HYMPAVZI
PFIZER
marstacimabe
Anticorpo monoclonal. Tratamento da hemofilia.
Apresentações.
HympavziTM 150 mg/mL, solução injetável, em embalagens contendo 1 caneta preenchida.
VIA DE ADMINISTRAÇÃO:
VIA SUBCUTÂNEA
USO ADULTO E PEDIÁTRICO ACIMA DE 12 ANOS DE IDADE
Composição.
Cada caneta preenchida de HympavziTM contém 150 mg de marstacimabe em 1,0 mL de solução.
Excipientes: sacarose, histidina, edetato dissódico di-hidratado, polissorbato 80, cloridrato de histidina monoidratado e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
HympavziTM (marstacimabe) é indicado para:
-Profilaxia de rotina para prevenir ou reduzir a frequência de episódios de sangramento em pacientes com 12
anos de idade ou mais (acima de 35kg de peso corporal) com hemofilia A severa (deficiência congênita de
FVIII, FVIII < 1%) sem inibidores de FVIII, e
-Profilaxia de rotina para prevenir ou reduzir a frequência de episódios de sangramento em pacientes com 12
anos de idade ou mais (acima de 35kg de peso corporal) com hemofilia B severa (deficiência congênita de
FIX, FIX < 1%) sem inibidores de FIX.
2. RESULTADOS DE EFICÁCIA
Estudos clínicos em pacientes adultos e adolescentes com hemofilia A sem inibidores de fator VIII (FVIII) ou hemofilia B sem inibidores de fator IX (FIX)
Pacientes (com idade ≥12 anos e ≥35 kg) com hemofilia A sem inibidores e hemofilia B sem inibidores (estudo B7841005/BASIS)
O estudo pivotal de Fase 3 (BASIS) foi um estudo multicêntrico, aberto, cruzado, de via única, em 116 homens adultos e adolescentes (com 12 anos ou mais e ≥35 kg) com hemofilia A grave sem inibidores de FVIII ou hemofilia B grave sem inibidores de FIX que anteriormente receberam tratamento sob demanda (N = 33) ou profilático (N = 83) com FVIII ou FIX. Pacientes com tratamento prévio ou atual ou histórico de doença arterial coronariana, trombose venosa ou arterial ou doença isquêmica foram excluídos do estudo.
A população do estudo foi caracterizada por um fenótipo de sangramento grave. A média das taxas anualizadas de sangramento (TASs) para sangramentos tratados foi de 39,86 e 7,90 na Fase Observacional para as coortes sob demanda e profilaxia, respectivamente, antes de mudarem para a profilaxia semanal com HympavziTM . Todos os pacientes (100%) na coorte sob demanda tinham uma ou mais articulações-alvo no início do estudo, e 36% tinham três ou mais articulações-alvo no início do estudo. No grupo de profilaxia de rotina, 56,6% dos pacientes tinham uma ou mais articulações-alvo no início do estudo, e 15,7% tinham três ou mais articulaçõesalvo no início do estudo.
Após uma fase observacional de 6 meses na qual os pacientes receberam terapia baseada em fator profilático de rotina ou sob demanda, os pacientes receberam uma dose inicial de ataque de 300 mg de HympavziTM seguida de doses de manutenção de 150 mg de HympavziTM uma vez por semana por 12 meses. A dose foi escalonada para 300 mg de HympavziTM uma vez por semana após 6 meses para pacientes pesando ≥50 kg que tiveram dois ou mais sangramentos excessivos. Quatorze (12,1%) dos 116 pacientes que receberam HympavziTM por pelo menos 6 meses foram submetidos ao escalonamento de dose de sua dose de manutenção.
A idade média entre os grupos de tratamento foi de 32,4 anos (mín. 13, máx. 66); 16,4% dos pacientes tinham 12 a < 18 anos e 83,6% tinham ≥18 anos, 100% eram do sexo masculino. Neste estudo, 48,3% dos pacientes eram Brancos, 50,0% eram Asiáticos, 0,9% eram Pretos ou Afro-Americanos e 0,9% não apresentavam informações raciais; 10,3% dos pacientes identificados como Hispânicos ou Latinos. Todos os pacientes eram não inibidores (hemofilia A a 78,4%, hemofilia B a 21,6%).
O objetivo primário de eficácia do estudo foi comparar a profilaxia de HympavziTM durante a Fase de Tratamento Ativo versus a terapia profilática de rotina baseada em fatores na Fase Observacional, conforme medido pela TAS de sangramentos tratados. Outros principais objetivos de eficácia do estudo incluíram a avaliação da profilaxia de HympavziTM em comparação com a terapia profilática de rotina baseada em fatores, medida pelas incidências de sangramentos espontâneos, sangramentos articulares, sangramentos nas articulações-alvo e sangramentos totais, bem como a avaliação da qualidade de vida relacionada à saúde (HRQoL) dos pacientes.
A Tabela 1 mostra os resultados de eficácia da profilaxia com HympavziTM em comparação com a terapia profilática de rotina baseada em fatores. HympavziTM demonstrou não inferioridade e superioridade em relação ao tratamento profilático de rotina baseada em fatores, conforme medida pela TAS de sangramentos tratados.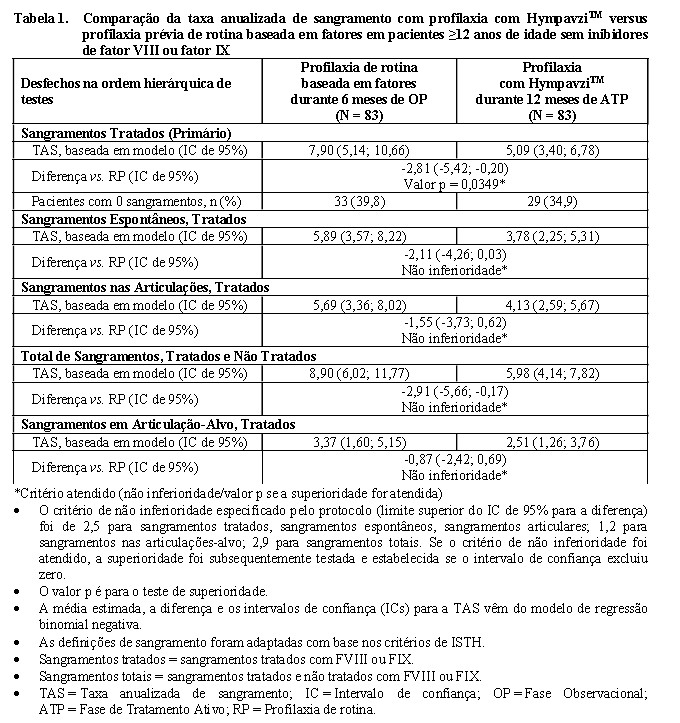
Em uma análise de apoio, HympavziTM também demonstrou superioridade sobre a terapia baseada em fatores sob demanda em incidências de sangramentos tratados, sangramentos espontâneos, sangramentos articulares, sangramentos totais e sangramentos nas articulações-alvo.
Análise interina do Estudo B7841007
Na extensão aberta do estudo central de Fase 3, 87 pacientes receberam marstacimabe nas doses estabelecidas durante a participação no estudo de B7841005 (ou seja, 150 mg ou 300 mg via subcutânea uma vez por semana) por até mais 16 meses (média de 7 meses), em que se demonstrou que o marstacimabe manteve a eficácia a longo prazo ( > 12 meses).
Foram realizadas análises descritivas para avaliar a profilaxia com HympavziTM ao longo do tempo. A média baseada em modelo e os outros resumos descritivos para a TAS para sangramentos tratados são mostrados na Tabela 2.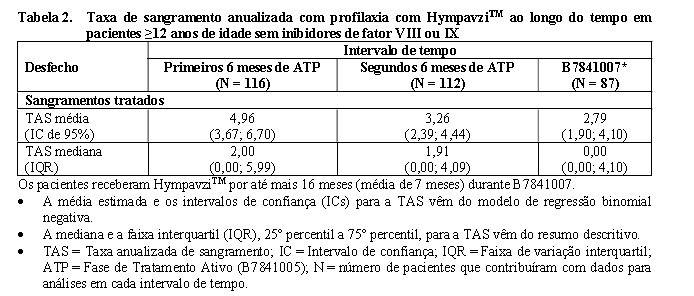
Medidas de desfechos relacionados à saúde (Estudo B7841005)
O Estudo central de Fase 3 avaliou os resultados de qualidade de vida relacionados à hemofilia relatados pelo paciente com o Questionário de Qualidade de Vida Específica para Hemofilia (Haem-A-QoL), para o qual o Escore de Domínio de Saúde Física (ou seja, inchaços dolorosos, presença de dor articular, dor com movimento, dificuldade para caminhar longas distâncias e necessidade de mais tempo para se arrumar) e Escore Total (resumo de todas as pontuações) foram desfechos definidos pelo protocolo de interesse. Para medir a alteração no estado de saúde, o Escore do Índice a Escala Analógica Visual (EVA) do Questionário de cinco dimensões e cinco níveis da EuroQoL (EQ-5D-5L) foram examinados.
As Tabelas 3 e 4 fornecem uma comparação da alteração em relação à avaliação inicial em 6 meses entre HympavziTM profilático semanalmente durante a Fase de Tratamento Ativo e a terapia profilática de rotina baseada em fator durante a Fase Observacional no Escore do Domínio da Saúde Física e no Escore Total na Haem-A-QoL, e Escore do Índice e Escala Visual Analógica na EQ-5D-5L, respectivamente. A melhora observada com a profilaxia semanal de HympavziTM foi não inferior à observada com a terapia profilática de rotina baseada em fatores nesses desfechos.

Imunogenicidade
Assim como com todas as proteínas terapêuticas, existe o potencial de uma resposta imune em pacientes tratados com marstacimabe. A incidência observada da formação de anticorpos antidroga (ADA) é altamente dependente da sensibilidade e especificidade do ensaio. As diferenças nos métodos de ensaio impedem comparações significativas da incidência de ADA nos estudos descritos abaixo com a incidência de ADA em outros estudos, incluindo aqueles relacionados ao marstacimabe.
Durante o período de tratamento de 12 meses no estudo B7841005 central de Fase 3, 23 dos 116 (19,8%) pacientes tratados com marstacimabe avaliáveis para ADA desenvolveram ADAs. Os ADAs foram transitórios em 61% (14/23) e persistentes em 39% (9/23) dos pacientes positivos para ADA, indicativo de um perfil transitório de ADA na maioria dos pacientes. Os títulos de ADA foram resolvidos em 22/23 (95,7%) pacientes até o final do estudo. Anticorpos neutralizantes (NAbs) foram desenvolvidos em 6/116 (5,2%) pacientes tratados com marstacimabe avaliáveis para ADA durante o estudo. Os NAbs foram transitórios em todos os pacientes e nenhum paciente foi considerado positivo para NAb no final do estudo. Embora concentrações médias de marstacimabe ligeiramente mais baixas (média aritmética: ~ 24%-32% menor; média geométrica: ~ 24%-50% menor) tenham sido relatadas em pacientes ADA-positivos em comparação com pacientes ADA-negativos, as concentrações se sobrepuseram amplamente entre esses dois grupos e não houve efeito clinicamente significativo identificado de ADAs, incluindo NAbs, na segurança ou eficácia do marstacimabe ao longo da duração do tratamento de 12 meses. No geral, o perfil de segurança de marstacimabe foi semelhante entre os pacientes com e sem ADAs (incluindo NAbs).
No estudo OLE de Fase 3, apenas um dos 44 pacientes avaliáveis para ADA que continuou a receber marstacimabe por pelo menos 6 meses foi persistentemente positivo para ADAs.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Mecanismo de ação
O marstacimabe é um anticorpo monoclonal IgG1 humano direcionado contra o domínio Kunitz 2 (K2) do inibidor da via do fator tecidual (TFPI), o inibidor primário da cascata extrínseca de coagulação. O TFPI inicialmente liga-se e inibe o sítio ativo do fator Xa por meio de seu segundo domínio Kunitz (K2) do inibidor. Assim, neutralizar a atividade do TFPI pode servir para melhorar a via extrínseca e reduzir ou eliminar a necessidade de reposição de FVIII ou FIX.
Efeitos farmacodinâmicos
O marstacimabe, de acordo com seu mecanismo de ação anti-TFPI, provoca um aumento no TFPI total e em biomarcadores derivados da geração de trombina, como fragmentos de protrombina 1+2, pico de trombina e Ddímero, quando administrado em pacientes com hemofilia. Essas mudanças são reversíveis após a descontinuação do tratamento. Aumentos esporádicos ou transitórios nos fragmentos de dímero D e protrombina 1+2 acima dos valores fisiológicos foram relatados no estudo de Fase 3, sem preocupações de segurança associadas. A terapia com marstacimabe não causa alterações clinicamente significativas em medidas padrão de coagulação, incluindo o tempo de tromboplastina parcial ativada (TTPA) e o tempo de protrombina (TP).
Propriedades farmacocinéticas
A farmacocinética (PK) do marstacimabe foi determinada por meio de análise não compartimental em participantes sadios e também em pacientes com hemofilia A e B, bem como usando uma análise farmacocinética populacional em um banco de dados composto por 213 participantes (150 pacientes com hemofilia e 63 participantes sadios) que receberam doses de marstacimabe subcutâneas (30 mg a 450 mg) ou intravenosas (150 e 440 mg) uma vez por semana.
O marstacimabe exibiu farmacocinética não linear com exposição sistêmica à marstacimabe, medida por ASC e Cmáx, aumentando de maneira maior que proporcional à dose. Esse comportamento farmacocinético não linear é causado pela disposição do fármaco mediada pelo alvo (TDPM) e pela eliminação não linear dependente da concentração de marstacimabe, que ocorre quando o marstacimabe se liga ao TFPI endotelial.
A razão média de acúmulo no estado de equilíbrio para marstacimabe foi de aproximadamente 4 a 5, em relação à primeira exposição à dose após a dosagem subcutânea semanal de 150 mg e 300 mg. Espera-se que as concentrações no estado de equilíbrio de marstacimabe sejam atingidas em aproximadamente 60 dias, ou seja, pela 8ª ou 9ª dose subcutânea quando administrada uma vez por semana. Para 150 mg de marstacimabe subcutâneo uma vez por semana, as estimativas populacionais de Cmín,ss, Cmáx,ss e Cméd,ss médias para adultos e adolescentes pesando pelo menos 35 kg são mostradas na Tabela 5.
Absorção
Após múltiplas administrações subcutâneas de marstacimabe em pacientes com hemofilia, o Tmáx mediano variou de 23 a 59 horas. A biodisponibilidade do marstacimabe após a administração subcutânea foi estimada em cerca de 71% por modelagem farmacocinética populacional. Não foram observadas diferenças clinicamente relevantes na biodisponibilidade do marstacimabe entre o braço, a coxa e o abdômen.
Distribuição
O volume de distribuição no estado de equilíbrio de marstacimabe em pacientes com hemofilia foi de 8,6 L com base em uma análise farmacocinética populacional. Essa distribuição extravascular limitada sugere que o marstacimabe é restrito ao espaço intravascular.
Metabolismo
Não foram conduzidos estudos do metabolismo com marstacimabe. Assim como outras proteínas terapêuticas com pesos moleculares acima do limite de filtração glomerular, espera-se que marstacimabe passe por catabolismo proteolítico e depuração mediada por receptor. Além disso, com base no TDPM, espera-se que
marstacimabe também seja eliminado por depuração mediada pelo alvo devido à formação do complexo marstacimabe/TFPI.
Excreção
Não foram conduzidos estudos de excreção com o marstacimabe. Com base no peso molecular, espera-se que marstacimabe seja submetido à degradação catabólica e não se espera que seja eliminado pelos rins. O marstacimabe é eliminado por meio de mecanismos lineares e não lineares. Após múltiplas doses subcutâneas e com base em uma análise PK populacional, a depuração linear de marstacimabe foi de aproximadamente 0,019 L/h. A meia-vida média efetiva no estado de equilíbrio (dependente tanto da acumulação quanto da eliminação) de marstacimabe foi estimada em aproximadamente 16 a 18 dias para adultos e adolescentes e nos grupos de dose. Com base na análise farmacocinética populacional, espera-se que 90% do marstacimabe seja eliminado ao final de aproximadamente 1 mês após a última dose (o tempo médio para que 50% do medicamento seja eliminado é de aproximadamente 7 a 10 dias).
Populações especiais
Peso corporal, faixa etária, raça e tipo de hemofilia
Embora o peso seja uma covariável importante para descrever a farmacocinética do marstacimabe, não há ajuste de dose recomendado para o peso em pacientes pesando ≥35 kg. A depuração de marstacimabe (CL/F) foi 29% menor em adolescentes (12 a < 18 anos de idade) em comparação com adultos (18 anos ou mais). Após o ajuste para o peso, a CL (L/h/kg) em adolescentes foi estimada em aproximadamente 3% mais baixa em comparação com a de adultos, indicando que o peso é responsável pela maioria das diferenças na CL. Essa diferença na PK não se traduziu em uma diferença clinicamente relevante nos níveis do pico de trombina do marcador farmacodinâmico derivado entre os dois grupos. O impacto da raça (Asiáticos vs. não-Asiáticos) e do tipo de hemofilia na farmacocinética do marstacimabe não foi clinicamente relevante na população de pacientes. A depuração ajustada ao peso do marstacimabe foi 32% maior em pacientes Asiáticos em comparação com pacientes não-Asiáticos. Essa diferença não é considerada clinicamente relevante. Não há dados suficientes para avaliar potenciais diferenças na exposição ao marstacimabe em outras raças ou etnias.
Estudos clínicos com marstacimabe não incluíram um número suficiente de pacientes com 65 anos ou mais para determinar se existem diferenças na exposição em comparação com pacientes mais jovens.
Pacientes com insuficiência renal
A depuração renal não é considerada importante para a eliminação de anticorpos monoclonais (mAbs) devido ao seu tamanho grande e filtragem ineficiente através do glomérulo. Não foram realizados estudos clínicos para avaliar o efeito da insuficiência renal na farmacocinética de marstacimabe.
Todos os pacientes com hemofilia A e B na análise farmacocinética populacional apresentaram função renal normal (N = 129; eTFG ≥90 mL/min/1,73 m2) ou insuficiência renal leve (N = 21; eTFG de 60 a 89 mL/min/1,73 m2). A insuficiência renal leve não afetou a farmacocinética de marstacimabe. Não há dados disponíveis sobre o uso de marstacimabe em pacientes com comprometimento renal moderado ou grave.
Pacientes com insuficiência hepática
Não foram realizados estudos clínicos para avaliar o efeito da insuficiência hepática na farmacocinética do marstacimabe, pois geralmente não é considerado clinicamente relevante para mAbs.
Todos os pacientes com hemofilia A e B nos estudos clínicos apresentaram função hepática normal (N = 135; bilirrubina total e AST ≤LSN) ou insuficiência hepática leve (N = 15; bilirrubina total 1× a ≤1,5× LSN ou AST > LSN). A insuficiência hepática leve não afetou a farmacocinética de marstacimabe. Não há dados disponíveis sobre o uso de marstacimabe em pacientes com insuficiência hepática moderada ou grave.
Dados de segurança pré-clínicos
Dados não clínicos em animais normais hemostáticos não demonstraram nenhum risco especial para humanos com base em estudos de toxicidade de dose repetida de até 6 meses de duração em ratos ou 3 meses de duração em macacos-cinomolgos, incluindo desfechos de farmacologia de segurança e desfechos de toxicidade reprodutiva, em exposições de pelo menos 201 vezes e 219 vezes, respectivamente, a exposição da ASC em uma dose clínica de 300 mg subcutânea por semana.
Os efeitos não adversos de marstacimabe na cascata de coagulação em ratos e macacos-cinomolgos incluíram diminuição do fibrinogênio, aumento do D-dímero, alterações no tempo de protrombina e tempo parcial de tromboplastina ativada, e apenas em ratos, trombos/êmbolos microscópicos no local da injeção e nos pulmões.
Infiltração não adversa reversível de células mistas, hemorragia e necrose foram observadas nos locais de injeção em ratos após administração subcutânea de marstacimabe.
Comprometimento de fertilidade
O marstacimabe não afetou a fertilidade quando administrado como dose repetida em ratos machos em doses de até 1000 mg/kg/dose e uma margem de exposição de 212 vezes a exposição da ASC em uma dose clínica de 300 mg por via subcutânea semanalmente. Nenhum efeito foi observado em órgãos reprodutores masculinos ou femininos nos estudos de toxicidade de dose repetida em ratos e macacos-cinomolgos em doses de 1000 mg/kg/dose e 500 mg/kg/dose e margens de exposição de, pelo menos, 201 vezes e 219 vezes, respectivamente, a exposição da ASC a uma dose clínica de 300 mg por via subcutânea semanalmente.
Toxicidade de desenvolvimento
Não há dados de animais disponíveis informando o risco associado ao marstacimabe.
Genotoxicidade e carcinogenicidade
Não foram realizados estudos para avaliar o marstacimabe quanto ao potencial carcinogênico ou mutagênico.
4. CONTRAINDICAÇÕES
Hipersensibilidade ao marstacimabe ou a qualquer componente da formulação do produto.
5. ADVERTÊNCIAS E PRECAUÇÕES
Eventos tromboembólicos
HympavziTM é um antagonista do inibidor da via do fator tecidual (TFPI) e pode aumentar o risco de complicações tromboembólicas. Eventos trombóticos venosos foram relatados em estudos clínicos com HympavziTM, resultando em embolia em um participante saudável (vide item 9. Reações Adversas). Esses eventos ocorreram em indivíduos com múltiplos fatores de risco para tromboembolia. O uso de outros produtos inibidores da via do fator antitecidual (anti-TFPI) tem sido associado ao desenvolvimento de complicações tromboembólicas em pacientes expostos a agentes hemostáticos adicionais em estreita proximidade. HympavziTM não foi estudado em pacientes com histórico de eventos tromboembólicos anteriores (vide item 2. Resultados de Eficácia).
Os seguintes pacientes podem apresentar risco aumentado de eventos tromboembólicos com o uso de HympavziTM:
• pacientes com histórico de doença arterial coronariana, trombose venosa ou arterial ou doença isquêmica,
• pacientes com fatores de risco conhecidos para tromboembolismo, incluindo, entre outros, condições genéticas pró-trombóticas (por exemplo, Fator V de Leiden), pacientes com períodos prolongados de imobilização, obesidade e tabagismo,
• pacientes que estejam apresentando uma doença aguda grave com aumento da expressão do fator tecidual (como infecção grave, sepse, trauma, lesões por esmagamento, câncer).
Interrompa a profilaxia com HympavziTM se forem encontrados achados diagnósticos consistentes com tromboembolismo e gerencie conforme clinicamente indicado.
Os produtos de fator VIII e fator IX foram administrados com segurança para o tratamento de sangramentos excessivos em pacientes que receberam HympavziTM. Se os produtos de fator VIII ou fator IX forem indicados em um paciente que esteja recebendo profilaxia com HympavziTM, será recomendada a dose efetiva mínima do produto de fator VIII ou fator IX, de acordo com a bula do produto.
Considere o benefício e o risco de usar HympavziTM em pacientes com fatores de risco conhecidos para tromboembolismo.
Reações de hipersensibilidade
Reações cutâneas de erupção e prurido que podem refletir hipersensibilidade ao medicamento ocorreram em pacientes tratados com HympavziTM (vide item 9. Reações Adversas). Se os pacientes tratados com HympavziTM desenvolverem uma reação de hipersensibilidade grave, oriente-os a interromper o uso de HympavziTM e procurar tratamento de emergência imediatamente.
Fertilidade, gravidez e lactação
Mulheres com potencial para engravidar
Mulheres em idade fértil que estão recebendo HympavziTM devem utilizar contracepção eficaz durante o tratamento e por pelo menos 1 mês após o término do tratamento com HympavziTM (vide item 3. Características Farmacológicas).
Fertilidade
O risco de infertilidade em machos e fêmeas com potencial reprodutivo não foi estudado em humanos. O marstacimabe não afetou a fertilidade quando administrado em ratos machos (vide item 3. Características Farmacológicas).
Gravidez
Não há estudos clínicos de marstacimabe em mulheres grávidas. Não há dados de animais disponíveis informando o risco associado ao marstacimabe. Não se sabe se HympavziTM pode causar danos fetais quando administrado a uma mulher grávida ou se ele pode afetar a capacidade de reprodução. HympavziTM deve ser usado durante a gravidez apenas se o potencial benefício para a mãe superar o risco para o feto.
HympavziTM é um medicamento classificado na categoria C de risco de gravidez. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Lactação
Estudos de lactação não foram realizados em seres humanos ou animais. Não se sabe se marstacimabe é excretado no leite humano. Sabe-se que a IgG humana está presente no leite humano. Deve-se decidir pela interrupção da amamentação ou pela interrupção/abstenção da terapia com HympavziTM , levando em consideração o benefício da amamentação para a criança e o benefício da terapia para a mulher.
O uso deste medicamento no período da lactação depende da avaliação e acompanhamento do seu médico.
Efeitos na capacidade de dirigir veículos e operar máquinas
Não foram realizados estudos do efeito sobre a capacidade de dirigir ou operar máquinas. Não foram observados efeitos sobre a habilidade para dirigir e operar máquinas.
6. INTERAÇÕES MEDICAMENTOSAS
Não foram realizados estudos clínicos de interação medicamentosa com marstacimabe.
Como um anticorpo monoclonal, espera-se que o marstacimabe seja eliminado pelo catabolismo após endocitose pelo sistema fagocítico mononuclear. Uma vez que a eliminação de mAbs não ocorre por vias não catabólicas, como enzimas metabólicas hepáticas (ou seja, enzimas do citocromo P450) ou por transportadores de fármacos renais/hepáticos de moléculas pequenas, as interações farmacocinéticas com medicamentos concomitantes que são eliminados por essas vias são improváveis. O efeito indireto de um biológico, como o marstacimabe, na expressão das enzimas do citocromo P450 também não é esperado.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenar em geladeira (de 2 °C a 8 °C). Não congelar. Não agitar. Manter na embalagem até o final do uso. Manter na embalagem original para proteger da luz. O prazo de validade do medicamento é de 24 meses a partir da data de fabricação.
HympavziTM pode ser removido do armazenamento em geladeira e armazenado em sua embalagem original por um único período de, no máximo, 7 dias em temperatura ambiente (até 30 °C). O produto não deve ser devolvido para armazenamento em geladeira. Antes do final deste período de armazenamento em temperatura ambiente, o produto deve ser usado ou descartado.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original. Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
Características físicas e organolépticas: HympavziTM é uma solução incolor a amarelo claro.
8. POSOLOGIA E MODO DE USAR
O tratamento deve ser iniciado sob a supervisão de um médico/profissional de saúde experiente no tratamento da hemofilia.
Posologia
A dose recomendada para pacientes com 12 anos de idade ou mais, pesando pelo menos 35 kg, é uma dose de ataque inicial de 300 mg por injeção subcutânea, seguida de 150 mg por injeção subcutânea uma vez por semana.
Duração do tratamento
HympavziTM é destinado ao tratamento profilático de longo prazo.
Ajustes de dose durante o tratamento
Um ajuste da dose para a injeção subcutânea de 300 mg semanalmente pode ser considerado em pacientes com peso ≥50 kg quando o controle de eventos de sangramento for considerado inadequado pelo profissional de saúde. Não há dados suficientes para recomendar doses acima de 300 mg por semana.
Dose esquecida
Para pacientes em dose de manutenção de 150 mg
Se uma dose for esquecida, administre o mais rápido possível antes do dia da próxima dose programada e, em seguida, retome o cronograma de dosagem semanal usual de 150 mg por via subcutânea (mesmo esquema anterior à dose esquecida ou novo esquema com base na data de administração da dose esquecida).
Se a dose tiver sido esquecida por mais de 13 dias após a última dose, então uma dose de ataque de 300 mg por injeção subcutânea deve ser administrada, seguida pela retomada de 150 mg por injeção subcutânea uma vez por semana.
Para pacientes em dose de manutenção de 300 mg
Se uma ou mais doses forem esquecidas, administre uma dose o mais rápido possível e, em seguida, retome o cronograma de dosagem semanal usual de 300 mg por via subcutânea (mesmo esquema anterior à dose esquecida ou novo esquema com base na data de administração da dose esquecida).
Mudança para HympavziTM
Mudança da terapia de substituição do fator profilático para HympavziTM: Antes do início de HympavziTM, os pacientes devem descontinuar o tratamento com concentrados de fator de coagulação (concentrados de fator VIII ou fator IX). Os pacientes podem iniciar HympavziTM a qualquer momento após a descontinuação dos concentrados de fator de coagulação.
Mudança de medicamentos para hemofilia não baseada em fatores para HympavziTM: Não há dados de estudos clínicos disponíveis para orientar a conversão de pacientes de medicamentos não baseados em fatores para Hympavzi. Embora um período de washout não tenha sido estudado, uma abordagem é permitir um período de washout adequado (pelo menos 5 meias-vidas) do agente anterior com base na meia-vida rotulada antes de iniciar o tratamento com HympavziTM. Pode ser necessário suporte hemostático com concentrados de fator de coagulação durante a mudança de outros medicamentos para hemofilia não baseados em fatores para HympavziTM .
Populações especiais
Orientação sobre o uso com tratamentos para sangramentos excessivos
Os produtos de fator VIII e fator IX podem ser administrados para o tratamento de sangramentos excessivos em pacientes que recebem HympavziTM. Doses adicionais de HympavziTM não devem ser usadas para tratar eventos de sangramentos excessivos. Os profissionais de saúde devem discutir com todos os pacientes e/ou cuidadores sobre a dose e o cronograma dos concentrados do fator de coagulação a serem usados, se necessário, enquanto recebem profilaxia com HympavziTM, incluindo o uso da dose efetiva mais baixa possível do concentrado do fator de coagulação (vide item 5. Advertências e Precauções). Consulte as informações do produto para saber o fator de coagulação concentrado usado.
Insuficiência hepática
Nenhum ajuste de dose é recomendado em pacientes com insuficiência hepática leve (vide item 3. Características Farmacológicas). O marstacimabe não foi estudado em pacientes com insuficiência hepática moderada ou grave.
Insuficiência renal
Nenhum ajuste de dose é recomendado em pacientes com insuficiência renal leve (vide item 3. Características Farmacológicas). O marstacimabe não foi estudado em pacientes com insuficiência renal moderada ou grave.
População idosa
Nenhum ajuste de dose é recomendado em pacientes acima de 65 anos de idade. Há dados limitados em pacientes acima de 65 anos de idade.
População pediátrica
A segurança e a eficácia de HympavziTM em pacientes pediátricos < 12 anos de idade não foram estabelecidas. A segurança e a eficácia de marstacimabe em adolescentes com peso corporal < 35 kg não foram estabelecidas. Não há dados disponíveis.
Gerenciamento no ambiente perioperatório
A segurança e a eficácia de HympavziTM não foram formalmente avaliadas no ambiente cirúrgico. Os pacientes apresentaram pequenos procedimentos cirúrgicos sem interromper a profilaxia com HympavziTM em estudos clínicos.
Para uma cirurgia de grande porte, HympavziTM deve ser descontinuado de 6 a 12 dias antes e o tratamento iniciado conforme o padrão de cuidado local com concentrado de fator de coagulação, além de medidas para gerenciar o risco de trombose venosa, que pode ser elevado no período perioperatório. Consulte as informações do produto referentes ao concentrado do fator de coagulação para obter orientações de dosagem em pacientes com hemofilia submetidos a cirurgia de grande porte. A retomada da terapia com HympavziTM deve considerar o estado clínico geral do paciente, incluindo a presença de fatores de risco tromboembólicos pós-cirúrgicos, o uso de outros produtos hemostáticos e outros medicamentos concomitantes (vide subitem Dose esquecida acima).
Tratamento em pacientes com doença grave aguda
Há experiência limitada com o uso de HympavziTM em pacientes com doença aguda grave. Razões para considerar uma interrupção temporária da dose de HympavziTM incluem a ocorrência de doença aguda grave (por exemplo, infecção grave, sepse, trauma) na qual pode haver aumento da ativação da coagulação e que o médico/profissional de saúde considere que possa aumentar os riscos associados à administração de HympavziTM (vide item 5. Advertências e Precauções). O tratamento de doença aguda grave deve ser gerenciado de acordo com o padrão de cuidados local, e a continuação do tratamento com HympavziTM nessa situação deve ser avaliada em relação aos potenciais riscos envolvidos. A terapia com HympavziTM pode ser retomada assim que o paciente se recuperar clinicamente (vide subitem Dose esquecida acima).
Método de administração
HympavziTM destina-se apenas para injeção subcutânea.
HympavziTM é destinado ao uso sob a orientação de um profissional de saúde. Após o treinamento adequado na técnica de injeção subcutânea, um paciente ou cuidador pode injetar HympavziTM se um médico/profissional de saúde determinar que é apropriado.
Antes da administração subcutânea, HympavziTM pode ser retirado da geladeira e deixado à temperatura ambiente na embalagem por 15 a 30 minutos, protegido da luz solar direta. HympavziTM não deve ser aquecido usando uma fonte de calor, como água quente ou micro-ondas. Após retirar HympavziTM da geladeira, ele deve ser utilizado dentro de 7 dias ou descartado (vide item 7. Cuidados de armazenamento do medicamento).
HympavziTM deve ser administrado por injeção subcutânea, uma vez por semana, a qualquer hora do dia. Os locais recomendados para a injeção são o abdômen e a coxa. Outros locais são aceitáveis, se necessário. A administração de HympavziTM nas nádegas (somente caneta preenchida) deve ser realizada apenas por um cuidador ou profissional de saúde.
Para a dose de ataque de 300 mg, cada uma das duas injeções de 150 mg do HympavziTM deve ser administrada em locais diferentes.
Recomenda-se alternar o local da injeção a cada aplicação. HympavziTM não deve ser administrado em áreas ósseas ou em áreas onde a pele esteja machucada, vermelha, sensível ou rígida, ou em áreas com cicatrizes ou estrias. HympavziTM não deve ser injetado em uma veia.
Durante o tratamento com HympavziTM , outros medicamentos para administração subcutânea devem, de preferência, ser injetados em diferentes locais anatômicos.
Precauções especiais para descarte e manuseio
O produto é apenas para uso único.
Não agite.
Para uma injeção mais confortável, permita que o produto atinja a temperatura ambiente na embalagem por cerca de 15 a 30 minutos protegido da luz solar direta.
Inspecione a solução visualmente antes do uso. HympavziTM é uma solução clara e incolor a amarelo claro. Não use se o medicamento estiver turvo, amarelo escuro ou contiver flocos ou partículas.
HympavziTM não contém conservantes; portanto, porções não usadas devem ser descartadas. Não use após a data de validade.
Os produtos não utilizados ou os resíduos devem ser descartados de acordo com as exigências locais.
Incompatibilidades
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros.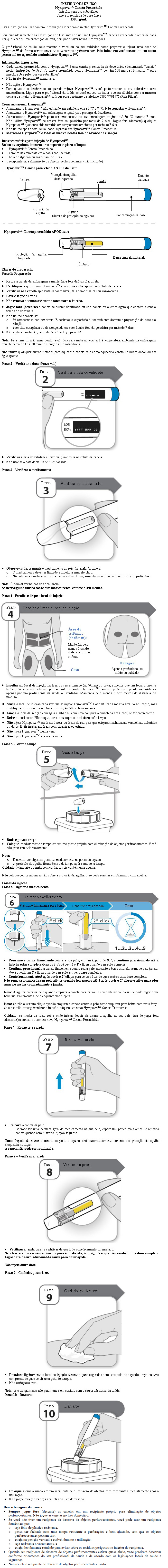
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
O perfil geral de segurança do HympavziTM baseia-se em dados de estudos clínicos. A reação adversa medicamentosa mais grave relatada nos estudos clínicos com o HympavziTM foi trombose (vide abaixo e o item
5. Advertências e Precauções).
As reações adversas relatadas com mais frequência após o tratamento com HympavziTM foram reações no local da injeção (RLIs) (11,2%).
Lista tabulada de reações adversas
Os dados de segurança são baseados em dados agrupados do estudo de Fase 3 de segurança e eficácia (BASIS) e seu estudo de extensão aberta (OLE) em andamento. O estudo multicêntrico de Fase 3 foi realizado em pacientes adolescentes e adultos com hemofilia A ou B grave (atividade do fator de coagulação < 1%) entre 12 e < 75 anos de idade, sem inibidores que comparassem a terapia baseada em fator à profilaxia com HympavziTM (vide item 2. Resultados de Eficácia). Os dados do estudo de Fase 3, fase de tratamento ativo de 12 meses, refletem a exposição de 116 pacientes do sexo masculino com hemofilia A ou B sem inibidores ao HympavziTM administrado uma vez por semana. Noventa e sete (83,6%) pacientes eram adultos (18 anos de idade ou mais) e 19 (16,4%) eram adolescentes (12 anos até < 18 anos). Um total de 87 dos 116 pacientes que concluíram o período de tratamento de 12 meses subsequentemente se inscreveram no estudo OLE. A duração mediana da exposição foi de 518,5 dias (faixa de variação de 28 a 847 dias).
A Tabela 6 apresenta as reações adversas ao medicamento (RAMs) em pacientes com 12 anos de idade ou mais com hemofilia A ou hemofilia B sem inibidores (N = 116) que receberam o regime de dosagem recomendado de HympavziTM .
Descrição das reações adversas selecionadas
Eventos tromboembólicos
Dois eventos trombóticos venosos foram relatados em estudos clínicos com marstacimabe. Um paciente com hemofilia A no estudo de extensão aberto desenvolveu trombose venosa profunda após aproximadamente 3 anos de exposição ao HympavziTM. O tratamento com HympavziTM foi suspenso e anticoagulante foi administrado. O paciente apresentava múltiplos fatores contribuintes potenciais, incluindo mutação genética heterozigótica do Fator V de Leiden e fatores de estilo de vida (ou seja, períodos de imobilização prolongada e tabagismo).
No estudo de Fase 1, um participante saudável com sarcoidose previamente não diagnosticada desenvolveu trombose venosa profunda e embolia pulmonar 9 dias após receber uma dose única de 300 mg de HympavziTM e aproximadamente 4 semanas após receber uma segunda dose de uma vacina contra COVID-19 baseada em vetor (não mRNA) (não mais disponível para uso). O HympavziTM foi descontinuado definitivamente, o tratamento anticoagulante foi administrado e as reações adversas foram resolvidas.
Reações no local da injeção
No total, 13 (11,2%) pacientes tratados com HympavziTM relataram RLIs. A maioria das RLIs observadas em estudos clínicos de HympavziTM foi transitória e relatada como leve a moderada em termos de gravidade. Nenhuma ocorrência de reação no local da injeção levou a um ajuste da dose ou descontinuação do medicamento.
Erupção cutânea
Na população não inibidora, um (0,9%) paciente relatou erupção cutânea não grave (Grau 1).
Na população inibidora de um estudo clínico em andamento em que 35 pacientes com hemofilia com inibidores são tratados com HympavziTM, um (2,9%) paciente com hemofilia B grave e histórico de reação alérgica ao fator IX exógeno apresentou erupção cutânea grave com início em aproximadamente 9 meses. O paciente necessitou de um curso prolongado de corticosteroides orais para resolução, e o tratamento com marstacimabe foi descontinuado.
População pediátrica
A população pediátrica estudada compreende um total de 19 pacientes adolescentes (de 12 a < 18 anos de idade). O perfil de segurança de HympavziTM foi globalmente consistente entre adolescentes e adultos.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo ao Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
A dose mais alta de marstacimabe administrada em ensaios clínicos foi de 450 mg por via subcutânea, uma vez por semana, em pacientes adultos com peso > 50 kg por até 3 meses (N = 6). Este foi um grupo pequeno e o efeito de exposições elevadas a longo prazo é desconhecido. Doses superiores a 150 mg por semana para adolescentes de 12 a 17 ano