IMDELLTRA
AMGEN
tarlatamabe
Anticorpo biespecífico.
Apresentações.
IMDELLTRA (tarlatamabe) é um pó liofilizado fornecido da seguinte forma:
•embalagem de 1 mg que contém 1 frasco-ampola de IMDELLTRA 1 mg e 2 frasco-ampolas de Solução Estabilizante IV de 7 ml.
•embalagem de 10 mg que contém 1 frasco-ampola de IMDELLTRA 10 mg e 2 frascos-ampola de Solução Estabilizante IV de 7 ml.
Não utilize a solução estabilizante IV (SEIV) para reconstituição do IMDELLTRA.
VIA INTRAVENOSA
USO ADULTO
Composição.
Cada frasco-ampola de uso único contém:
Informações técnicas.
1. INDICAÇÕES
IMDELLTRA é indicado para o tratamento de pacientes adultos com câncer de pulmão de pequenas células em estágio extenso (CPPCEE) com progressão da doença durante ou após quimioterapia à base de platina.
Esse medicamento foi registrado por meio de um procedimento especial, conforme previsão da Resolução RDC n° 205, de 28 de dezembro de 2017, considerando a raridade da doença para a qual está indicado e a condição séria debilitante que representa. Dados complementares e provas adicionais ainda serão submetidos à Anvisa, após a concessão do registro do medicamento. A revisão desses novos dados pela Anvisa poderá implicar a alteração das informações descritas nesta bula, ou mesmo a alteração do status de registro do medicamento.
2. RESULTADOS DE EFICÁCIA
O IMDELLTRA é uma molécula biespecífica ativadora de células T que se liga seletivamente à DLL3 (expressa em células tumorais) e CD3 (expressa em células T). IMDELLTRA é produzido usando a tecnologia de DNA recombinante em células de ovário de hamster chinês (CHO). IMDELLTRA consiste em 982 aminoácidos e tem um peso molecular de aproximadamente 105 quilodaltons.
Classe farmacológica: molécula biespecífica ativadora de células T Código ATC: L01FX33
A estrutura de domínio do tarlatamabe é mostrada na figura abaixo.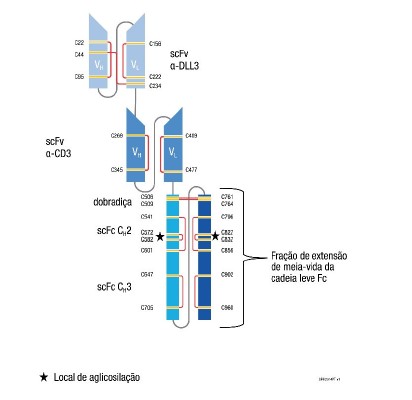
A eficácia do IMDELLTRA foi demonstrada em pacientes incluídos no estudo clínico fase 2, aberto, multicêntrico e de múltiplas coortes, o estudo DeLLphi-301. Os pacientes elegíveis deveriam ter CPPC em estágio extenso com progressão da doença após receberem quimioterapia à base de platina e pelo menos uma linha de terapia anterior, status performance ECOG de 0 a 1 e pelo menos uma lesão mensurável, conforme definido pelos Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST v1.1). Os estudos clínicos excluíram pacientes com metástases cerebrais sintomáticas, evidência de doença pulmonar intersticial ou pneumonite não infecciosa e imunodeficiência ativa
A parte 1 do estudo DeLLphi-301 foi uma comparação de dose que randomizou 176 pacientes em uma proporção 1:1 para receber 10 ou 100 mg de tarlatamabe (como uma infusão IV de 60 minutos). Em análise preliminar pré-especificada, 30 pacientes por grupo foram avaliados para determinar a dose selecionada para a parte 2. A parte 2 foi uma expansão de dose que incluiu 100 pacientes (parte 1 e 2 combinadas) na dose selecionada de 10 mg.
Um total de 99 pacientes recebeu IMDELLTRA por via intravenosa em uma dose inicial de 1 mg no Dia 1 do Ciclo 1, seguido de 10 mg nos Dias 8, 15 e a cada 2 semanas depois disso, até a progressão da doença ou toxicidade inaceitável.
Os pacientes foram elegíveis para continuar recebendo IMDELLTRA depois da progressão radiográfica, se continuassem a ter benefício clínico no parecer do investigador e não apresentassem toxicidades significativas ou inaceitáveis. As avaliações tumorais foram realizadas a cada 6 semanas pelas primeiras 48 semanas e, posteriormente, a cada 12 semanas.
As principais medidas dos resultados de eficácia foram taxa de resposta objetiva (ORR) e duração da resposta (DOR), conforme avaliadas pela Revisão Central Independente Cega (BICR), de acordo com o RECIST v1.1.
Para pacientes que receberam IMDELLTRA a 10 mg, os dados demográficos iniciais e as características da doença da população do estudo foram: idade mediana de 64 anos (intervalo: 35 a 82); 48,5% dos pacientes com 65 anos ou mais e 10% dos pacientes com 75 anos ou mais; 71,7% do sexo masculino; 57,6% brancos e 41,4% asiáticos; 26,3% ECOG PS de 0 e 73,7% ECOG PS de 1; 2% tinham doença M0 e 98% tinham doença M1; e 22,2% tinham histórico de metástases cerebrais. 100% receberam terapia prévia com platina, 20,2% receberam terapia prévia com topotecano, 69,7% receberam terapia prévia anti PD-L1; 65,7% receberam duas linhas de terapia prévia e 32,3% receberam três ou mais linhas de terapia. 8,1% nunca fumaram, 73,7% eram ex-fumantes e 18,2% eram fumantes atuais. O tempo para a progressão após terapia com platina em primeira linha foi descrito em 69/99 pacientes. O tempo para a progressão foi < 90 dias em 27/69 (39,1%) dos pacientes e ≥ 90 dias em 42/69 (60,9%) dos pacientes.
ORR e DOR foram, em geral, semelhantes à população total nos subgrupos que tiveram < 90 ou ≥ 90 dias até a progressão após terapia de primeira linha com platina.
Os resultados de eficácia estão resumidos na Tabela 1.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O IMDELLTRA é uma molécula biespecífica ativadora de células T que se liga a DLL3 expresso na superfície das células tumorais e CD3 expresso na superfície das células T. A ligação biespecífica do tarlatamabe às células T e às células tumorais DLL3 positivas desencadeia a ativação das células T, produzindo citocinas inflamatórias e liberando proteínas citotóxicas, o que resulta na lise das células tumorais.
A resposta farmacodinâmica foi caracterizada pela elevação transitória de citocinas e biomarcadores imunológicos.
Farmacodinâmica
A resposta farmacodinâmica após uma única infusão de tarlatamabe foi caracterizada pela ativação e redistribuição de células T e elevação transitória de citocinas. A redistribuição periférica de células T (ou seja, adesão de células T ao endotélio do vaso sanguíneo e/ou transmigração para o tecido) ocorreu dentro de 24 horas após dose inicial de tarlatamabe de 1 mg no Dia 1. As contagens de células T diminuíram em até 6 horas após infusão e retornaram aos níveis iniciais na maioria dos pacientes, antes da próxima infusão no Dia 8. As citocinas séricas IL-2, IL-6, IL-8, IL-10, IFN-c e TNF-a foram transitoriamente elevadas após dose inicial de tarlatamabe de 1 mg no Dia 1. Os níveis de citocinas atingiram o pico nos primeiros 2 dias após início da infusão de tarlatamabe e, em geral, retornaram aos níveis iniciais antes da próxima infusão no Dia 8. Nas infusões posteriores, a elevação de citocinas ocorreu em um número menor de pacientes e com menor intensidade em comparação à infusão inicial no Dia 1.
Propriedades farmacocinéticas
O pico de concentração sérica (Cmáx), a concentração sérica mínima (Cmínima) e a área sob a curva de concentração sérica versus tempo no estado de equilíbrio (AUCtau) do tarlatamabe aumentaram proporcionalmente no intervalo de dose avaliado de 0,003 mg a 100 mg a cada 2 semanas e 200 mg a cada 3 semanas (20 vezes a dose recomendada). O estado de equilíbrio aproximado das exposições séricas de tarlatamabe foi atingido no Dia 28.
Distribuição
O valor mediano geométrico (CV%) para o volume de distribuição no estado de equilíbrio é de 8,6 L (18,3%).
Metabolismo
A via metabólica do tarlatamabe não foi caracterizada. Como outras terapias proteicas, espera-se que o tarlatamabe seja degradado em pequenos peptídeos e aminoácidos por meio de vias catabólicas.
Eliminação
A depuração sistêmica estimada (CV% entre pacientes) foi de 0,65 L/dia (44%) e a meia-vida de eliminação terminal foi de aproximadamente 11,2 dias em pacientes com CPPC.
Populações especiais
Não foram observadas diferenças clinicamente significativas na depuração do tarlatamabe com base na idade, peso corporal, sexo, raça, comprometimento renal leve ou moderado (TFGe ≥ 30 ml/min) ou comprometimento hepático leve (bilirrubina total ≤ limite superior do normal (LSN) e AST > LSN) a comprometimento hepático moderado (bilirrubina total > 1,5 a 3 × LSN, qualquer AST).
Efeitos de tarlatamabe nos substratos de CYP450
O tarlatamabe causa a liberação transitória de citocinas que podem suprimir as enzimas CYP450 e pode resultar em um aumento na exposição de substratos de CYP concomitantes durante e até 14 dias depois da ocorrência da síndrome de liberação de citocinas
Imunogenicidade
A incidência observada de anticorpos anti-medicamentos é altamente dependente da sensibilidade e da especificidade do ensaio. As diferenças nos métodos de ensaio impedem comparações significativas da incidência de anticorpos anti-medicamentos nos estudos descritos abaixo com a incidência de anticorpos anti-tarlatamabe em outros estudos, incluindo os do IMDELLTRA ou de outros produtos acopladores de células T a DLL3. No Estudo DeLLphi-300 e no Estudo DeLLphi-301, a incidência de desenvolvimento de anticorpos anti-tarlatamabe foi de 4,9% (7/144) em pacientes que receberam a dose de 10 mg. No Estudo DeLLphi-301 de fase 2, que empregou o ensaio de neutralização, nenhum dos pacientes desenvolveu anticorpos neutralizantes. O status positivo de anticorpo anti-tarlatamabe foi associado a uma diminuição na exposição ao IMDELLTRA, porém não teve impacto clinicamente relevante sobre a eficácia, na dose recomendada. Não foi observada associação entre o desenvolvimento de anticorpo antitarlatamabe e reações adversas.
4. CONTRAINDICAÇÕES
IMDELLTRA é contraindicado em pacientes com hipersensibilidade conhecida ao tarlatamabe ou a qualquer componente da formulação do produto.
5. ADVERTÊNCIAS E PRECAUÇÕES
Síndrome da liberação de citocinas (SLC)
A administração de IMDELLTRA foi associada à SLC, que pode ser grave ou de risco à vida. Em estudos clínicos com dados de segurança agrupados de 160 pacientes com CPPC nos estudos DeLLphi-300 e DeLLphi-301, que receberam a dose de IMDELLTRA a 10 mg, a SLC ocorreu em 53,8% dos pacientes, com eventos de Grau 1 em 32,5%, Grau 2 em 20,0% dos pacientes, Grau 3 em 0,6% dos pacientes e eventos de Grau 4 em 0,6% dos pacientes. Nenhum paciente apresentou eventos Grau 5 de SLC.
Eventos graves de SLC foram reportados em 23,1% dos pacientes. Depois da primeira dose de IMDELLTRA, 41,3% dos pacientes apresentaram algum grau de SLC, com 28,8% dos pacientes apresentando algum grau de SLC depois da segunda dose. A maioria dos eventos de SLC ocorreu após as duas primeiras doses, com 8,8% dos pacientes apresentando SLC depois da terceira dose em diante. Depois da infusão do Dia 1, 15,6% dos pacientes apresentaram SLC ≥ Grau 2. Depois da infusão do Dia 8, 4,4% dos pacientes apresentaram SLC ≥ Grau 2. O tempo mediano da primeira dose de IMDELLTRA até os primeiros sintomas da SLC foi de 2 dias (intervalo: 1 a 25 dias).
Em pacientes tratados com IMDELLTRA a 10 mg incluídos no Estudo DeLLphi-301 (n=133), a SLC ocorreu em 52,6% dos pacientes, incluindo Grau 1 em 31,6%, Grau 2 em 20,3% e Grau 3 em 0,8% dos pacientes. Nenhum paciente apresentou eventos de Grau 4 ou Grau 5. A maioria dos pacientes apresentou SLC depois das primeiras duas doses de IMDELLTRA, com 9,8% apresentando SLC a partir ou depois da terceira dose. Depois da infusão do Dia 1, 16,5% dos pacientes apresentaram SLC ≥ Grau 2. Depois da infusão do Dia 8, 3,0% dos pacientes apresentaram SLC ≥ Grau 2. O tempo mediano para o início do primeiro sintoma de SLC foi de 15,5 horas. Para os eventos de Grau 1 que progrediram para maior ou igual a Grau 2, o tempo mediano entre o evento de Grau 1 e os eventos de Grau 2 foi de 22,1 horas.
A SLC pode estar associada aos sintomas que incluem pirexia, hipotensão, hipóxia, taquicardia, cefaleia, calafrios, náusea e vômitos. A maioria desses eventos não levaram à descontinuação do IMDELLTRA nos estudos clínicos. Administre IMDELLTRA em uma clínica equipada para monitorar e gerenciar a SLC. Certifique-se de que os pacientes estejam euvolêmicos antes de iniciar as infusões. Monitore atentamente os pacientes quanto aos sinais e sintomas de SLC durante o início do tratamento com IMDELLTRA. Inicie a terapia de acordo com o cronograma de dosagem de IMDELLTRA para reduzir o risco de SLC. Monitore os pacientes quanto à SLC e suspenda ou descontinue permanentemente o IMDELLTRA com base na gravidade.
Síndrome de neurotoxicidade associada a células efetoras imunes (ICANS)
A administração de IMDELLTRA foi associada à ICANS, que pode ser grave ou de risco à vida. A ICANS pode ocorrer até várias semanas após administração do IMDELLTRA.
Em estudos clínicos com dados de segurança agrupados de 160 pacientes com CPPC incluídos nos estudos DeLLphi-300 e DeLLphi-301, que receberam IMDELLTRA a 10 mg, a ICANS foi relatada em 9,4% dos pacientes. Os eventos adversos que podem ser associados à ICANS incluem cefaleia, encefalopatia, confusão, delírio, convulsão, ataxia, neurotoxicidade e tremor. O tempo mediano da primeira dose de IMDELLTRA até o início da ICANS foi de 30 dias (intervalo: 1 a 154 dias). O tempo mediano até a resolução da ICANS foi de 33 dias (intervalo de 1 a 93 dias).
Os pacientes devem ser monitorados atentamente quanto a sinais e sintomas de ICANS durante o tratamento com IMDELLTRA. Ao primeiro sinal de ICANS, avalie imediatamente o paciente e forneça terapia de suporte com base na gravidade. Suspenda ou descontinue permanentemente o IMDELLTRA com base na gravidade, de acordo com as recomendações, e considere o tratamento adicional de acordo com as diretrizes práticas atuais. Aconselhe os pacientes a não dirigir e não se envolver em ocupações ou atividades perigosas, como operar máquinas pesadas ou possivelmente perigosas, no caso de qualquer sintoma neurológico, até que eles desapareçam.
Neutropenia
A administração de IMDELLTRA foi associada à neutropenia. Em estudos clínicos com dados de segurança agrupados para 160 pacientes com CPPC incluídos nos estudos DeLLphi-300 e DeLLphi-301, que receberam IMDELLTRA a 10 mg, a neutropenia ocorreu em 14,4% dos pacientes, incluindo eventos igual ou maior que Grau 3 em 6,3% dos pacientes e eventos de Grau 4 em 2,5% dos pacientes. O tempo mediano da primeira dose de IMDELLTRA até o primeiro sintoma da neutropenia foi de 43 dias (intervalo: 3 a 244 dias). A neutropenia que levou à interrupção da dose ocorreu em 0,6% dos pacientes e nenhuma levou à descontinuação do tratamento. Os eventos foram, em sua maioria, não graves e foram resolvidos espontaneamente ou com o uso de G-CSF. Os pacientes devem ser monitorados atentamente quanto aos sinais e sintomas de neutropenia durante o tratamento com o IMDELLTRA.
Infecções
O IMDELLTRA pode causar infeções sérias, incluindo infecções de risco à vida e fatais. Na população de segurança agrupada, [vide 9. REAÇÕES ADVERSAS], infecções, incluindo infecções oportunistas, ocorreram em 41% dos pacientes que receberam IMDELLTRA. Ocorreram infecções de Grau 3 ou Grau 4 em 13% dos pacientes. As infecções mais frequentes foram COVID-19 (9%, a maioria durante a pandemia de COVID-19), infecção do trato urinário (10%), pneumonia (9%), infecção do trato respiratório (3,2%) e infecção por candida (3,2%). Monitore os pacientes quanto a sinais e sintomas de infecção antes e durante o tratamento com IMDELLTRA e administre conforme clinicamente indicado. Suspenda ou descontinue permanentemente o IMDELLTRA com base na gravidade [vide 8. POSOLOGIA E MODO DE USO].
Hepatotoxicidade
IMDELLTRA pode causar hepatotoxicidade.Na população de segurança agrupada [vide 9. REAÇÕES ADVERSAS], ocorreu a elevação de ALT em 42%, ocorrendo a elevação de ALT com Grau 3 ou Grau 4 em 2,1% dos pacientes tratados com IMDELLTRA. A elevação de AST ocorreu em 44% dos pacientes, ocorrendo a elevação de AST com Grau 3 ou Grau 4 em 3,2%. A elevação de bilirrubina ocorreu em 15% dos pacientes, com a elevação de bilirrubina com Grau 3 ou Grau 4 em 1,6% dos pacientes [vide 9. REAÇÕES ADVERSAS]. A elevação das enzimas hepáticas pode ocorrer com ou sem SLC concomitante. Monitore as enzimas hepáticas e a bilirrubina antes do tratamento com IMDELLTRA, antes de cada dose e conforme clinicamente indicado. Suspenda ou descontinue permanentemente o IMDELLTRA com base na gravidade [vide 8. POSOLOGIA E MODO DE USO].
Este medicamento pode causar hepatotoxicidade.
Por isso, requer uso cuidadoso, sob vigilância médica estrita e acompanhado por controles periódicos da função hepáticaantes de cada dose e conforme clinicamente indicado
Hipersensibilidade
IMDELLTRA pode causar reações graves raras de hipersensibilidade. Os sinais e sintomas clínicos de hipersensibilidade podem incluir, dentre outros, erupção cutânea e broncoespasmo. Monitore os pacientes quanto a sinais e sintomas de hipersensibilidade durante o tratamento com IMDELLTRA e controle conforme clinicamente indicado. Suspenda ou considere a descontinuação permanente de IMDELLTRA com base na gravidade [vide 8. POSOLOGIA E MODO DE USO].
Carcinogenicidade
Nenhum estudo de carcinogenicidade ou genotoxicidade foi realizado com o tarlatamabe.
Comprometimento da fertilidade
Não foram realizados estudos para avaliar os efeitos do tarlatamabe na fertilidade.
Populações especiais
Gravidez
Com base em seu mecanismo de ação, o IMDELLTRA pode causar dano fetal quando administrado em mulheres grávidas. Não há dados disponíveis sobre o uso de IMDELLTRA em mulheres grávidas.
Toxicidade para a reprodução e desenvolvimento
Não há dados disponíveis sobre o uso de tarlatamabe em mulheres grávidas. Com base em seu mecanismo de ação, o IMDELLTRA pode causar dano fetal quando administrado em mulheres grávidas. Em um estudo do desenvolvimento embrionário e fetal murino, foi verificado que o substituto murino do tarlatamabe é capaz de atravessar a barreira placentária. No entanto, não houve efeitos do substituto murino do tarlatamabe, designado muS757, observado nos resultados da gravidez ou nos parâmetros fetais em nenhum parâmetro materno, incluindo pesos corporais maternos médios ou ganhos de peso corporal. Sabe-se que as proteínas compostas de domínios de fragmentos cristalizáveis (Fc) derivados da imunoglobulina G (IgG) atravessam a barreira placentária e, como o IMDELLTRA contém um domínio Fc derivado de IgG1, ele tem o potencial de ser transferido da mãe para o feto em desenvolvimento. Além disso, não houve achados macroscópicos relacionados ao muS757 ou efeitos sobre qualquer parâmetro ovariano, uterino ou da ninhada em qualquer nível de dose, e a administração do muS757 não produziu nenhuma malformação ou variação externa, visceral ou esquelética no feto.
Categoria B para gravidez - Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
Não há dados sobre a presença de tarlatamabe no leite humano, sobre o efeito na criança amamentada ou sobre os efeitos na produção de leite. Sabe-se que a IgG materna está presente no leite humano. Os efeitos da exposição gastrointestinal local e da exposição sistêmica limitada da criança amamentada ao IMDELLTRA são desconhecidos. Devido ao potencial de reações adversas graves em uma criança amamentada, aconselhe as pacientes a não amamentar durante o tratamento com IMDELLTRA e por 2 meses após a última dose.
Uso contraindicado na amamentação ou na doação de leite humano:
Este medicamento é contraindicado durante o aleitamento ou doação de leite, pois é excretado no leite humano e pode causar reações indesejáveis no bebê. Seu médico ou cirurgião-dentista deve apresentar alternativas para o seu tratamento ou para a alimentação do bebê.
Mulheres e homens com potencial reprodutivo
O IMDELLTRA pode causar danos ao feto quando administrado a uma mulher grávida.
Teste de gravidez
Verifique o status da gravidez em mulheres com potencial reprodutivo, antes de iniciar o IMDELLTRA.
Contracepção
Mulheres
Aconselhe as mulheres com potencial reprodutivo a usar métodos contraceptivos eficazes durante o tratamento com IMDELLTRA e por 2 meses após a última dose do IMDELLTRA.
Fertilidade
Não há estudos clínicos para avaliar o efeito do IMDELLTRA na fertilidade.
Pediatria
A segurança e a eficácia do IMDELLTRA em pacientes pediátricos não foi estabelecida.
Geriatria
Dos 160 pacientes com CPPC que receberam IMDELLTRA a 10 mg como monoterapia, 52,5% tinham 65 anos ou mais e 12,5% tinham 75 anos ou mais. Em estudos clínicos, não foram observadas diferenças gerais na farmacocinética, na segurança ou na eficácia do IMDELLTRA entre pacientes geriátricos (≥ 65 anos de idade) e pacientes mais jovens. Não é necessário o ajuste de dose para pacientes geriátricos.
Comprometimento hepático
Com base nos resultados farmacocinéticos da população, não é necessário o ajuste de dose em pacientes com comprometimento hepático leve ou moderado. O IMDELLTRA não foi estudado em pacientes com comprometimento hepático grave.
Comprometimento renal
Com base nos resultados farmacocinéticos da população, não é necessário o ajuste da dose em pacientes com comprometimento renal leve ou moderado. O IMDELLTRA não foi estudado em pacientes com comprometimento renal grave.
Efeitos sobre a capacidade de dirigir e operar máquinas
Não foram realizados estudos dos efeitos do IMDELLTRA sobre a capacidade de dirigir e operar máquinas. No entanto, devido a possíveis eventos neurológicos associados a ICANS, depois da infusão do IMDELLTRA, aconselhe os pacientes a não dirigir e não se envolver em ocupações ou atividades perigosas, como operar máquinas pesadas ou possivelmente perigosas, no caso de qualquer sintoma neurológico, até que eles desapareçam.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos formais de interação medicamentosa com IMDELLTRA. O início do tratamento com IMDELLTRA causa liberação transitória de citocinas que podem suprimir as enzimas CYP450 e pode resultar em exposições elevadas dos substratos de CYP concomitantes. Em pacientes que estejam recebendo concomitantemente substratos de CYP450, particularmente aqueles com um índice terapêutico estreito, monitore eventos adversos conhecidos. Ajuste a dose do medicamento concomitante conforme necessário.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazene os frasco-ampolas de IMDELLTRA e da Solução Estabilizante IV na embalagem original em geladeira (2 °C a 8 °C) e protegidos da luz até o momento do uso. Não congelar.
Prazo de validade: 36 meses.
As informações na Tabela 3 indicam o tempo de armazenamento da bolsa de infusão preparada com IMDELLTRA.
IMDELLTRA para injeção é fornecido como um pó liofilizado branco a levemente amarelado, estéril, de dose única e sem conservantes, com uma dose administrável de 1 ou 10 mg; deve ser diluído em uma bolsa intravenosa (IV) com a Solução Estabilizante IV (SEIV) e soro fisiológico a 0,9%.
Número de lote, data de fabricação e prazo de validade: consulte embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USO
Informações importantes sobre a dosagem
- Administre IMDELLTRA de acordo com o esquema de dosagem gradual da Tabela 3 para reduzir a incidência e a gravidade da síndrome de liberação de citocinas (SLC).
-Para o Ciclo 1, administre os medicamentos concomitantes recomendados na Tabela 3 antes e depois das infusões de IMDELLTRA do Ciclo 1 para reduzir o risco de reações de SLC.
-O IMDELLTRA só deve ser administrado por um profissional de saúde qualificado com suporte médico adequado para gerenciar reações graves, como a SLC e a toxicidade neurológica, incluindo a síndrome de neurotoxicidade associada a células efetoras imunológicas (ICANS).
-Devido ao risco de SLC e toxicidade neurológica, incluindo ICANS, monitore os pacientes desde o início da infusão de IMDELLTRA por 22 a 24 horas no Dia 1 do Ciclo 1 e no Dia 8 do Ciclo 1 em um ambiente de saúde apropriado.
-Recomende que os pacientes fiquem a no máximo 1 hora de um centro de saúde apropriado por um total de 48 horas a partir do início da infusão com IMDELLTRA após as doses do Dia 1 do Ciclo 1 e do Dia 8 do Ciclo 1, acompanhados por um cuidador.
-Antes da administração de IMDELLTRA, avalie o hemograma completo, as enzimas hepáticas e a bilirrubina antes de cada dose e conforme clinicamente indicado.
- Certifique-se de que o paciente esteja bem hidratado antes da administração de IMDELLTRA.
Posologia
- Administre IMDELLTRA como uma infusão intravenosa durante uma hora.
-O esquema de administração de dose gradual recomendado para IMDELLTRA é fornecido na Tabela 3. Administre seguindo a dosagem gradual para reduzir a incidência e a gravidade da SLC
- Após o esquema de dosagem gradual, administre IMDELLTRA quinzenalmente (a cada 2 semanas) até a progressão da doença ou toxicidade inaceitável.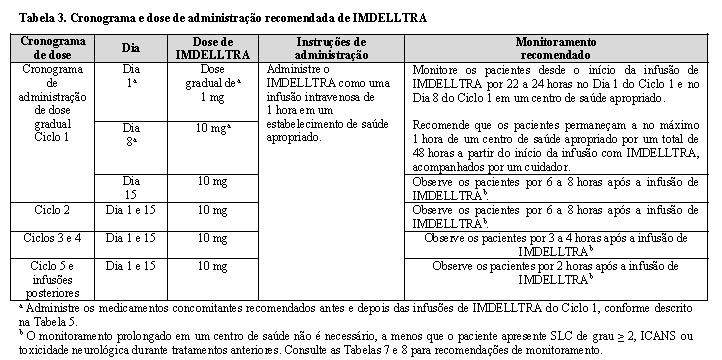
Observação: vide a Tabela 6 para obter recomendações sobre o reinício do IMDELLTRA depois de atrasos na dose.
Administração
-O cateter intravenoso (IV) para administração de medicamentos concomitantes pode ser usado para administrar a infusão de IMDELLTRA.
-Para garantir a permeabilidade, lave o cateter IV durante 3 a 5 minutos usando cloreto de sódio a 0,9% para injeção.
-Administre o IMDELLTRA reconstituído e diluído como uma infusão intravenosa em uma taxa de fluxo constante usando uma bomba de infusão. A bomba deve ser programável, bloqueável, não elastomérica e ter um alarme.
-A Tabela 4 fornece a duração e a taxa de infusão.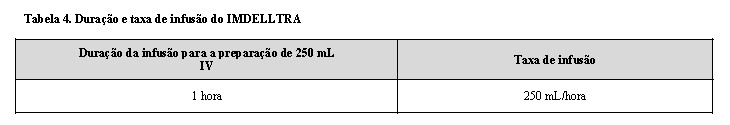
Medicamentos concomitantes recomendados para a administração de IMDELLTRA no Ciclo 1
Administre os medicamentos concomitantes recomendados para a administração de IMDELLTRA durante o Ciclo 1, conforme apresentado na Tabela 5, para reduzir o risco da síndrome de liberação de citocinas.
Reinício de IMDELLTRA após atraso da administração da dose
Se uma dose de IMDELLTRA for atrasada, reinicie a terapia com base na recomendação listada na Tabela 6 e retome o esquema de administração da dose de acordo. Administre os medicamentos concomitantes recomendados, conforme indicado na Tabela 5.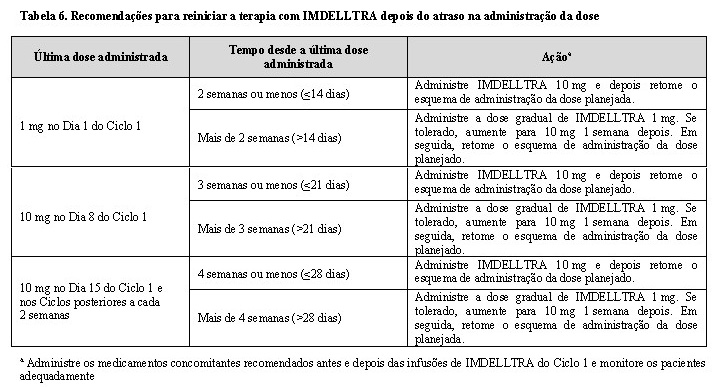
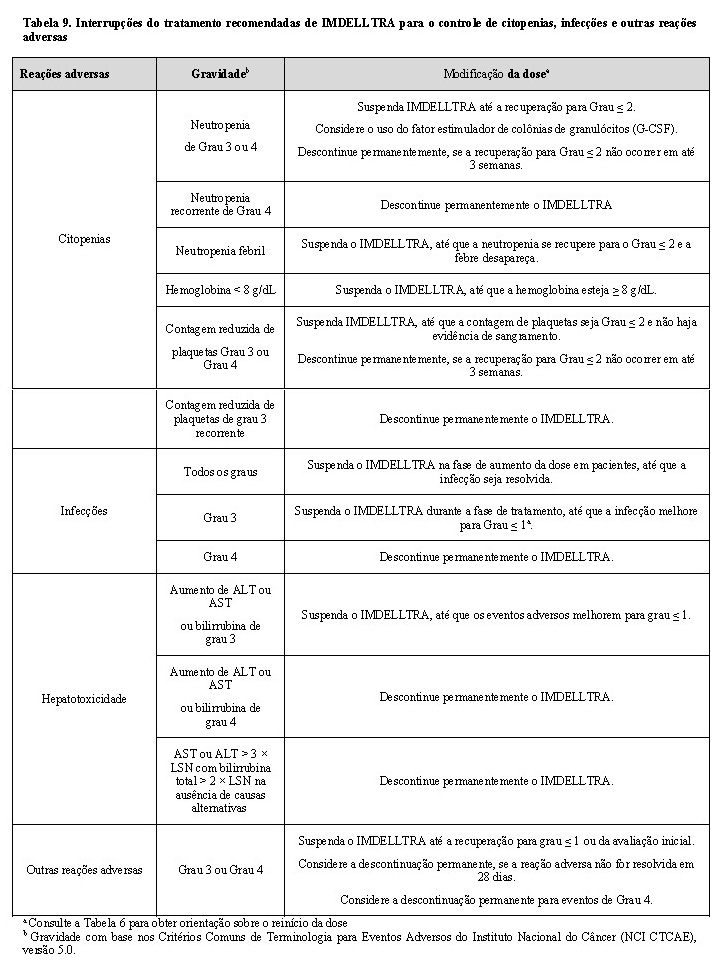
Modificações de dose recomendadas e controle das reações adversas
Não é recomendada a redução da dose administrada de IMDELLTRA. Consulte a Tabela 7 e a Tabela 8 para ver as ações recomendadas para o tratamento da SLC, toxicidade neurológica, incluindo ICANS, respectivamente, e a Tabela 9 para citopenias, infecções e outras reações adversas.
Síndrome da liberação de citocinas (SLC)
Realize o diagnóstico da SLC com base na apresentação clínica. Avalie e trate outras causas de febre, hipóxia e hipotensão.
Se houver suspeita de SLC, faça o tratamento de acordo com as recomendações da Tabela 7. Monitore os pacientes com SLC de grau 2 ou superior (por ex., hipotensão que não responde a fluidos ou hipóxia que requer oxigênio complementar) com telemetria cardíaca contínua e oximetria de pulso.
Em caso de SLC grave ou de risco à vida, recomende administrar tocilizumabe ou terapia equivalente e monitoramento intenso (por ex. UTI) para terapia de suporte. Realize exames laboratoriais para monitorar a coagulação intravascular disseminada (CID), os parâmetros hematológicos, bem como a função pulmonar, cardíaca, renal e hepática.
A Tabela 7 fornece as diretrizes para a classificação e modificação da dosagem e tratamento da síndrome de liberação de citocinas.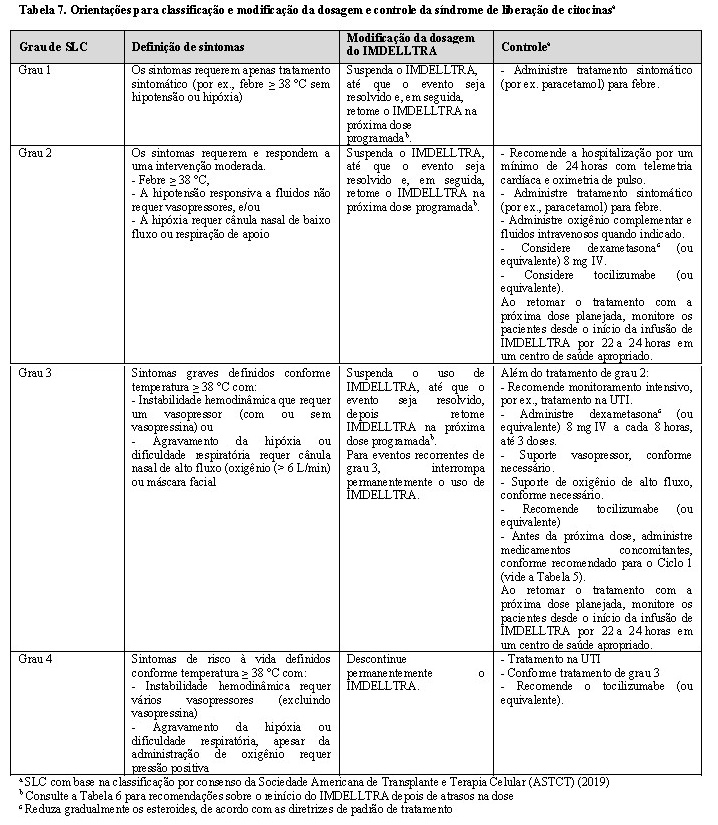
Toxicidade neurológica incluindo ICANS
Ao primeiro sinal de toxicidade neurológica, incluindo ICANS, suspenda o IMDELLTRA e considere uma avaliação neurológica. Exclua outras causas de sintomas neurológicos. Forneça terapia de suporte, que pode incluir cuidados intensivos, para toxicidades neurológicas graves ou com risco de vida, inclusive ICANS. Trate a ICANS e a toxicidade neurológica de acordo com as recomendações da Tabela 8 e considere o tratamento adicional, de acordo com as diretrizes práticas atuais.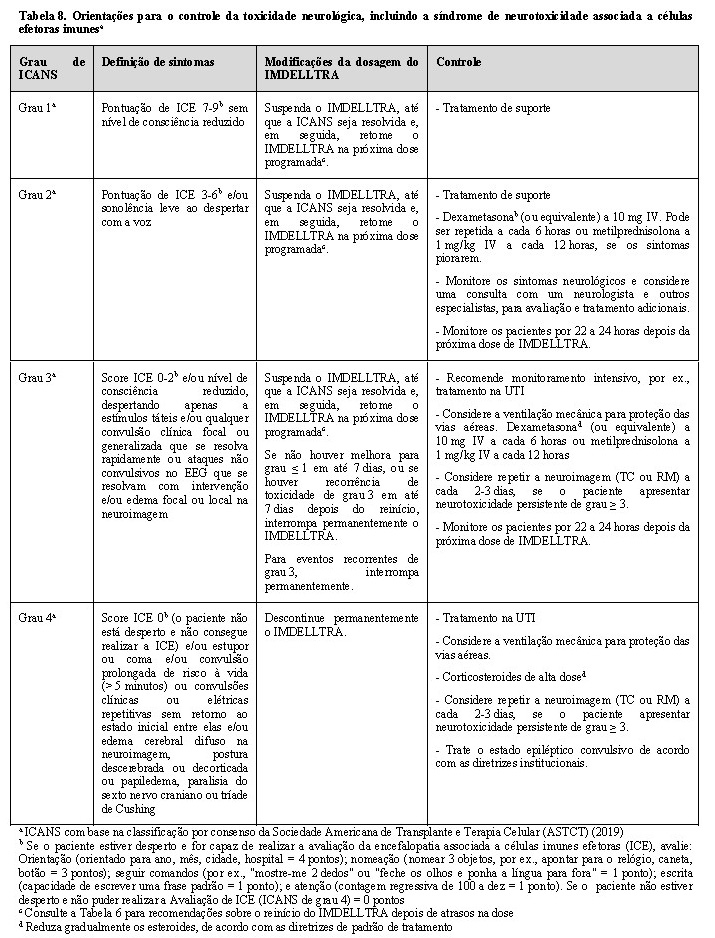
Preparação
Informações sobre compatibilidade de materiais
-As bolsas IV compostas de acetato de etil vinila (EVA), poliolefina e cloreto de polivinila (PVC) demonstraram ser compatíveis com IMDELLTRA, nas condições de administração especificadas.
-Materiais de cateteres e de linhas intravenosas compostos de poliolefina, PVC e poliuretano demonstraram ser compatíveis com IMDELLTRA nas condições de administração especificadas.
-O uso do dispositivo de transferência de sistema fechado (CSTD) não é recomendado devido ao risco potencial de erro de medicação. A Amgen não realizou testes de compatibilidade de CSTDs adaptadores de frasco-ampola com IMDELLTRA.
Etapa 1: Reconstituir IMDELLTRA com água estéril para injeção
-A Tabela 10 fornece a quantidade necessária de água estéril para injeção para reconstituir os frascos-ampola de IMDELLTRA 1 mg e 10 mg.
Não use o Estabilizador de solução IV (SEIV) para reconstituir IMDELLTRA.
O estabilizador de solução IV (SEIV) é usado para revestir a bolsa intravenosa antes da adição de IMDELLTRA reconstituído, para evitar a adsorção de IMDELLTRA às bolsas IV e aos tubos IV.
-Usando uma agulha e uma seringa preenchidas com a quantidade necessária de água estéril, injete a água estéril no frasco-ampola de vidro. Evite injetar a água diretamente sobre o pó, para evitar a formação de espuma.
-Misture suavemente o conteúdo para misturar. Não agite.
-Inspecione os medicamentos parenterais quanto à presença de partículas e descoloração, antes da administração. Verifique se a solução está límpida a opalescente, incolor a levemente amarelada. Não use se a solução estiver turva ou com partículas.
-Dilua ainda mais o IMDELLTRA reconstituído.
-O IMDELLTRA reconstituído deve ser diluído em até 4 horas após a reconstituição ou descartado. Prepare a bolsa de infusão: Etapas 2 a 5
Etapa 2: Retirada de cloreto de sódio a 0,9% para injeção
- Usando uma bolsa pré-preenchida de 250 mL de cloreto de sódio a 0,9% para injeção, retire a quantidade de cloreto de sódio especificada na Tabela 11 e descarte.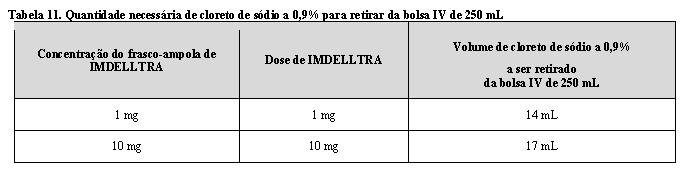
Etapa 3: Adicione a solução estabilizante IV à bolsa de infusão
-Injete 13 mL de solução estabilizante IV (SEIV) na bolsa de infusão de 250 mL de cloreto de sódio a 0,9%, consulte a Tabela 12.
-Misture suavemente o conteúdo da bolsa de infusão para evitar a formação de espuma. Não agite.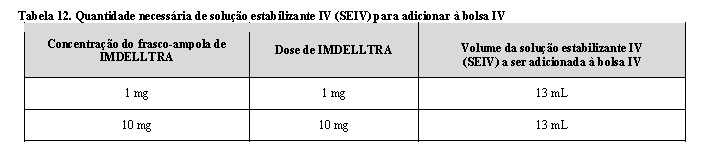
Etapa 4: Dilua o IMDELLTRA reconstituído na bolsa de infusão
- Transfira o volume necessário de IMDELLTRA reconstituído listado na Tabela 13 para a bolsa de infusão (contendo o estabilizador de solução IV).
OBSERVAÇÃO: as concentrações finais para os frascos-ampola de diferentes concentrações NÃO são as mesmas após a reconstituição e a diluição adicional.
- Misture suavemente os conteúdos da bolsa. Não agite.
Etapa 5: Remoção de ar da bolsa IV
Remova o ar da bolsa IV preparada usando uma seringa vazia para evitar a formação de espuma.
Etapa 6: Preparação do tubo IV
-Prepare o tubo IV com cloreto de sódio a 0,9% para injeção ou com o produto final preparado.
-Consulte a Tabela 14 para saber o tempo máximo de armazenamento da infusão preparada com IMDELLTRA.
Requisitos de armazenamento da bolsa de infusão preparada com IMDELLTRA
-Administre IMDELLTRA reconstituído e diluído imediatamente.
-A Tabela 14 exibe o tempo máximo de armazenamento para a bolsa de infusão preparada com IMDELLTRA.
-O tempo máximo de armazenamento inclui a duração total desde o momento da reconstituição do frasco-ampola de IMDELLTRA até o final da infusão.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
A segurança do IMDELLTRA foi avaliada em 160 pacientes com câncer de pulmão de pequenas células (CPPC) que receberam 10 mg como monoterapia. A duração média da exposição ao IMDELLTRA foi de 14,14 semanas (intervalo: 0,1 a 93,4).
As reações adversas mais comuns foram síndrome de liberação de citocinas (53,5%), pirexia (36,9%), disgeusia (30%), diminuição de apetite (29,4%), constipação (27,5%), fadiga (26,9%), anemia (25%) e astenia (21,9%).
Lista tabulada de reações adversas
Reações adversas relatadas em estudos clínicos do IMDELLTRA são apresentadas na Tabela 15 abaixo. A frequência é fornecida por categoria MedDRA: muito comum (≥ 1/10), comum (≥ 1/100 a < 1/10), incomum (≥ 1/1.000 a < 1/100), rara (≥ 1/10.000 e < 1/1.000), muito rara ( < 10.000). Em cada classe de sistema de órgãos, as reações adversas são apresentadas em ordem decrescente de gravidade.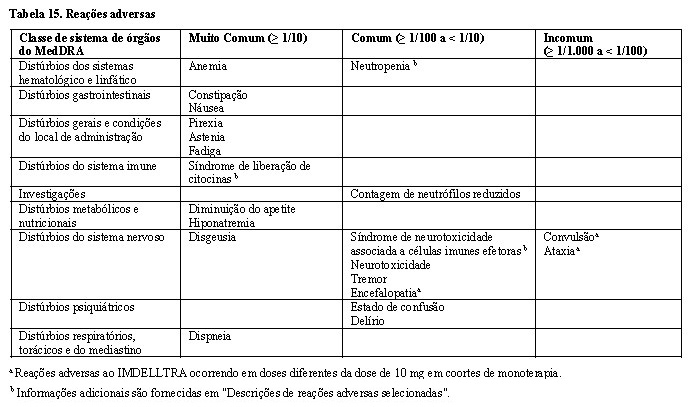
Descrição de reações adversas selecionadas
Síndrome da liberação de citocinas (SLC)
Em estudos clínicos com dados de segurança agrupados de 160 pacientes com CPPC nos estudos DeLLphi-300 e DeLLphi-301, que receberam a dose de IMDELLTRA a 10 mg, a SLC ocorreu em 53,8% dos pacientes com eventos de grau 1 em 32,5%, grau 2 em 20% dos pacientes, grau 3 em 0,6% dos pacientes e grau 4 em 0,6% dos pacientes. Nenhum paciente apresentou eventos grau 5. Foram relatados eventos graves de SLC em 23,1% dos pacientes. Depois da primeira dose de IMDELLTRA, 41,3% dos pacientes apresentaram algum grau de SLC, com 28,8% dos pacientes apresentando algum grau de SLC depois da segunda dose. A maioria dos eventos de SLC ocorreu após as primeiras doses, com 8,8% dos pacientes apresentando SLC depois da terceira dose ou mais tarde. Depois da infusão do Dia 1, 15,6% dos pacientes apresentaram SLC ≥ grau 2. Depois da infusão do Dia 8, 4,4% dos pacientes apresentaram SLC ≥ grau 2. O tempo mediano da primeira dose de IMDELLTRA até os primeiros sintomas da SLC foi de 2 dias (intervalo: 1 a 25 dias)
Em pacientes tratados com IMDELLTRA a 10 mg incluídos no Estudo DeLLphi-301 (n=133), a SLC ocorreu em 52,6% dos pacientes, incluindo grau 1 em 31,6%, grau 2 em 20,3% e grau 3 em 0,8% dos pacientes. Nenhum paciente apresentou eventos de graus 4 ou 5. A maioria dos pacientes apresentou SLC depois das primeiras duas doses de IMDELLTRA com 9,8% apresentando SLC depois da terceira dose ou mais tarde. Depois da infusão do Dia 1, 16,5% dos pacientes apresentaram SLC ≥ grau 2. Depois da infusão do Dia 8, 3,0% dos pacientes apresentaram SLC ≥ grau 2 Para os eventos de grau 1 que progrediram para grau 2 ou mais, o tempo mediano entre o evento de grau 1 e os eventos de grau 2 foi de 22,1 horas.
Síndrome de neurotoxicidade associada a células imunes efetoras (ICANS)
Em estudos clínicos com dados de segurança agrupados de 160 pacientes com CPPC incluídos nos estudos DeLLphi-300 e DeLLphi-301, que receberam IMDELLTRA a 10 mg, a ICANS foi relatada em 9,4% dos pacientes. O tempo mediano da última dose de IMDELLTRA até o primeiro início da ICANS foi de 30 dias (intervalo: 1 a 154 dias). O tempo mediano até a resolução de ICANS foi de 33 dias (intervalo: 1 a 93 dias).
Neutropenia
Em estudos clínicos com dados de segurança agrupados para 160 pacientes com CPPC incluídos nos estudos DeLLphi-300 e DeLLphi301, que receberam IMDELLTRA a 10 mg, a neutropenia ocorreu em 14,4% dos pacientes incluindo eventos de grau 3 ou maior em 6,3% dos pacientes e eventos de grau 4 em 2,5% dos pacientes. O tempo mediano da primeira dose de IMDELLTRA até o primeiro sintoma da neutropenia foi de 43 dias (intervalo: 3 a 244 dias). A neutropenia que levou à interrupção da dose ocorreu em 0,6% dos pacientes e nenhuma levou à descontinuação do tratamento.
Hipersensibilidade
Foram relatadas reações de hipersensibilidade em pacientes tratados com IMDELLTRA, incluindo eventos graves raros. Os sinais e sintomas clínicos de hipersensibilidade podem incluir, dentre outros, erupção cutânea e broncoespasmo. Monitore os pacientes quanto a sinais e sintomas de hipersensibilidade durante o tratamento com IMDELLTRA e administre conforme indicado clinicamente. Suspenda ou considere a descontinuação permanente de IMDELLTRA, com base na gravidade.
Interações com outros medicamentos e outras formas de interação
Não foram realizados estudos formais de interação medicamentosa com IMDELLTRA. O início do tratamento com IMDELLTRA causa liberação transitória de citocinas que podem suprimir as enzimas CYP450 e podem resultar em exposições aumentadas dos substratos de CYP concomitantes. Em pacientes que estejam recebendo concomitantemente substratos de CYP450, particularmente aqueles com um índice terapêutico estreito, monitore os eventos adversos conhecidos. Ajuste a dose do medicamento concomitante, conforme necessário.
Imunogenicidade
A incidência observada de anticorpos anti-medicamentos é altamente dependente da sensibilidade e da especificidade do ensaio. As diferenças nos métodos de ensaio impedem comparações significativas da incidência de anticorpos anti-medicamento nos estudos descritos abaixo com a incidência de anticorpos anti-tarlatamabe em outros estudos, incluindo as de tarlatamabe ou de outros produtos ativadores de células T ligantes de DLL3.
No Estudo DeLLphi-300 e no Estudo DeLLphi-301, a incidência de desenvolvimento de anticorpos anti-tarlatamabe foi de 4,7% (7/149) em pacientes que receberam a dose de 10 mg. No Estudo DeLLphi-301 de fase 2, que empregou o ensaio de neutralização, nenhum dos pacientes desenvolveu anticorpos neutralizantes. O status positivo de ligação do anticorpo anti-tarlatamabe não teve impacto clinicamente relevante sobre a eficácia e a segurança.
Notificação de suspeitas de reações adversas
É importante notificar suspeitas de reações adversas após a autorização do medicamento. Ela permite o monitoramento contínuo da relação risco/benefício do medicamento. Solicita-se aos profissionais de saúde que relatem qualquer suspeita de reação adversa.
Em caso de eventos adversos, notifique pelo sistema VigiMed, disponível no Portal da Anvisa. Informe também a empresa por meio de seu serviço de atendimento ao cliente.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Não há experiência clínica com superdosagem do IMDELLTRA. Doses de até 100 mg a cada duas semanas e 200 mg a cada três semanas foram administradas em estudos clínicos. Em caso de superdosagem, o paciente deve ser tratado sintomaticamente e medidas de suporte devem ser instituídas, conforme necessário.
Em caso de intoxicação, ligue para 0800 722 6001, se precisar de mais orientações.
Dizeres legais.
VENDA SOB PRESCRIÇÃOUSO RESTRITO A ESTABELECIMENTOS DE SAÚDE
Registro: 1.0244.0023