproximadamente 8 vezes maiores que a exposição humana observada na dose recomendada (200 mg 2x/dia).
Em um estudo conduzido em ratos, não houve efeitos sobre o acasalamento ou a fertilidade com o tratamento com Intelence™ até 500 mg/kg/dia e níveis de exposição equivalentes aos observados em humanos na dose clinicamente recomendada. Não houve teratogenicidade com a etravirina em ratos (1.000 mg/kg) e coelhos (375 mg/kg) em exposições equivalentes às observadas em humanos na dose clínica recomendada. Em uma avaliação de desenvolvimento pré e pós-natal em ratos, a etravirina não teve efeito sobre o desenvolvimento dos filhotes durante a lactação ou após o desmame quando a mãe recebeu até 500 mg/kg e nas exposições equivalentes às observadas na dose clínica recomendada.
A etravirina foi avaliada para o potencial carcinogênico pela administração oral por gavagem em camundongos e ratos até a semana 104. Doses diárias de 50, 200 e 400 mg/Kg foram administradas em camundongos e doses de 70, 200 e 600 mg/Kg foram administradas em ratos. A etravirina não foi carcinogênica em ratos e em camundongos machos. Um aumento na incidência de adenomas hepatocelulares e carcinomas foi observado em camundongos fêmeas. A administração de etravirina não causou um aumento estatisticamente significante na incidência de qualquer outra neoplasia benigna ou maligna em camundongos ou ratos. Os achados hepatocelulares observados em camundongos fêmeas são geralmente considerados para serem específicos de roedores, associados com indutor de enzima hepática e de relevância limitada aos humanos. Nas mais altas doses testadas, a exposição sistêmica (baseada na AUC) à etravirina foi 0,6 vezes (camundongos) e entre 0,2 e 07 vezes (ratos), relativa àquela observada em humanos na dose terapêutica recomendada (200 mg 2x/dia).
A etravirina foi negativa nos ensaios de mutação reversa de Ames in vitro, aberração cromossômica in vitro em linfócitos humanos e clastogenicidade in vitro em linfoma de camundongos, testada na ausência e na presença de um sistema de ativação metabólica. A etravirina não induziu dano cromossômico no teste de micronúcleos in vivo em camundongos.
Contraindicações.
Hipersensibilidade à etravirina ou qualquer um dos excipientes.
Advertências e precauções.
Advertências
Os pacientes devem ser avisados de que a terapia anti-retroviral atual não cura o HIV e de que ainda não se comprovou que ela previne a transmissão do HIV a outras pessoas através do sangue ou do contato sexual. As precauções adequadas devem continuar a ser utilizadas.
Estão sendo conduzidos estudos clínicos em crianças e adolescentes infectadas pelo HIV-1 (idades entre 6 e 17 anos, inclusive).
Reações cutâneas e de hipersensibilidade graves
Foram relatadas reações cutâneas fatais e potencialmente fatais com o uso de Intelence™. Os relatos de Síndrome de Stevens-Johnson e necrólise epidérmica tóxica foram raros ( < 0,1%). Reações de hipersensibilidade também foram relatadas e caracterizadas como erupção cutânea, achados clínicos e, em menor frequência, como disfunção de órgão, incluindo falência hepática.
Mais frequentemente, a erupção cutânea foi de leve a moderada, e ocorreu principalmente na segunda semana de tratamento e foi infrequente após a quarta semana. A erupção cutânea foi principalmente auto-limitante e, em geral, resolvida dentro de 1-2 semanas de tratamento contínuo (veja o item Reações Adversas).
Interrompa o tratamento com Intelence™ imediatamente, se sinais ou sintomas de reações cutâneas graves ou reações de hipersensibilidade se desenvolverem (incluindo, mas não limitado a, erupção cutânea grave ou erupção cutânea acompanhada de febre, mal-estar generalizado, fadiga, dores musculares ou nas articulações, bolhas, lesões orais, conjuntivite, hepatite e eosinofilia). A avaliação clínica, incluindo transaminases hepáticas, deve ser efetuada e o tratamento apropriado instituído. A demora na interrupção do tratamento com Intelence™ após o início da reação cutânea grave pode resultar em reações que colocam a vida do paciente em risco.
Idosos
A experiência em pacientes geriátricos é limitada. Nos estudos de Fase III, 6 pacientes > 65 anos e 53 com 56-64 anos receberam Intelence™. O tipo e a incidência de eventos adversos nos pacientes > 55 anos foram semelhantes aos observados nos pacientes mais jovens (veja os itens Posologia e Propriedades Farmacocinéticas).
Pacientes com condições coexistentes
Doença hepática
Não é necessário ajustar a dose em pacientes com insuficiência hepática leve ou moderada (pontuação A ou B de Child-Pugh). A farmacocinética de Intelence™ ainda não foi estudada em pacientes com insuficiência hepática grave (pontuação C de Child-Pugh) (veja os itens Posologia e Propriedades Farmacocinéticas).
Doença renal
Como a depuração renal da etravirina é insignificante ( < 1,2%), não é de se esperar a ocorrência de uma redução da depuração corpórea total em pacientes com insuficiência renal. Não são necessárias precauções especiais nem ajustes da dose em pacientes com insuficiência renal. Como a etravirina apresenta alta taxa de ligação a proteínas plasmáticas, é improvável que seja significativamente removida por hemodiálise ou diálise peritoneal (veja os itens Posologia e Propriedades Farmacocinéticas).
Redistribuição da Gordura
A terapia anti-retroviral combinada vem sendo associada à redistribução da gordura do corpo (lipodistrofia) nos pacientes infectados pelo HIV. As consequências a longo prazo desses eventos são atualmente desconhecidas no momento. O conhecimento sobre o mecanismo é incompleto. Levantou-se a hipótese de conexão entre lipomatose visceral e IPs, e lipodistrofia e inibidores da transcriptase reversa análogos de nucleosídeos (ITRNs) O maior risco de lipodistrofia está associado a fatores individuais, como idade avançada, e fatores relacionados ao medicamento, como duração mais prolongada do tratamento anti-retroviral e distúrbios metabólicos associados. O exame clínico deve incluir a avaliação de sinais físicos da redistribuição da gordura (veja o item Reações Adversas)
Síndrome da reconstituição imunológica
Nos pacientes infectados com o HIV com deficiência imunológica grave na ocasião da instituição da terapia anti-retroviral combinada, pode ocorrer uma reação inflamatória a patógenos oportunistas assintomáticos ou residuais que pode causar condições clínicas sérias ou piora dos sintomas. Caracteristicamente, essas reações foram observadas nas primeiras semanas ou meses do início da terapia anti-retroviral combinada. Exemplos relevantes são retinite por citomegalovírus, infecções por micobactérias generalizadas e/ou focais e pneumonia por Pneumocystis jiroveci. Todos os sintomas inflamatórios devem ser avaliados e o tratamento deve ser instituído quando necessário (veja o item Reações Adversas).
Interações com medicamentos
Para informações sobre as interações com medicamentos, veja o item Interações Medicamentosas.
Efeitos sobre a capacidade de dirigir veículos e utilizar máquinas
Não foram conduzidos estudos para avaliar os efeitos de Intelence™ sobre a capacidade de dirigir veículos ou operar máquinas. Não há evidências de que Intelence™ altere a capacidade do paciente de dirigir veículos e operar máquinas, no entanto, o perfil de reações adversas medicamentosas de Intelence™ deve ser levado em consideração (veja o item Reações Adversas).
Gravidez (Categoria B) e Lactação
Gravidez
Não há estudos adequados e bem-controlados com a etravirina em mulheres grávidas. Os estudos em animais não demonstraram evidências de toxicidade ao desenvolvimento ou efeito sobre a função reprodutiva e a fertilidade (veja o item Dados pré-clínicos de segurança)
Intelence™ deve ser usado durante a gravidez apenas se o benefício potencial justificar o risco potencial.
Lactação
Não se sabe se a etravirina é excretada no leite humano. Devido ao potencial de transmissão do HIV e ao potencial de eventos adversos nos lactentes, as mães devem ser orientadas a não amamentar se estiverem tomando Intelence™.
Fertilidade
Não estão disponíveis dados do efeito da etravirina sobre a fertilidade em humanos. Em ratos, não houve efeito sobre o acasalamento ou fertilidade no tratamento com Intelence™ (veja o item Dados pré-clínicos de segurança).
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
USO EM IDOSOS, CRIANÇAS E OUTROS GRUPOS DE RISCO
Idosos: estão disponíveis informações limitadas nessa população (veja os itens Advertências e Propriedades Farmacocinéticas).
Crianças (menos de 12 anos) e adolescentes (12 a 17 anos): não se recomenda o tratamento com Intelence™ em crianças e adolescentes. A segurança e a eficácia de Intelence™ nessas populações ainda estão em estudo (veja o item Propriedades Farmacocinéticas).
Insuficiência hepática: não é necessário ajustar a dose em pacientes com insuficiência hepática leve ou moderada (pontuação A ou B de Child-Pugh). A farmacocinética de Intelence™ ainda não foi estudada em pacientes com insuficiência hepática grave (pontuação C de Child-Pugh) (veja os itens Advertências e Propriedades Farmacocinéticas).
Insuficiência renal: não é necessário ajustar a dose em pacientes com insuficiência renal (veja os itens Advertências e Propriedades Farmacocinéticas)
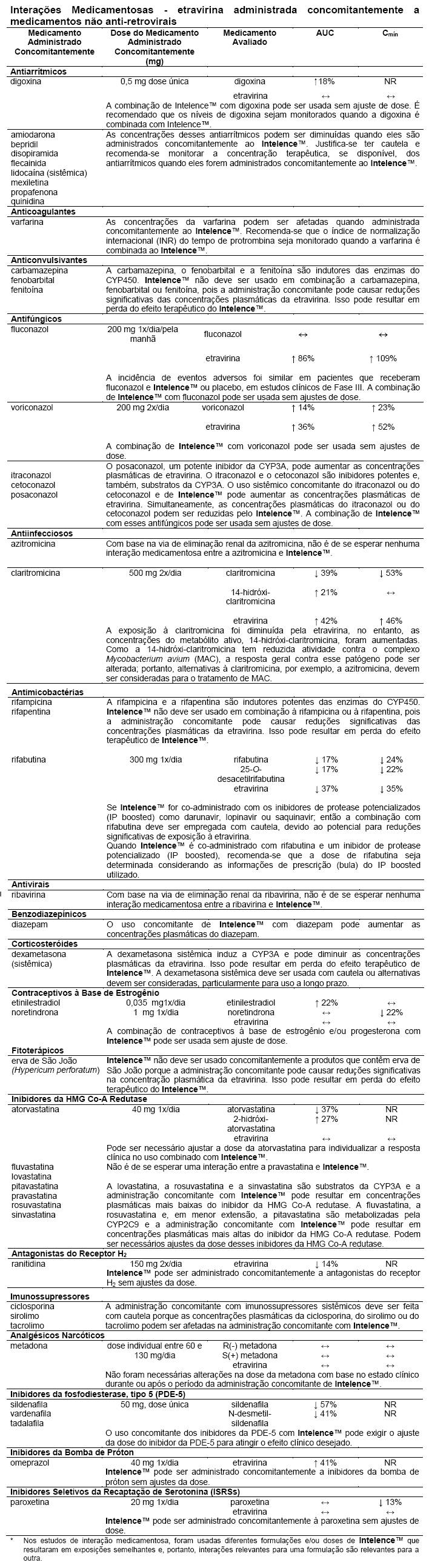
Interações medicamentosas.
Medicamentos que afetam a exposição à etravirina
A etravirina é metabolizada pelo citocromo P450 (CYP)3A, CYP2C9 e CYP2C19, seguida da glicuronidação dos metabólitos pela uridina difosfato glicuronosil transferase (UDPGT). Os medicamentos que induzem a CYP3A, a CYP2C9 ou a CYP2C19 podem aumentar a depuração da etravirina que resulta na redução das concentrações plasmáticas da etravirina. A administração concomitante de Intelence™ e medicamentos que inibem a CYP3A, a CYP2C9 ou a CYP2C19 podem diminuir a depuração da etravirina e podem resultar em aumento das concentrações plasmáticas da etravirina.
Medicamentos que são afetados pelo uso da etravirina
A etravirina é um indutor fraco da CYP3A. A administração concomitante de Intelence™ com medicamentos metabolizados principalmente pelo CYP3A pode resultar na redução das concentrações plasmáticas desses medicamentos, o que poderia diminuir ou encurtar os seus efeitos terapêuticos. A etravirina é um inibidor fraco da CYP2C9 e da CYP2C19. A etravirina é também um inibidor fraco da P-glicoproteína, mas não um substrato. A administração concomitante com medicamentos metabolizados principalmente pela CYP2C9 ou pela CYP2C19 ou transportados pela P-glicoproteína pode resultar no aumento das concentrações plasmáticas desses medicamentos, o que poderia aumentar ou prolongar os seus efeitos terapêuticos ou o perfil de eventos adversos.
As interações conhecidas e teóricas com alguns medicamentos anti-retrovirais e não-anti-retrovirais são apresentadas nas tabelas a seguir.
Tabela de Interação *
As interações entre a etravirina e os medicamentos administrados concomitantemente são apresentadas nas tabelas a seguir (aumento é indicado como "↑", diminuição como "↓", inalterado como "↔", não realizado como "NR", uma vez por dia como "1x/dia", uma vez por dia pela manhã como "1x/dia/pela manhã" e duas vezes por dia como "2x/dia").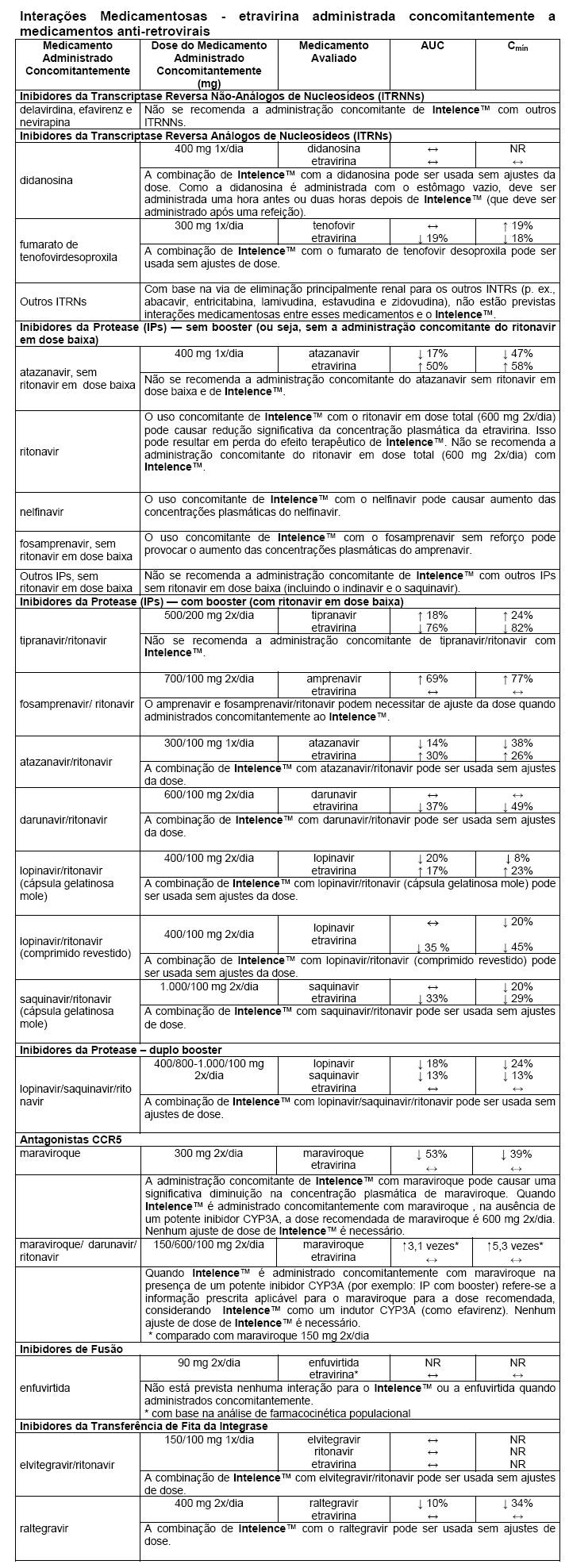
Cuidados de armazenamento.
Conserve o frasco de Intelence™ em temperatura ambiente (entre 15°C e 30°C).
Armazenar os comprimidos no frasco original. Manter o frasco hermeticamente fechado para proteger o produto da umidade. Não remover os sachês de dessecante.
Posologia e modo de usar.
Modo de usar e cuidados de conservação depois de aberto
Os comprimidos de Intelence™ devem ser tomados por via oral, duas vezes ao dia, junto com uma refeição.
Intelence™ deve sempre ser administrado em associação a outros medicamentos anti-retrovirais.
Armazenar os comprimidos no frasco original. Manter o frasco hermeticamente fechado para proteger o produto da umidade. Não remover os sachês de dessecante.
Posologia
Adultos: a dose recomendada da Intelence™ é de 200 mg (dois comprimidos de 100 mg) administrados por via oral duas vezes por dia (2x/dia), após uma refeição (veja o item Propriedades Farmacocinéticas). Os pacientes devem ser informados para engolir os comprimidos inteiros com um líquido, como água. Os pacientes incapazes de engolir os comprimidos de Intelence™ inteiros podem dissolvê-los em um copo de água. Uma vez que os comprimidos são dissolvidos, os pacientes devem agitar bem o conteúdo e bebê-lo imediatamente. Deve-se adicionar água ao copo várias vezes, e logo após, deve-se ingerir todo o conteúdo para assegurar que toda a dose seja consumida.
Crianças (menos de 12 anos) e adolescentes (12 a 17 anos): não se recomenda o tratamento com Intelence™ em crianças e adolescentes. A segurança e a eficácia de Intelence™ nessas populações ainda estão em estudo (veja o item Propriedades Farmacocinéticas).
Idosos: estão disponíveis informações limitadas nessa população (veja os itens Advertências e Propriedades Farmacocinéticas).
Insuficiência hepática: não é necessário ajustar a dose em pacientes com insuficiência hepática leve ou moderada (pontuação A ou B de Child-Pugh). A farmacocinética de Intelence™ ainda não foi estudada em pacientes com insuficiência hepática grave (pontuação C de Child-Pugh) (veja os itens Advertências e Propriedades Farmacocinéticas).
Insuficiência renal: não é necessário ajustar a dose em pacientes com insuficiência renal (veja os itens Advertências e Propriedades Farmacocinéticas).
Se o paciente esquecer de tomar a dose de Intelence™ em até 6 horas do horário que geralmente deveria ter sido tomada, o paciente deve ser aconselhado a tomar Intelence™ após uma refeição assim que possível e, depois, tomar a dose seguinte de Intelence™ no horário regular programado. Se esquecer de tomar a dose da Intelence™ mais de 6 horas depois do horário que geralmente deveria ter sido tomada, o paciente deve ser aconselhado a não tomar a dose esquecida e simplesmente voltar ao esquema de administração usual.
Reações adversas.
Reações Adversas ocorridas durante Estudos Clínicos
A avaliação de segurança baseia-se em todos os dados de 1.203 pacientes nos estudos DUET-1 e DUET-2, que são estudos de Fase III controlados por placebo em pacientes adultos infectados pelo HIV-1 com experiência no tratamento anti-retroviral, em que 599 deles receberam Intelence™ (200 mg 2x/dia). Nesses estudos agrupados, a exposição mediana nos pacientes dos grupos Intelence™ e placebo foi de 52,3 e 51,0 semanas, respectivamente.
As reações adversas a medicamentos relatadas (≥ 5%) com maior frequência de gravidade de no mínimo Grau 2 foram erupção cutânea (10,0% no grupo Intelence™ e 3,5% no grupo placebo), diarréia (7,0% no grupo Intelence™ e 11,3% no grupo placebo), hipertrigliceridemia (6,3% no grupo de Intelence™ e 4,3% no grupo placebo) e náusea (5,2% no grupo Intelence™ e 4,8% no grupo placebo) (vide tabela a seguir).
A maioria das reações adversas medicamentosas relatadas durante o tratamento com Intelence™ foram de Graus 1 a 2. Foram relatadas reações adversas medicamentosas de Grau 3 ou 4 em 22,2% e 17,2% dos pacientes tratados com Intelence™ e placebo, respectivamente. As reações adversas medicamentosas de Grau 3 ou 4 mais frequentemente relatadas foram hipertrigliceridemia (4,2% no grupo Intelence™ e 2,3% no grupo placebo), hipercolesterolemia (2,2% no grupo Intelence™ e 2,3% no grupo placebo), falência renal (2,0% no grupo Intelence™ e 1,2% no grupo placebo) e anemia (1,7% no grupo Intelence™ e 1,3% no grupo placebo). Para anormalidades clínico-laboratoriais ocorridas durante o tratamento (Grau 3 ou 4) relatadas em ≥ 2% dos pacientes tratados com Intelence™, vide tabela "Anormalidades Laboratoriais Ocorridas Durante o Tratamento". Todas as outras reações adversas medicamentosas de Graus 3 e/ou 4 foram relatadas em menos de 1,5% dos pacientes tratados com Intelence™. 5,2% dos pacientes no grupo Intelence™ descontinuaram o tratamento devido a reações adversas medicamentosas em comparação a 2,6% dos pacientes no grupo placebo. A reação adversa mais comum que resultou em descontinuação foi erupção cutânea (2,2% no grupo Intelence™ versus 0% no grupo placebo).
Erupção cutânea foi mais frequentemente leve a moderada, geralmente macular a maculopapular ou eritematosa, ocorreu principalmente na segunda semana de terapia e foi infrequente após a quarta semana. Erupção cutânea foi na maioria das vezes auto-limitante e, em geral, resolvida em 1-2 semanas de tratamento contínuo (veja o item Advertências). A incidência de erupção cutânea foi maior em mulheres que em homens no grupo Intelence™ nos estudos DUET. No amplo banco de dados de Fase IIb/III (N = 1223), nenhuma diferença entre os sexos foi vista. Não houve diferença entre os sexos na gravidade ou na descontinuação do tratamento devido a erupção cutânea. Nos pacientes com história de erupção cutânea relacionada aos ITRNN, não houve aumento do risco aparente de desenvolvimento de erupção cutânea relacionada ao Intelence™ em comparação aos pacientes sem história de erupção cutânea relacionada a ITRNN.
As reações adversas medicamentosas de intensidade moderada ou maior (Grau ≥ 2) e relatadas em ≥ 1% dos pacientes tratados com Intelence™ estão resumidas na tabela a seguir. As reações adversas medicamentosas são apresentadas por classe de sistema/órgão (SOC) e frequência. As anormalidades laboratoriais consideradas reações adversas medicamentosas foram incluídas em uma tabela a seguir (vide Anormalidades Laboratoriais de Graus 3 a 4 Ocorridas Durante o Tratamento Relatadas em ≥ 2% dos Pacientes).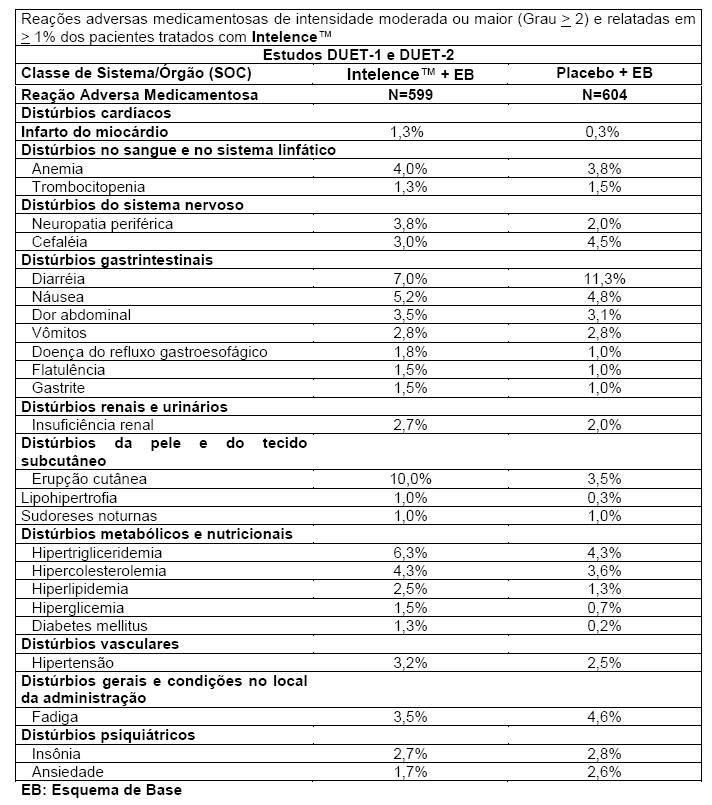
As reações adversas medicamentosas de intensidade moderada ou maior (Grau > 2) ocorridas durante o tratamento em menos de 1% dos pacientes que receberam Intelence™ foram:
- distúrbios cardíacos: angina pectoris, fibrilação atrial;
- distúrbios do sistema nervoso: parestesia, sonolência, convulsão, hipoestesia, amnésia, síncope, distúrbio de atenção, hipersonia, tremor;
- distúrbios oculares: visão turva;
- distúrbios do ouvido e do labirinto: vertigem;
- distúrbios respiratórios, torácicos e mediastinais: dispnéia de esforço, broncoespasmo;
- distúrbios gastrintestinais: distensão abdominal, pancreatite, constipação, boca seca, hematêmese, vômito seco, estomatite;
- distúrbios da pele e do tecido subcutâneo: prurigo, hiperhidrose, pele seca, inchaço facial;
- distúrbios metabólicos e nutricionais: anorexia, dislipidemia;
- distúrbios gerais e condições no local da administração: letargia;
- distúrbios do sistema imunológico: hipersensiblidade a medicamento, síndrome da reconstituição imunológica;
- distúrbios hepatobiliares: hepatomegalia, hepatite citolítica, esteatose hepática, hepatite;
- distúrbios do sistema reprodutor e nas mamas: ginecomastia;
- transtornos psiquiátricos: distúrbios do sono, sonhos anormais, estado confusional, desorientação, nervosismo, pesadelos.
Outras reações adversas medicamentosas de intensidade no mínimo moderada observadas em outros estudos foram lipodistrofia adquirida, edema angioneurótico, eritema multiforme e AVC hemorrágico, cada uma relatada em mais de 0,5% dos pacientes. Síndrome de Stevens-Johnson (rara; < 0,1%) e necrólise epidérmica tóxica (muito rara; < 0,01%) foram relatadas durante o desenvolvimento clínico com Intelence™.
Anormalidades laboratoriais
As anormalidades clínico-laboratoriais ocorridas durante o tratamento (Grau 3 ou 4), consideradas reações adversas à medicamentos, relatadas em ≥ 2% dos pacientes tratados com Intelence™ são apresentadas na tabela a seguir.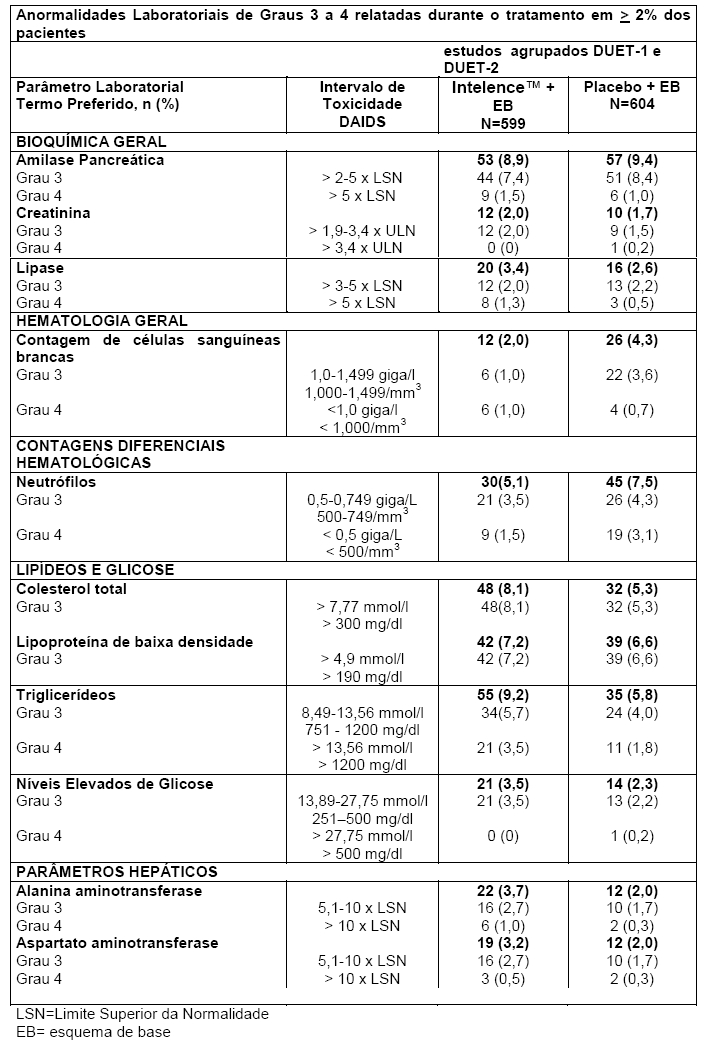
Lipodistrofia
A terapia anti-retroviral combinada é associada à redistribuição da gordura corpórea (lipodistrofia) nos pacientes infectados pelo HIV, incluindo perda da gordura