HUMIRATM
ABBVIE
adalimumabe
Anti-reumático.
Apresentações.
HUMIRA® (adalimumabe) solução injetável de:
- 40 mg em seringa com 0,8 mL de dose única pronta para uso: embalagens com 2 blisters contendo, cada um, 1 seringa pronta para uso e 1 envelope com lenço umedecido em álcool.
VIA SUBCUTÂNEA
USO ADULTO E PEDIÁTRICO ACIMA DE 13 ANOS
Composição.
Cada seringa contém: adalimumabe 40 mg, Excipientes* qsp 0,8 mL, * cloreto de sódio, fosfato de sódio monobásico diidratado, fosfato de sódio dibásico diidratado, citrato de sódio, ácido cítrico monoidratado, manitol, polissorbato e água para injeção.
Indicações.
Este medicamento é destinado ao tratamento de:
Artrite Reumatóide
HUMIRA® (adalimumabe) é destinado para reduzir os sinais e sintomas, induzir uma resposta clínica e remissão clínica maior, inibir a progressão dos danos estruturais e melhorar a capacidade física em pacientes adultos com artrite reumatóide (AR) ativa de intensidade moderada a grave, que apresentaram resposta inadequada a uma ou mais drogas antirreumáticas modificadoras do curso da doença (DMCD).
HUMIRA® (adalimumabe) é destinado ao tratamento da artrite reumatóide grave, ativa e progressiva em pacientes não tratados com metotrexato previamente.
HUMIRA® (adalimumabe) pode ser utilizado isoladamente ou em combinação com metotrexato ou outra DMCD.
Artrite Psoriásica
HUMIRA® (adalimumabe) é destinado para reduzir os sinais e sintomas da artrite psoriásica (APs). O medicamento demonstrou reduzir a taxa de progressão das lesões articulares periféricas, conforme medido por raio-X em pacientes com subtipos poliarticular simétrico da doença, e melhora da função física. HUMIRA® (adalimumabe) pode ser utilizado isoladamente ou em combinação a fármacos antirreumáticos modificadores do curso da doença (DMCD).
Espondilite Anquilosante
HUMIRA® (adalimumabe) é destinado ao tratamento da espondilite anquilosante (EA) ativa em pacientes que responderam inadequadamente à terapia convencional.
Doença de Crohn
HUMIRA® (adalimumabe) é destinado para reduzir sinais e sintomas e induzir e manter a remissão clínica em pacientes adultos com doença de Crohn (DC) ativa de intensidade moderada a grave, que apresentaram resposta inadequada à terapia convencional. HUMIRA® (adalimumabe) também é destinado para reduzir sinais e sintomas e induzir remissão clínica em pacientes que perderam resposta ou são intolerantes ao infliximabe.
Psoríase em placas
HUMIRA® (adalimumabe) é destinado ao tratamento de psoríase em placas crônica moderada a grave em pacientes adultos com indicação de terapia sistêmica ou fototerapia e quando outras terapias sistêmicas forem menos apropriadas.
Artrite Idiopática Juvenil Poliarticular
HUMIRA® (adalimumabe) é destinado para reduzir sinais e sintomas de moderados a graves da artrite idiopática juvenil poliarticular (AIJ) ativa em pacientes acima de 13 anos de idade. HUMIRA® (adalimumabe) pode ser utilizado isoladamente ou em combinação com metotrexato. HUMIRA® (adalimumabe) é uma das opções terapêuticas para o tratamento da AIJ após a utilização de pelo menos um DMARD.
Resultados de eficácia.
- Artrite reumatóide
HUMIRA® (adalimumabe) foi avaliado em mais de 3000 pacientes com artrite reumatóide em ensaios clínicos. Alguns pacientes foram tratados por até 60 meses. A eficácia e a segurança de HUMIRA® (adalimumabe) foram avaliados em cinco ensaios clínicos controlados, duplo-cegos e randomizados.1-5
O estudo I (ARMADA)1 avaliou 271 pacientes com artrite reumatóide moderada a grave, com mais de 18 anos de idade, que falharam ao tratamento com pelo menos uma droga modificadora da doença (DMCD), com resposta insuficiente ao metotrexato em doses constantes de 12,5 a 25mg/semana (ou 10mg caso o paciente fosse intolerante ao metotrexato). Os pacientes apresentavam articulações edemaciadas ≥ 6 e articulações doloridas ≥ 9 e com RA diagnosticada de acordo com o critério ACR. Os pacientes receberam placebo ou 20, 40 ou 80mg de HUMIRA® (adalimumabe) a cada 2 semanas, por 24 semanas, por via subcutânea (SC).
O estudo II (DE011)2 avaliou 544 pacientes com artrite reumatóide moderada a grave, com mais de 18 anos de idade, que falharam ao tratamento com pelo menos uma DMCD (metotrexato, sulfassalazina, hidroxicloroquina, ouro oral ou injetável, d-penicilamina, azatioprina). Os pacientes apresentaram articulações edemaciadas ≥ 10 e articulações doloridas ≥ 12 e também diagnosticados de acordo com o critério ACR.Os pacientes foram divididos em 5 grupos: placebo semanal, HUMIRA® (adalimumabe) 20mg + placebo semanal, HUMIRA® (adalimumabe) 40mg + placebo semanal, HUMIRA® (adalimumabe) 20mg + placebo a cada 2 semanas, HUMIRA® (adalimumabe) 40mg + placebo a cada 2 semanas. Todos os pacientes receberam os tratamentos por via SC.
A duração do estudo foi de 26 semanas.
O estudo III (DE019)3 avaliou 619 pacientes com artrite reumatóide moderada a grave, com mais de 18 anos de idade, com resposta insuficiente ao metotrexato em doses constantes semanais de 12,5 a 25mg/semana (ou 10mg caso o paciente fosse intolerante ao metotrexato). Diferente do estudo I, os pacientes com RA do estudo III não apresentavam falhas ao tratamento com pelo menos uma droga modificadora da doença (DMCD). Os pacientes foram divididos em grupos: placebo a cada 2 semanas, HUMIRA® (adalimumabe) 20mg + placebo a cada 2 semanas e HUMIRA® (adalimumabe) 40mg + placebo a cada 2 semanas. Todos os pacientes receberam os tratamentos por via SC. A duração do estudo foi de 52 semanas. Após este período, os pacientes puderam entrar em um período de extensão aberto no qual avaliou-se o uso de HUMIRA® (adalimumabe) 40mg a cada 2 semanas, por via SC, por até 60 meses.6
O estudo IV (STAR)4 avaliou 636 pacientes com artrite reumatóide moderada a grave, com mais de 18 anos de idade. A população do estudo incluiu pacientes que nunca haviam usado DMCDs ou que estavam em tratamento com DMCDs estável por no mínimo 28 dias. Estes tratamentos incluíram leflunomida, hidroxicloroquina, sulfassalazina e/ou sais de ouro. Os pacientes foram randomizados para receberem HUMIRA® (adalimumabe) 40mg ou placebo, por via SC, a cada 2 semanas, por 24 semanas.
O estudo V (PREMIER)5 avaliou 799 pacientes com artrite reumatóide de início recente (duração média dos sintomas de menos de 9 meses), moderada a grave, que nunca haviam usado metotrexato. O estudo avaliou a eficácia, a segurança e a progressão radiológica da destruição articular de HUMIRA® (adalimumabe) 40mg + metotrexato a cada 2 semanas, HUMIRA® (adalimumabe) 40mg + placebo a cada 2 semanas e monoterapia com metroxato, por 104 semanas. Todos os tratamentos foram por via SC.
As medidas de desfecho primárias dos estudos I, II e III e a medida de desfecho secundária do estudo IV foram a porcentagem de pacientes que atingiu respostas ACR20 nas semanas 24 ou 26 (diminuição de 20% dos critérios do American Collegge of Rheumatology). A medida de desfecho primária do estudo V foi a porcentagem de pacientes que atingiu respostas ACR50 (diminuição de 50% nos critérios do American College of Rheumatology) na semana 52. Os estudos 3 e 5 também tiveram a inibição da progressão da doença (medida por exames de raios-X) como medida de desfecho co-primária na semana 52. O estudo III também avaliou mudanças em escores de qualidade de vida como medida de desfecho co-primária.
Os principais resultados de eficácia destes estudos são apresentados a seguir.
Respostas ACR
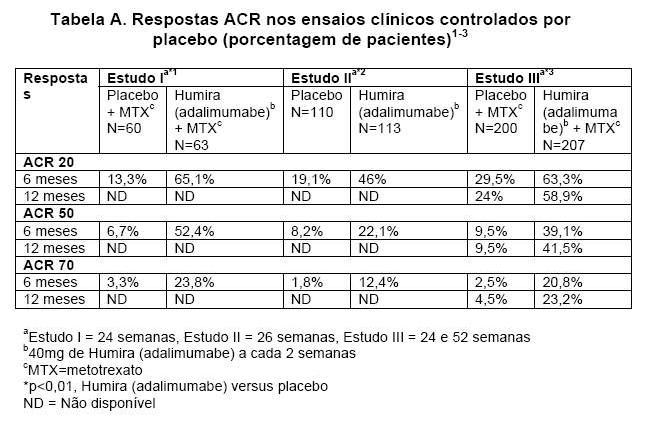
No estudo IV, a resposta ACR 20 dos pacientes tratados com HUMIRA® (adalimumabe) foi significativamente melhor do que os pacientes tratados com placebo (p < 0,001).4
Nos estudos I-IV, todos os componentes individuais dos critérios de resposta ACR (número de articulações dolorosas, número de articulações edemaciadas, avaliação da atividade da doença e da dor pelo médico, avaliação da atividade da doença e da dor pelo paciente, escores do índice de incapacidade [HAQ - Health Assessment Questionnaire] e valores de PCR [proteína C reativa] em mg/dl) melhoraram em 241,3,4 ou 26 semanas2, quando comparados ao placebo. No estudo III, estas melhoras foram mantidas ao longo de 52 semanas.3
Além disto, as taxas de respostas ACR foram mantidas na maioria dos pacientes seguidos na fase de extensão aberta do estudo III.114/207 pacientes continuaram com HUMIRA® (adalimumabe) 40mg SC a cada 2 semanas por 60 meses. Destes, 65%, 58% e 35% apresentaram respostas ACR 20/50/70, respectivamente, no mês 60.6
Nos estudos I-V, os pacientes tratados com HUMIRA® (adalimumabe) atingiram melhores respostas ACR 20 e 50, quando comparados ao placebo, de forma estatisticamente significante, após 1 ou 2 semanas após o início do tratamento.1-5
No estudo V, o tratamento combinado de HUMIRA® (adalimumabe) com metotrexato, em pacientes com artrite reumatóide inicial, levou a respostas ACR maiores e mais rápidas do que as monoterapias com HUMIRA® (adalimumabe) ou metotrexato, na semana 52, mantidas na semana 104 (Tabela B).
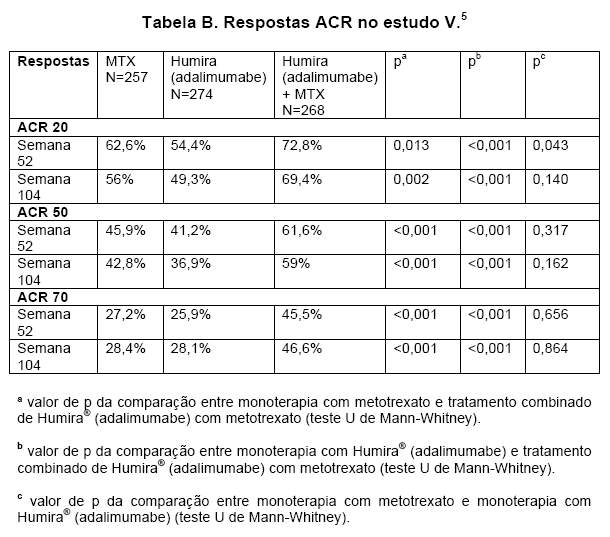
Na semana 52, 42,9% dos pacientes que receberam o tratamento combinado de HUMIRA® (adalimumabe) com metotrexato atingiram remissão clínica (DAS 28 < 2,6), comparados a 20,9% dos pacientes que receberam monoterapia com metotrexato e 23,4% dos que receberam monoterapia com HUMIRA® (adalimumabe). O tratamento combinado de HUMIRA® (adalimumabe) com metotrexato foi superior às monoterapias com metotrexato e HUMIRA® (adalimumabe) (ambos p < 0,001) em atingir baixa atividade da doença em pacientes com artrite reumatóide recentemente diagnosticada de moderada a grave. A resposta entre ambas monoterapias foi semelhante (p=0,447).5
Progressão radiográfica
No estudo III, no qual os pacientes tratados com HUMIRA® (adalimumabe) apresentaram uma duração média da artrite reumatóide de aproximadamente 11 anos, o dano articular estrutural foi avaliado radiograficamente e expresso por meio da mudança no escore total de Sharp modificado e seus componentes (escores de erosão e de alargamento dos espaços articulares). Os pacientes tratados com HUMIRA® (adalimumabe) e metotrexato apresentaram significativamente menos progressão radiográfica do que os pacientes tratados apenas com metotrexato, após 6 e 12 meses (Tabela C).3
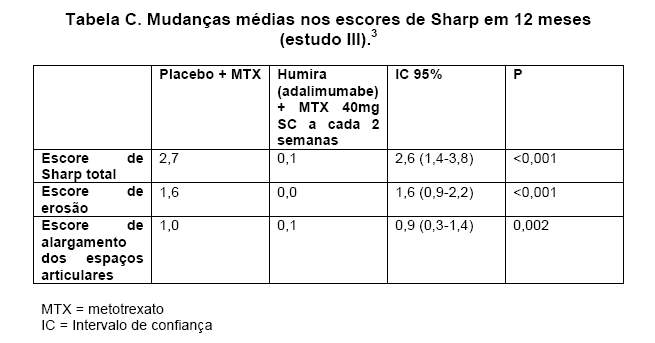
Dados da fase de extensão indicaram que a redução na taxa de progressão do dano estrutural é mantido por 60 meses em um subgrupo de pacientes. 113/207 pacientes originalmente tratados com HUMIRA® (adalimumabe) 40mg SC a cada 2 semanas foram avaliados após 5 anos. Destes, 66 pacientes não mostraram nenhuma progressão do dano estrutural, definida por mudança no escore total de Sharp de zero ou menos.6
No estudo V, o dano estrutural foi avaliado radiograficamente e também expresso por meio das mudanças no escore total de Sharp modificado e seus componentes, de acordo com a Tabela D.
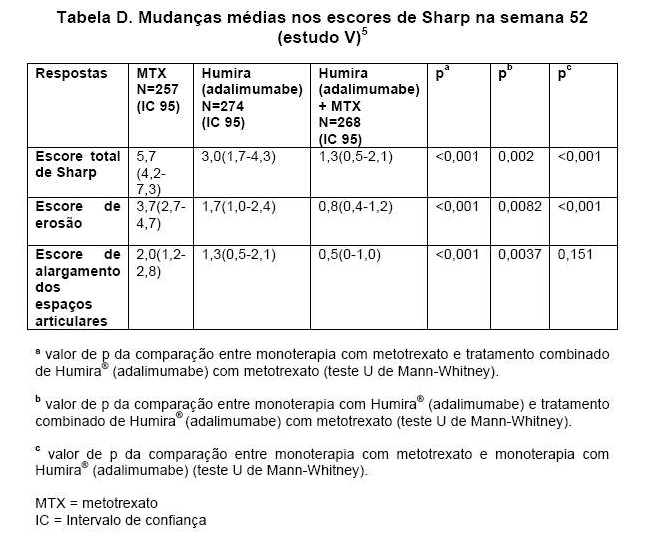
Após 52 e 104 semanas de tratamento, a porcentagem de pacientes sem progressão (mudança no escore total de Sharp modificado < 0,5) foi significativamente maior no grupo de tratamento combinado de HUMIRA® (adalimumabe) mais metotrexato (63,8% e 61,2%, respectivamente), quando comparado ao grupo que recebeu monoterapia com HUMIRA® (adalimumabe) (50,7%, p < 0,002, e 44,5%, p < 0,001, respectivamente) e monoterapia com metotrexato (37,4% e 33,5%), respectivamente, ambos p < 0,001).5
Qualidade de vida e função física
Qualidade de vida e função física foram avaliados pelo HAQ (Health Assessment Questionnaire), em todos os estudos de HUMIRA® (adalimumabe), com placebo como comparador, sendo uma medida de desfecho co-primária no estudo III. Todos os grupos tratados com HUMIRA® (adalimumabe) apresentaram melhora significativamente maior que o placebo no índice de incapacidade do HAQ, após 6 meses, o mesmo acontecendo no estudo III após 52 semanas. Nestes estudos, uma melhora do componente físico do Short Form 36 (SF-36) também suporta estes achados. No estudo V, a melhora do índice de incapacidade do HAQ e do componente físico do SF-36 foi significativamente maior para o grupo tratado com HUMIRA® (adalimumabe) e metotrexato, quando comparada aos grupos tratados com monoterapia com HUMIRA® (adalimumabe) e metotrexato (p < 0,001).1-4
Uma diminuição significativa da fadiga, medida pelo escore FACIT (Functional Assessment of Chronic Illness Therapy) foi observada nos estudos I, III e IV, onde tal instrumento foi usado.1,3,5 No estudo III, a melhora da função física foi mantida por até 60 meses da fase de extensão aberta. A qualidade de vida foi medida até a semana 156 (36 meses) e a melhora foi mantida por este período.3
- Artrite Psoriásica
HUMIRA® (adalimumabe) 40mg SC a cada duas semanas, foi avaliado em pacientes com artrite psoriásica moderada a grave em 2 estudos controlados por placebo. No estudo I, foram observados 313 pacientes adultos com resposta inadequada a anti-inflamatórios não-esteroidais (AINES), por 24 semanas.7 No estudo 2, 100 pacientes com resposta inadequada a DMCDs foram observados por 12 semanas.8 Os pacientes de ambos os estudos puderam entrar em uma fase aberta, onde todos receberam HUMIRA® (adalimumabe) 40mg SC a cada 2 semanas, por até 144 semanas.9,10 As respostas ACR no estudo I foram semelhantes com e sem tratamento concomitante com metotrexato (aproximadamente 50% dos pacientes foram tratados concomitantemente com metotrexato) (Tabela E).
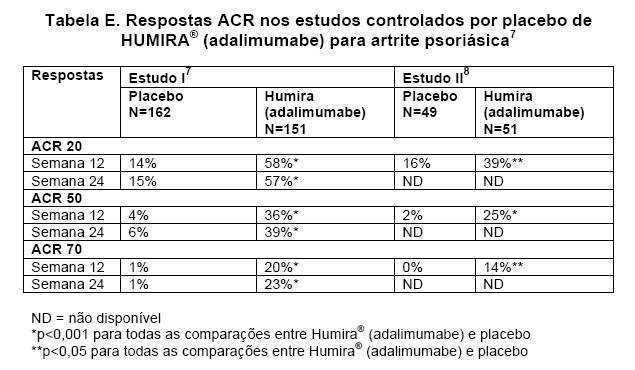
As respostas ACR foram mantidas na fase de extensão aberta por até 104
semanas.9
As mudanças radiográficas também foram avaliadas nos estudos de artrite psoriásica. Radiografias de mãos, punhos e pés foram obtidas no início do estudo e nas semanas 24 (fase duplo-cega do estudo I)7 e semana 48 (fase aberta).10 Um escore de Sharp modificado (mTSS), que incluiu as articulações interfalangianas distais, foi usado para medir a progressão radiográfica. HUMIRA® (adalimumabe) reduziu a taxa de progressão dodano articular periférico, quando comparado com o placebo (mudança média do mTSS = 0,8 ±2,42 no grupo placebo na semana 24 comparado a 0,1±1,95 no grupo tratado com HUMIRA® (adalimumabe) na semana 48, p < 0,001).9,10. Nos pacientes tratados com HUMIRA® (adalimumabe) sem progressão radiográfica do início do estudo até a semana 48 (n=102), 84% continuaram a demonstrar ausência de progressão por até 144 semanas de tratamento.9,10
Os pacientes tratados com HUMIRA® (adalimumabe) demonstraram melhora significativa na função física, avaliada pelo HAQ e pelo SF-36 comparados aos pacientes que receberam placebo, na semana 24.7 A melhora da função física continuou durante a fase de extensão aberta até a semana 136.9
- Espondilite Anquilosante
HUMIRA® (adalimumabe) 40mg SC a cada duas semanas foi avaliado 2 estudos duplo-cegos, placebo-controlados, de 24 semanas, em pacientes com espondilite anquilosante ativa, sem resposta adequada ao tratamento convencional.11-13 O período cego foi seguido de uma fase de extensão aberta, na qual os pacientes receberam apenas HUMIRA® (adalimumabe) 40mg SC a cada 2 semanas.14
No estudo I, com 315 pacientes, os resultados apresentaram melhora significativa dos sinais e sintomas da espondilite anquilosante nos pacientes tratados com HUMIRA® (adalimumabe), quando comparados aos tratados com placebo. Uma resposta significativa foi observada na semana 2, e mantida ao longo de 24 semanas (Tabela F).11
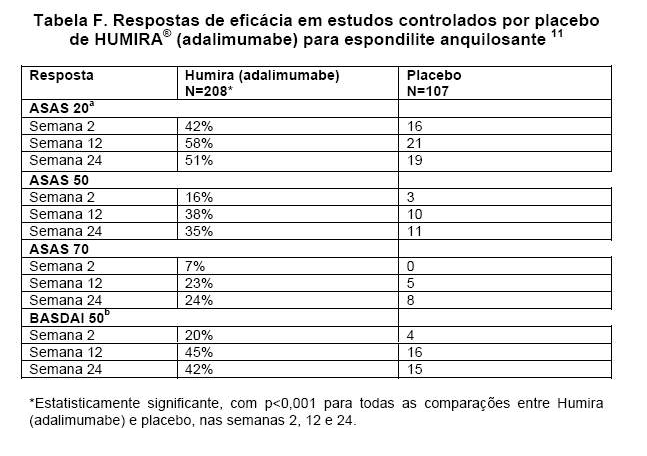
A melhora nas respostas ASAS e nos escores BASDAI foi mantida por até 2 anos.14
Pacientes tratados com HUMIRA® (adalimumabe) apresentaram melhora significativamente maior nos escores de dor, fadiga e rigidez,15 e nos escores de qualidade de vida (SF-36 e ASQoL - Questionário de Qualidade de Vida para Espondilite Anquilosante), quando comparados aos que receberam placebo, na semana 24.16
Tendências semelhantes (nem todas estatisticamente significantes) foram observadas no estudo II, realizado com 82 pacientes adultos com espondilite anquilosante ativa.12,13
- Doença de Crohn
A segurança e eficácia de HUMIRA® (adalimumabe) foram avaliadas em mais de 1400 pacientes com doença de Crohn (DC) ativa, moderada a grave (Crohn's Disease Activity Index (CDAI) ≥ 220 e ≤ 450) em estudos duplo-cegos, randomizados, controlados por placebo. Nestes estudos foi permitido o uso concomitante de doses estáveis de aminossalicilatos, corticosteróides e/ou agentes imunomoduladores. A indução de remissão clínica (definida como CDAI < 150) foi avaliada em dois estudos, Estudo I18 de DC (M02-403) e Estudo II19 de DC (M04-691). No Estudo I18 de DC, 299 pacientes virgens de antagonistas de TNF foram randomizados para um de quatro grupos de tratamento: placebo nas semanas 0 e 2, 160 mg de HUMIRA® (adalimumabe) na semana 0 e 80 mg na semana 2, 80 mg na semana 0 e 40 mg na semana 2, e 40 mg na semana 0 e 20 mg na semana 2. No Estudo II19 de DC, 325 pacientes que tinham perdido resposta ou eram intolerantes ao infliximabe foram randomizados para receber ou 160 mg HUMIRA® (adalimumabe) na semana 0 e 80 mg na semana 2 ou placebo nas semanas 0 e 2. Em ambos estudos os resultados clínicos foram avaliados na semana 4.
Uma maior porcentagem de pacientes tratados com 160/80 mg de HUMIRA® (adalimumabe) alcançou a indução de remissão clínica, em comparação com o placebo na semana 4, independentemente de os pacientes serem virgens de tratamento com bloqueadores de TNF (Estudo 18 de DC), ou terem perdido resposta ou terem sido intolerantes ao infliximabe (Estudo II19 de DC) - Tabela G.
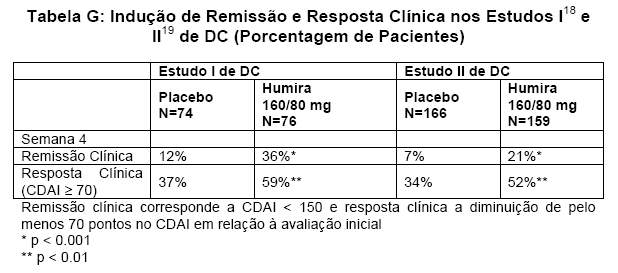
A manutenção da remissão clínica foi avaliada no Estudo III20 de DC (M02404). No Estudo III20 de DC, 854 pacientes receberam de forma aberta 80 mg de HUMIRA® (adalimumabe) na semana 0 e 40 mg na semana 2. Na semana 4 os pacientes foram randomizados para receber 40 mg em semanas alternadas, 40 mg todas as semanas, ou placebo com uma duração total do estudo de 56 semanas. Pacientes com resposta clinica (CR-70 = diminuição do CDAI ≥ 70) na semana 4 foram estratificados e analisados separadamente daqueles sem resposta clínica na semana 4.
No Estudo III20 de DC (CHARM), na semana 4, 58% (499/854) dos pacientes apresentavam resposta clínica e foram avaliados na análise primária. Os índices de manutenção da remissão e de resposta clínica estão representados na Tabela H. Os índices de remissão clínica permaneceram relativamente constantes independentemente de uma exposição prévia a um antagonista de TNF.

Nos Estudos I18 e II19 de DC, foi observada melhora estatisticamente significante na pontuação total do questionário específico para a doença inflamatória intestinal (IBDQ) alcançada na semana 4 nos pacientes randomizados para HUMIRA® (adalimumabe) 80/40 mg e 160/80 mg comparada a placebo. A melhora também foi vista nas semanas 26 e 56 no Estudo III de DC entre os grupos de tratamento adalimumabe comparados com o grupo placebo. No Estudo III, houve também uma diminuição estatisticamente significante de hospitalização e cirurgias relacionadas à doença quando comparada com o placebo na Semana 5621.
No Estudo I18, 117/276 pacientes com DC e 272/777 pacientes do Estudo II19 e III20 foram acompanhados por pelo menos 3 anos em terapia aberta com HUMIRA® (adalimumabe). Respectivamente 88 (75,2%) e 189 (69,5%) pacientes, continuaram com remissão clínica. A resposta clínica foi mantida em 107 (91,5%) e 248 (91,2%) pacientes, respectivamente. Os 117/854 pacientes (a partir de estudo DC III) apresentaram fístulas drenadas tanto na seleção como no baseline. Para a avaliação da cicatrização da fístula, os dados de ambas as doses de adalimumabe utilizados no estudo foram agrupados. A proporção de pacientes com cicatrização da fístula na semana 26 foi estatística e significativamente maior em pacientes tratados com adalimumabe [21/70 (30,0%)] em comparação com placebo [6/47 (12,8%)]. A cicatrização completa da fístula foi mantida até a Semana 56 em 23/70 (32,9%) pacientes no grupo com adalimumabe e 6/47 (12,8%) no grupo placebo.
Um estudo endoscópico (M05-769) que envolveu 135 pacientes, indicou um efeito de HUMIRA® (adalimumabe) na cicatrização da mucosa. 27,4% dos pacientes tratados com HUMIRA® (adalimumabe) tinham cicatrização da mucosa na semana 12 comparados com 13,1% dos pacientes-placebo (p=0,056), e 24,2% dos pacientes tratados com HUMIRA® (adalimumabe) na semana 52 contra 0% dos pacientes-placebo (p < 0,001) 22.
- Psoríase
A segurança e eficácia de HUMIRA® (adalimumabe) foram avaliadas em estudos duplo-cegos, randomizados, realizados em pacientes adultos com psoríase crônica em placas (envolvimento ≥ 10% BSA e Psoriasis Area and Severity Index (PASI) ≥ 12 ou ≥ 10) que eram candidatos a terapia sistêmica ou fototerapia.
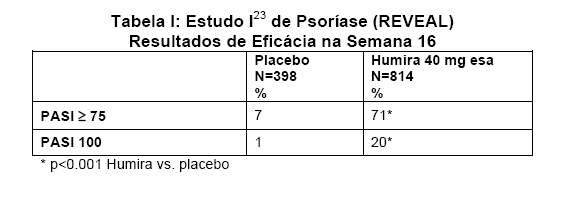
O Estudo I23 de Psoríase (M03-656) avaliou 1212 pacientes durante três períodos de tratamento. No período A, os pacientes receberam placebo ou HUMIRA ® (adalimumabe) na dose inicial de 80 mg seguida por 40 mg em semanas alternadas começando na semana 1, após a dose inicial. Após 16 semanas de terapia, os pacientes que alcançaram pelo menos uma resposta PASI 75 (melhora da pontuação PASI de pelo menos 75% em relação à avaliação inicial), entraram no período B e receberam de forma aberta 40 mg de HUMIRA® (adalimumabe) em semanas alternadas. Os pacientes que mantiveram resposta PASI ≥ 75 na semana 33 e que haviam sido originariamente randomizados para terapia ativa no Período A, foram novamente randomizados no Período C para receber 40 mg HUMIRA® (adalimumabe) em semanas alternadas ou placebo por mais 19 semanas. Considerando os três grupos de tratamento, a pontuação PASI média, na avaliação inicial, foi de 18.9 e a Avaliação Médica Global (Physician's Global Assessment -PGA) inicial variou de "moderada" (53% dos indivíduos incluídos), a "grave" (41%) e a "muito grave" (6%).
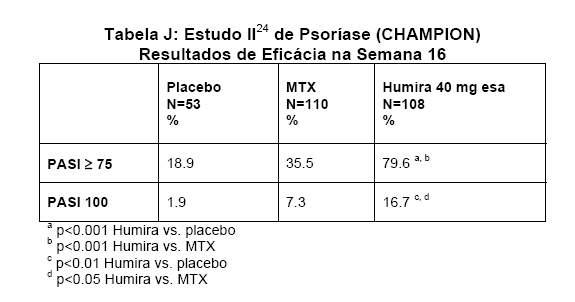
O Estudo II24 de Psoríase (M04-716) comparou a eficácia e segurança de HUMIRA® (adalimumabe) com metotrexato (MTX) e placebo em 271 pacientes. Os pacientes receberam placebo, uma dose inicial de MTX de 7.5 mg e, posteriormente, a dose era aumentada até a semana 12, com a dose máxima de 25 mg ou uma dose inicial de 80 mg de HUMIRA® (adalimumabe) seguida por 40 mg em semanas alternadas (iniciando uma semana após a dose inicial) durante 16 semanas. Não havia dados disponíveis comparando HUMIRA® (adalimumabe) e MTX durante 16 semanas de terapia. Os pacientes recebendo MTX que atingissem uma resposta PASI ³ 50 na semana 8 e/ou 12 não recebiam futuros aumentos de dose. Considerando os três grupos de tratamento, a pontuação PASI média, na avaliação inicial, foi de 19.7 e a Avaliação Médica Global (Physician's Global Assessment - PGA) inicial variou de "moderada" (48% dos indivíduos incluídos), a "grave" (46%) e a "muito grave" (6%).
Pacientes dos Estudos de Psoríase de Fase II e III foram eleitos a participar de um estudo clínico de extensão aberto (M03-658), onde HUMIRA® (adalimumabe) foi administrado por pelo menos mais 108 semanas.
Nos Estudo I23 e II24 de Psoríase, o desfecho primário foi a proporção de pacientes que atingiram uma resposta PASI 75 na semana 16, em relação à avaliação inicial (ver Tabelas I e J).
Um total de 233 de pacientes que atingiram a resposta PASI 75 na semana 16 e na semana 33 receberam continuamente HUMIRA® (adalimumabe) por 52 semanas no Estudo I de Psoríase e continuaram com a terapia em um estudo de extensão aberto. Após administração adicional por mais 108 semanas (no total de 160 semanas), 74,7% dos pacientes atingiram a resposta PASI 75 e 59,0% dos pacientes atingiram a Avaliação Médica Global (Physician's Global Assessment -PGA) com resposta mínima ou nenhuma. Já no Estudo II de Psoríase, dos 94 pacientes, 58,1% atingiram a resposta PASI 75 e 46,2% atingiram a Avaliação Médica Global (Physician's Global Assessment -PGA) com resposta mínima ou sem psoríase.
Um total de 347 estáveis e que responderam ao tratamento participaram de uma avaliação de retirada e retratamento em um estudo de extensão aberto. O tempo médio de recidiva (para PGA "moderado" ou pior) foi de aproximadamente 5 meses. Nenhum destes paciente relatou efeito rebote durante o período de retirada. Um total de 76,5% (218/285) dos pacientes que iniciaram o período de retratamento obtiveram uma resposta da PGA "sem psoríase" ou "mínima", após 16 semanas de retratamento, independentemente de recaída durante a retirada (69,1% [123/178] e 88,8% [95/107] para pacientes que recaíram e que não recaíram durante o período de suspensão, respectivamente).
Na semana 16 foram observadas melhoras estatisticamente significantes no Dermatology Life Quality Index (DLQI), em relação aos valores basais quando comparadas com placebo (Estudos I25 e II26) e MTX (Estudo II26). No Estudo I25 também foram evidenciadas melhoras no componente físico e mental da pontuação do SF-36 de forma estatisticamente significante, quando comparado ao placebo.
- Artrite Idiopática Juvenil Poliarticular
A segurança e eficácia de Humira foram avaliados em um estudo17 multicêntrico, randomizado, duplo-cego, de grupos paralelos, em 171 crianças (de 4 a 17 anos de idade) com artrite idiopática juvenil poliarticular (AIJ). No fase aberta introdutória (OL LI), os pacientes foram divididos em 2 grupos, os tratados com MTX (metotrexato) e os não tratados com MTX. Os pacientes que estavam no grupo dos não tratados com MTX, eram pacientes que nunca tinham recebido MTX ou que haviam suspendido o seu uso por pelo menos 2 semanas antes da administração da droga do estudo. Os pacientes mantiveram as doses regulares de AINEs e/ou prednisona ( < 0,2 mg/Kg/ dia ou 10 mg/dia no máximo). Na fase OL LI, todos os pacientes receberam 24 mg/m2 até no máximo 40 mg de Humira, a cada 14 dias por 16 semanas. A distribuição dos pacientes por idade e dose mínima, média e máxima recebida durante a fase OL LI está descrita na tabela K.

Pacientes que demonstraram uma resposta pediátrica ACR 30 na 16° semana foram elegíveis para serem randomizados para a fase duplo-cego e receberam Humira 24 mg/ m2 até o máximo de 40 mg ou placebo, a cada 14 dias por um período adicional de 32 semanas ou até o agravamento da doença. O critério para o agravamento da doença foi definido como uma piora > 30% em relação à avaliação inicial em > 3 de 6 critérios principais do ACR pediátrico, > 2 articulações activas, e melhora > 30% em não mais que 1 dos 6 critérios. Após 32 semanas ou até o agravamento da doença, os pacientes foram eleitos para se inscreverem na fase de extensão aberta.
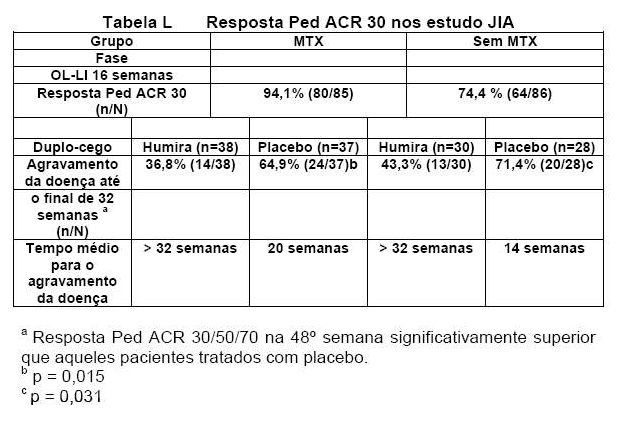
Entre aqueles que responderam até a 16° semana (n = 144), a resposta pediátrica ACR 30/50/90 foi mantida por até seis anos na fase OLE em pacientes que receberam Humira ao longo do estudo. No geral, 19 pacientes foram tratados por seis anos ou mais, sendo 11 dos 19 pacientes estando no grupo de faixa etária de 04 a 12 anos e os oito restantes, no grupo de faixa etária entre 13 e 17 anos.
As respostas gerais foram geralmente melhores e menos pacientes desenvolveram anticorpos quando tratados com a combinação de Humira e MTX comparados com Humira isoladamente. Considerando estes resultados, Humira é recomendado para o uso em combinação com MTX e para o uso como monoterapia em pacientes cujo o uso de MTX não é apropriado.
Referências Bibliográficas
REUMATOLOGIA
1. Weinblatt ME, Keystone EC, Furst DE, et al. Adalimumab, a fully human anti- tumor necrosis factor monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35-45.
2. van de Putte LB, Atkins C, Malaise M, et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann Rheum Dis. 2004;63(5):508- 16.
3. Keystone EC, Kavanaugh AF, Sharp JT, et al. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: a randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004;50(5):1400-11.
4. Furst DE, Schiff MH, Fleischmann RM, et al. Adalimumab, a fully human anti tumor necrosis factor-alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis). J Rheumatol. 2003;30(12):2563-71.
5. Breedveld FC, Weisman MH, Kavanaugh AF, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26-37.
6. Keystone EC, et al. Inhibition of radiographic progression in patients with longstanding rheumatoid arthritis treated with adalimumab plus methotrexate for 5 Years. Ann Rheum Dis. 2007;66(Suppl II):176.
7. Mease PJ, Gladman DD, Ritchlin CT, et al. A. Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52(10):3279-89.
8. Genovese MC, Mease PJ, Thomson GT, et al. Safety and efficacy of adalimumab in treatment of patients with psoriatic arthritis who had failed disease modifying antirheumatic drug therapy. J Rheumatol. 2007;34(5):1040-50.
9. Mease PJ, Ory P, Sharp JT, et al. Adalimumab for long-term treatment of psoriatic arthritis: two-year data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann Rheum Dis. 2008 Aug 6. [Epub ahead of print].
10. Gladman DD, Mease PJ, Ritchlin CT, et al. Adalimumab for long-term treatment of psoriatic arthritis: forty-eight week data from the adalimumab effectiveness in psoriatic arthritis trial. Arthritis Rheum. 2007;56(2):476-88.
11. van der Heijde D, Kivitz A, Schiff MH, et al. Efficacy and safety of adalimumab in patients with ankylosing spondylitis: Results of a multicenter, randomized, doubleblind, placebo-controlled trial. Arthritis Rheum. 2006;54(7):2136-46.
12. Lambert RG, Salonen D, Rahman P, et al. Adalimumab significantly reduces both spinal and sacroiliac joint inflammation in patients with ankylosing spondylitis: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2007;56(12):4005-14.
13. Maksymowych WP, Rahman P, Shojania K, et al. Beneficial effects of adalimumab on biomarkers reflecting structural damage in patients with ankylosing spondylitis. J Rheumatol. 2008;35(10):2030-7.
14. van der Heidje D, et al. Adalimumab effectiveness for the treatment of ankylosing spondylitis is maintained for up to 2 years: long-term results from the ATLAS trial. Ann Rheum Dis. Publicado online em 13/08/2008. doi:10.1136/ard.2007.087270
15. Revicki DA, Luo MP, Wordsworth P, et al. Adalimumab reduces pain, fatigue, and stiffness in patients with ankylosing spondylitis: results from the adalimumab trial evaluating long-term safety and efficacy for ankylosing spondylitis (ATLAS). J Rheumatol. 2008;35(7):1346-53.
16. Davis JC Jr, Revicki D, van der Heijde DM, et al. Health-related quality of life outcomes in patients with active ankylosing spondylitis treated with adalimumab: results from a randomized controlled study. Arthritis Rheum. 2007;57(6):1050-7.
17. Lovel DJ, Ruperto N, Goodman S, Reiff A, Jung L, Jarosova K, et al. Adalimumab with or without Methotrexate in Juvenile Rheumatoid Arthritis. N Engl J Med 2008;359:810-20
DOENÇA DE CROHN
18. Hanauer SB, Sandborn WJ, Rutgeerts P, et al. Human anti-tumor necrosis factor monoclonal antibody (adalimumab) in Crohn's disease: the CLASSIC-I trial. Gastroenterology. 2006;130(2):323-33.
19. Sandborn WJ, Rutgeerts P, Enns R, et al. Adalimumab induction therapy for Crohn disease previously treated with infliximab: a randomized trial. Ann Intern Med. 2007;146(12):829-38.
20. Colombel JF, Sandborn WJ, Rutgeerts P, et al. Adalimumab for maintenance of clinical response and remission in patients with Crohn's disease: the CHARM trial. Gastroenterology. 2007;132(1):52-65.
21. Feagan BG, Panaccione R, J Sandborn WJ, et al. Effects of Adalimumab Therapy on Incidence of Hospitalization and Surgery in Crohn's Disease: Results From the CHARM Study. Gastroenterology. 2008;135:1493-9.
22. Rutgeerts P, D'Haens GR, Van Assche GA, et al. Adalimumab Induces and Maintains Mucosal Healing in Patients with Moderate to Severe Ileocolonic Crohn's Disease - First Results of the Extend Trial. Gastroenterology. 2009;136 (5 Suppl 1):A-116.
PSORÍASE
23. Menter A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: A randomized, controlled phase III trial. J Am Acad Dermatol. 2008;58(1):106-15.
24. Saurat JH, Stingl G, Dubertret L, et al. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs.placebo in patients with psoriasis (CHAMPION). Br J Dermatol. 2008;158(3):558-66.
25. Revicki D, Willian MK, Saurat JH, et al. Impact of adalimumab treatment on health-related quality of life and other patient-reported outcomes: results from a 16week randomized controlled trial in patients with moderate to severe plaque psoriasis. Br J Dermatol. 2008;158(3):549-57.
26. Revicki DA, Willian MK, Menter A, et al..Impact of adalimumab treatment on patient-reported outcomes: results from a Phase III clinical trial in patients with moderate to severe plaque psoriasis. J Dermatolog Treat. 2007;18(6):341-50.
Caract. farmacológicas.
HUMIRA® (adalimumabe) é um anticorpo monoclonal recombinante da imunoglobulina humana (IgG1) contendo apenas sequências humanas de peptídeos. HUMIRA® (adalimumabe) foi desenvolvido a partir de técnica utilizando um fago contendo regiões variáveis de cadeias leves e pesadas totalmente humanas, o que confere especificidade ao fator de necrose tumoral (TNF), e sequências de cadeias pesadas e de cadeias leves capa (k) de IgG1 humana. HUMIRA® (adalimumabe) liga-se com alta afinidade e alta especificidade ao fator de necrose tumoral alfa (TNF-alfa), mas não à linfotoxina (TNF-beta). O adalimumabe é produzido por tecnologia de DNA recombinante em sistema de expressão de células de mamíferos. Consiste de 1330 aminoácidos e apresenta um peso molecular de aproximadamente 148 quilodaltons.
HUMIRA® (adalimumabe) é um medicamento de uso crônico e, portanto, o tempo estimado para início da ação terapêutica não é relevante. Considerando a monoterapia com dosagem de 40mg, as concentrações séricas mínimas duas semanas após a primeira dose são de 2,9 mg/mL, valor que excede a EC50 (1 mg/mL), sugerindo que as concentrações farmacológicas são atingidas após a primeira dose.
Farmacologia clínica
Mecanismo de ação
O adalimumabe liga-se especificamente ao TNF, neutralizando sua função biológica através do bloqueio de sua interação com os receptores de superfície de TNF (p55 e p75) presentes na superfície celular. O TNF é uma citocina de ocorrência natural, envolvida nas respostas inflamatórias e imunes normais. Níveis elevados de TNF são encontrados no líquido sinovial de pacientes com artrite reumatóide,, incluindo artrite idiopática juvenil poliarticular, artrite psoriásica e espondilite anquilosante, desempenhando um papel importante tanto na inflamação patológica quanto na destruição da articulação, características destas doenças. Níveis elevados de TNF também são encontrados nas placas psoriásicas. Nestas placas, o tratamento com HUMIRA® (adalimumabe) pode reduzir a espessura da epiderme e infiltração de células inflamatórias. A relação entre estas atividades farmacodinâmicas e o mecanismo de ação de HUMIRA® (adalimumabe) é desconhecida. O adalimumabe também modula respostas biológicas induzidas ou reguladas pelo TNF, incluindo alterações nos níveis de moléculas de adesão, responsáveis pela migração de leucócitos (ELAM-1, VCAM-1 e ICAM-1 com IC50 de 1-2 X 10-10 M).
Farmacodinâmica
Após o tratamento com HUMIRA® (adalimumabe), observou-se uma rápida diminuição em relação aos níveis basais dos marcadores de fase aguda da inflamação (proteína C-reativa, velocidade de hemossedimentação, e citocinas séricas como a IL-6) em pacientes com artrite reumatóide. Uma diminuição nos níveis de proteína C-reativa também foi observada em pacientes com artrite idiopática juvenil poliarticular ou doença de Crohn, bem como uma significativa redução na expressão de TNF e nos marcadores inflamatórios como o antígeno leucócitário humano (HLA-DR) e a mieloperoxidase (MPO) no cólon de pacientes com doença de Crohn. Observou-se também uma diminuição dos níveis séricos de metaloproteinases matriciais (MMP-1 e MMP-3), responsáveis pela remodelação tissular e pela destruição da cartilagem. Os pac
ientes com artrite reumatóide, artrite psoriásica e espondilite anquilosante frequentemente apresentam anemia leve a moderada e redução da contagem de linfócitos, bem como aumento do número de neutrófilos e de plaquetas. Os pacientes tratados com HUMIRA® (adalimumabe) geralmente apresentam melhora nesses parâmetros hematológicos de inflamação crônica.
Estimativas da EC50 do adalimumabe variando de 0,8 a 1,4 mcg/mL foram obtidas através da modelagem farmacocinética / farmacodinâmica de contagem de articulações inchadas, contagem de articulações doloridas e da resposta ACR 20 dos pacientes que participam dos estudos fase II e III.
Farmacocinética
Absorção: após administração de dose única de 40 mg de HUMIRA® (adalimumabe) por via subcutânea (SC) em 59 indivíduos adultos saudáveis, observou-se absorção e distribuição lenta do adalimumabe, com pico de concentração plasmática médio em cerca de cinco dias após a administração. A biodisponibilidade média absoluta do adalimumabe estimada a partir de três estudos após dose única subcutânea de 40 mg foi de 64%.
Distribuição e eliminação: a farmacocinética de dose única do adalimumabe foi determinada em vários estudos com doses intravenosas (IV) variando entre 0,25 a 10 mg/kg. O volume de distribuição variou de 4,7 a 6,0 litros, indicando que o adalimumabe se distribui de modo similar nos líquidos vascular e extravascular. O adalimumabe é eliminado lentamente, com depuração tipicamente abaixo de 12mL/h. A meia-vida média da fase terminal foi de aproximadamente duas semanas, variando de 10 a 20 dias. A depuração e a meia-vida permaneceram relativamente inalteradas no intervalo de doses estudado, e a meia-vida terminal foi semelhante após administração intravenosa e subcutânea. As concentrações do adalimumabe no líquido sinovial de vários pacientes com artrite reumatóide (AR) variou de 31 a 96% da concentração plasmática.
Farmacocinética no estado de equilíbrio: o acúmulo do adalimumabe foi previsível com base na meia-vida após administração SC de 40 mg de HUMIRA® (adalimumabe) a cada 14 dias em pacientes com AR atingindo, em média, concentrações mínimas no estado de equilíbrio de aproximadamente 5 mcg/mL (sem administração concomitante de metotrexato) e de 8 a 9 mcg/mL (com administração concomitante de metotrexato). Os níveis plasmáticos de vale do adalimumabe no estado de equilíbrio aumentaram quase proporcionalmente com a dose após administração SC de 20, 40 e 80 mg semanalmente ou a cada 14 dias. Em estudos de longa duração com administração por mais de dois anos, não houve evidência de alterações na depuração em função do tempo.
Em pacientes com psoríase, a concentração média no estado de equilíbrio é 5 mcg/mL durante o tratamento monoterápico de adalimumabe 40 mg a cada duas semanas.
A análise populacional de farmacocinética, com dados de mais de 1200 pacientes, revelou que a administração concomitante de metotrexato apresentou um efeito intrínseco sobre a depuração aparente do adalimumabe (ver Interações Medicamentosas). Conforme esperado, houve uma tendência a aumento da depuração aparente do adalimumabe com o aumento do peso corporal e com a presença de anticorpos anti-adalimumabe.
Foram identificados também outros fatores de menor importância: foi prevista maior depuração aparente em pacientes recebendo doses menores do que a dose recomendada, e em pacientes com altas concentrações de fator reumatóide ou de proteína C-reativa. Esses fatores não parecem ser clinicamente relevantes.
Em pacientes com doença de Crohn, com a dose inicial de 160 mg via SC na Semana 0, seguida de 80 mg na Semana 2, o adalimumabe atingiu nível sérico médio de aproximadamente 12 mcg/mL na Semana 2 e Semana 4. Nível médio do estado de equilíbrio (steady-state) de aproximadamente 7 mcg/mL foi observado na Semana 24 e Semana 56 em pacientes com doença de Crohn após receberem a dose de manutenção de 40 mg de HUMIRA® (adalimumabe) a cada 14 dias.
Populações especiais
Geriatria: a idade parece exercer um efeito mínimo sobre a depuração aparente do adalimumabe. Em análise populacional, a depuração média (ajustada segundo peso corpóreo), em pacientes de 40 a 65 anos (n=850) e ≥ 65 anos (n=287) foi de 0,33 e 0,30mL/h/kg, respectivamente. Pediatria: após a administração subcutânea de 24 mg/m2 (até no máximo de 40 mg) a cada 14 dias a pacientes com artrite idiopática juvenil poliarticular (AIJ) a média no estado de equilíbrio estável (valores medidos para da 20° semana à 48° semana) da concentração sérica de adalimumabe foi 5,6 ± 5,6 mg/mL (102% CV) na monoterapia com HUMIRA® (adalimumabe) e 10,9 ± 5,2 mg/mL (47,7% CV) com metotrexato concomitante. A média no estado de equilíbrio estável da concentração sérica de adalimumabe para sujeitos pesando < 30 kg recebendo 20 mg de HUMIRA® (adalimumabe) via subcutânea a cada 14 dias como monoterapia ou com metotrexato concomitante foi 6,8 mg/mL e 10,9 mg/mL, respectivamente. A média no estado de equilíbrio estável da concentração sérica de adalimumabe para sujeitos pesando > 30 kg recebendo 40 mg de HUMIRA® (adalimumabe) via subcutânea a cada 14 dias como monoterapia ou com metotrexato concomitante foi 6,6 mg/mL e 8,1 mg/mL, respectivamente.
Sexo: não foram observadas diferenças farmacocinéticas relacionadas ao sexo do paciente após correção para o peso corporal.
Etnia: não são esperadas diferenças na depuração de imunoglobulinas entre indivíduos de diferentes etnias. Com base em dados de pacientes não caucasianos, não foram observadas diferenças farmacocinéticas importantes para o adalimumabe.
Insuficiência renal e hepática: nenhum dado de farmacocinética está disponível em pacientes com insuficiência renal ou hepática.
Pacientes com artrite reumatóide: a farmacocinética foi a mesma em voluntários sadios e em portadores de artrite reumatóide.
Contraindicações.
HUMIRA® (adalimumabe) é contraindicado para o uso em pacientes com conhecida hipersensibilidade ao adalimumabe ou quaisquer componentes da fórmula do produto.
Advertências e precauções.
Infecções: foram relatadas infecções graves devido a bactérias, micobactérias, infecções fúngicas invasivas (histoplasmose disseminada ou extrapulmonar, aspergilose, coccidioidomicose), virais, parasitária ou outras infecções oportunistas que foram relatadas por pacientes que receberam agentes inibidores de TNF. Sepsis, raros casos de tuberculose, candidíase, listeriose e pneumocistose, também foram relatados em pacientes tratados com antagonistas do TNF, inclusive com HUMIRA® (adalimumabe). Outras infecções graves como pneumonia, pielonefrite, artrite séptica e septicemia foram relatadas em estudos clínicos. Hospitalização ou resultados fatais foram reportados associados com as infecções. Muitas das infecções graves ocorreram em pacientes tratados concomitantemente com imunossupressores, que, além da própria doença subjacente, podem predispor a infecções.
O tratamento com HUMIRA® (adalimumabe) não deve ser iniciado em pacientes com infecções ativas, incluindo infecções crônicas ou localizadas, até que as infecções estejam controladas. Em pacientes que foram expostos a tuberculose e pacientes que viajaram para áreas de alto risco de tuberculose ou de micoses endêmicas, como histoplasmose, coccidioidomicoses, ou blastomicoses, os riscos e benefícios do tratamento com HUMIRA® devem ser considerados antes de iniciar a terapia (ver Outras Infecções Oportunistas).
Assim como com outros antagonistas do TNF, antes, durante e após o tratamento com HUMIRA® (adalimumabe), os pacientes devem ser monitorados cuidadosamente quanto à presença de infecções, incluindo tuberculose.
Pacientes que desenvolverem nova infecção durante o tratamento com HUMIRA® (adalimumabe) devem ser monitorados cuidadosamente e submetidos a uma avaliação diagnóstica completa. A administração de HUMIRA® (adalimumabe) deve ser interrompida se o paciente desenvolver infecção grave ou sépsis, e deve ser iniciada uma terapia apropriada com antimicrobiano ou antifúngico até que a infecção esteja controlada.
Recomenda-se cautela quando se decidir utilizar HUMIRA® (adalimumabe) em pacientes com histórico de infecções de repetição ou com doença de base que possa predispor o paciente a infecções.
Tuberculose: assim como observado com outros antagonistas de TNF, foram relatados casos de tuberculose (frequentemente disseminada ou extrapulmonar) associados ao HUMIRA® (adalimumabe) durante os estudos clínicos. Embora os casos tenham ocorrido com todos os níveis de doses, a incidência de reativação de tuberculose foi particularmente aumentada com doses de HUMIRA® (adalimumabe) maiores do que as doses recomendadas. Antes de iniciar o tratamento com HUMIRA® (adalimumabe) todos os pacientes devem ser avaliados quanto à presença de tuberculose ativa ou inativa (latente). Esta avaliação deve incluir anamnese detalhada, com identificação de exposição prévia a pacientes com tuberculose ativa e tratamento prévio e/ou atual com imunossupressores. Testes de triagem apropriados (ex.: radiografia de tórax e teste tuberculínico - PPD) devem ser realizados. O tratamento de infecção por tuberculose latente deve ser iniciado anteriormente à terapia com HUMIRA® (adalimumabe). Quando o teste tuberculínico for realizado para detecção de tuberculose, a enduração do tamanho de 5 mm ou maior deve ser considerada positiva, mesmo se previamente vacinados com bacilo de Calmette-Guérin (BCG).
A possibilidade de tuberculose latente não detectada deve ser considerada especialmente em pacientes que imigraram de/ou viajaram a países com uma alta prevalência de tuberculose ou que tiveram contato próximo com pessoas que apresentem tuberculose ativa.
Se a tuberculose ativa for diagnosticada, o tratamento com HUMIRA® (adalimumabe) não deve ser iniciado.
Se for diagnosticada tuberculose latente, antes que o tratamento com HUMIRA® (adalimumabe) seja iniciado, deve-se iniciar a profilaxia antituberculose apropriada. Terapia antituberculose prévia ao tratamento com HUMIRA® (adalimumabe) também deve ser considerada em pacientes que apresentaram resultado negativo no teste para tuberculose latente, mas possuem fatores de risco para infecção por TB. A decisão de iniciar uma terapia antituberculose nestes pacientes somente deve ser tomada após avaliação do risco de infecção por tuberculose latente e do risco da terapia antituberculose. Se necessário, deve-se consultar um médico especialista em tratamento da tuberculose.
O tratamento antituberculose de pacientes com tuberculose latente reduz o risco da reativação em pacientes recebendo HUMIRA® (adalimumabe). No entanto, alguns pacientes, cujas triagens para tuberculose latente foram negativas, e que receberam HUMIRA® (adalimumabe), desenvolveram tuberculose ativa durante o tratamento com agentes bloqueadores TNF. Pacientes que utilizam HUMIRA® (adalimumabe) devem ser monitorados para sinais e sintomas de tuberculose ativa, particularmente porque os testes para infecção por tuberculose ativa podem dar resultados falso-negativos. O risco de resultado falso-negativo para o teste tuberculínico deve ser considerado especialmente em pacientes que estão severamente debilitados ou imunocomprometidos.
Os pacientes devem ser instruídos a procurar atendimento médico se apresentarem sinais/sintomas sugestivos de tuberculose (ex.: tosse persistente, perda de peso, febre baixa).
Outras Infecções Oportunistas: infecções oportunistas, incluindo infecções fúngicas invasivas, foram observadas em pacientes que receberam HUMIRA® (adalimumabe). Estas infecções não são consistentemente reconhecidas em pacientes que usam bloqueadores de TNF e isto leva ao atraso no início do tratamento apropriado, algumas vezes resultando em fatalidades.
Pacientes que usam bloqueadores de TNF são mais suscetíveis a infecções fúngicas sérias, tais como histoplasmose, coccidioidomicose, blastomicose, aspergilose, candidíase e outras infecções oportunistas. Aqueles que desenvolvem febre, mal-estar, perda de peso, sudorese, tosse, dispnéia e/ou infiltrados pulmonares, ou outras doenças sistêmicas sérias, com ou sem choque concomitante, devem prontamente procurar o médico para uma avaliação diagnóstica.
Para pacientes que residem ou viajam para regiões onde micoses são endêmicas, deve-se suspeitar de infecções fúngicas invasivas se eles desenvolverem sinais e sintomas de possível infecção fúngica sistêmica. Histoplasmose e outras infecções fúngicas invasivas são um risco para os pacientes e por esta razão o médico deve considerar o tratamento antifúngico empírico até que o patógeno seja identificado. O teste antígeno e anticorpo para histoplasmose pode ser negativo em alguns pacientes com infecção ativa. Quando possível, a decisão de administrar uma terapia antifúngica empírica nestes pacientes deve ser feita em conjunto com um médico especialista no diagnóstico e tratamento de infecções fúngicas invasivas e deve levar em consideração tanto o risco de uma infecção fúngica grave, como o risco da terapia antifúngica. Pacientes que desenvolvem uma infecção fúngica grave são também advertidos a interromper o uso de bloqueadores de TNF até que a infecção seja controlada.
Reativação da Hepatite B: o uso de inibidores de TNF foi associado à reativação do vírus da hepatite B (HBV) em pacientes portadores crônicos deste vírus. Em alguns casos, a ocorrência da reativação do HBV concomitantemente à terapia com inibidores de TNF foi fatal. A maioria destes relatos ocorreu em pacientes que receberam outros medicamentos supressores do sistema imunológico, que também podem contribuir para a reativação do HBV. Pacientes com risco de contrair infecção por HBV devem ser avaliados antes do início da terapia com inibidores de TNF. Deve-se ter cautela ao administrar inibidores de TNF em pacientes portadores do vírus da hepatite B. Pacientes portadores do HBV e que requerem terapia com inibidores de TNF devem ser cuidadosamente monitorados quanto a sinais e sintomas da infecção ativa por HBV durante a terapia e por alguns meses seguidos após o término da mesma. Não estão disponíveis dados de segurança e eficácia de pacientes portadores de HBV recebendo terapia antiviral concomitantemente à terapia com inibidores de TNF para prevenir a reativação do HBV. Em pacientes que desenvolvam a reativação do HBV, o uso de HUMIRA® (adalimumabe) deve ser suspenso e terapia antiviral adequada deve ser iniciada.
Eventos neurológicos: os antagonistas de TNF, incluindo HUMIRA® (adalimumabe), foram associados, em raros casos, com exacerbação de sintomas e/ou evidência radiológica de doença desmielinizante do sistema nervoso central, incluindo esclerose múltipla e doença desmielinizante periférica incluindo Síndrome de Guillain Barré. Deve-se ter cautela ao considerar o uso de HUMIRA® (adalimumabe) em pacientes com doenças desmielinizantes do sistema nervoso periférico ou central, de início recente ou pré-existentes.
Malignidades: em partes controladas de estudos clínicos com antagonistas de TNF, foi observado maior número de casos de linfoma entre os pacientes que receberam antagonistas de TNF do que entre os pacientes controle. O tamanho do grupo de controle e a duração limitada das partes controladas dos estudos não permitem chegar a conclusões concretas. Além disso, há maior risco de linfoma em pacientes com artrite reumatóide com doença inflamatória de longa duração, altamente ativa, o que complica a estimativa do risco. Durante os estudos abertos de longa duração com HUMIRA® (adalimumabe), a taxa total de malignidades foi similar ao que seria esperado para idade, sexo e raça na população geral. Com o conhecimento atual, um risco possível para o desenvolvimento dos linfomas ou outras malignidades nos pacientes tratados com um antagonista de TNF não pode ser excluído.
Malignidades, algumas fatais, foram relatadas entre crianças e adolescentes que foram tratados com agentes bloqueadores de TNF. Aproximadamente metade dos casos foram linfomas, incluindo linfomas de Hodgkin e não-Hodgkin. Os outros casos representam uma variedade de diferentes malignidades e incluem malignidades raras normalmente associadas à imunossupressão. As malignidades ocorreram em média em 30 meses de terapia. A maioria dos pacientes estava tomando concomitantemente imunossupressores.Os casos foram relatados após a comercialização e derivam de uma variedade de fontes incluindo registros e relatos espontâneos de pós-comercialização. Casos muito raros de linfoma hepatoesplênico de células T, um raro e agressivo linfoma que é frequentemente fatal, foram identificados em pacientes recebendo adalimumabe. A maioria dos pacientes foi previamente tratada com infliximabe, ou recebeu terapia concomitante com azatioprina ou 6-mercaptopurina para doença inflamatória intestinal. A associação causal entre este tipo de linfoma e adalimumabe não está clara.
Nenhum estudo foi conduzido incluindo pacientes com histórico de malignidade ou pacientes que continuaram o tratamento após o diagnóstico de malignidade durante o tratamento com HUMIRA® (adalimumabe). Assim, deve-se ter cautela adicional ao se considerar o tratamento com HUMIRA® (adalimumabe) nestes pacientes.
Todos os pacientes, em particular pacientes com histórico médico de extensa terapia imunossupressora ou pacientes com psoríase com histórico de tratamento com PUVA, devem ser examinados para a presença de câncer de pele não-melanoma antes e durante o tratamento com HUMIRA® (adalimumabe).
Casos de leucemia aguda e crônica foram relatados em associação ao uso de agentes bloqueadores de TNF na pós-comercialização em atrite reumatóide e outras indicações. Pacientes com artrite reumatóide podem estar expostos a um risco maior (até 2 vezes) do que a população geral para o desenvolvimento de leucemia, mesmo na ausência de terapia de bloqueador de TNF.
Alergia: durante estudos clínicos, reações alérgicas graves associadas ao uso de HUMIRA® (adalimumabe) foram raramente observadas. Em estudos de pós-comercialização, reações alérgicas graves, incluindo reação anafilática, foram raramente relatadas após o uso de HUMIRA® (adalimumabe). Se uma reação anafilática ou outra reação alérgica grave ocorrer, a administração de HUMIRA® (adalimumabe) deve ser interrompida imediatamente e deve-se iniciar o tratamento apropriado.
A tampa da agulha da seringa contém borracha natural (látex). Pacientes sensíveis ao látex podem ter reações alérgicas graves.
Eventos hematológicos: raros relatos de pancitopenia, incluindo anemia aplástica, foram observados com agentes bloqueadores de TNF. Eventos adversos do sistema hematológico, incluindo citopenia clinicamente significativa (por exemplo, trombocitopenia, leucopenia), foram relatados com HUMIRA® (adalimumabe). A relação causal destes relatos com HUMIRA® (adalimumabe) é incerta. Todos os pacientes devem ser orientados a procurar atenção médica imediatamente caso desenvolvam os sinais e sintomas sugestivos de discrasias sanguíneas (por exemplo, febre persistente, contusões, sangramento, palidez) durante o uso de HUMIRA® (adalimumabe).
A descontinuação da terapia com HUMIRA® (adalimumabe) deve ser considerada em pacientes com anormalidades hematológicas significativas confirmadas.
Uso com anacinra: infecções graves foram observadas em estudos clínicos com o uso simultâneo de anacinra e outro antagonista de TNF, etanercepte, sem benefício clínico adicional comparado com etanercepte isoladamente. Considerando-se a natureza dos eventos adversos observados na terapia combinada de etanercepte e anacinra, toxicidades similares podem também resultar da combinação de anacinra e outros antagonistas de TNF. Portanto, a combinação de adalimumabe e anacinra não é recomendada.
Uso com abatacepte: a administração concomitante de antagonistas de TNF e abatacepte tem sido associada a aumento do risco de infecções, incluindo infecções sérias, quando comparada a antagonistas de TNF isolados. Esta combinação não tem demonstrado aumento do benefício clínico. Logo, a combinação de antagonistas de TNF e abatacepte não é recomendada.
Imunossupressão: em um estudo de 64 pacientes com artrite reumatóide, tratados com HUMIRA® (adalimumabe), não houve evidência de diminuição da hipersensibilidade do tipo retardada, diminuição dos níveis de imunoglobulinas ou alterações na contagem de células T, B e NK, monócitos/macrófagos e neutrófilos.
Imunizações: em um estudo placebo-controlado, duplo-cego, randomizado, com 226 pacientes adultos com artrite reumatóide, tratados com HUMIRA® (adalimumabe), foram avaliadas as respostas dos anticorpos a vacinas concomitantes de pneumococcos e influenza. Níveis protetores de anticorpos contra antígenos pneumocócicos foram atingidos em 86% dos pacientes no grupo de HUMIRA® (adalimumabe) comparados a 82% no grupo placebo. Um total de 37% dos indivíduos tratados com HUMIRA® (adalimumabe) e de 40% dos indivíduos em placebo atingiram aumento de pelo menos 2 vezes em pelo menos 3 dos 5 antígenos pneumocócicos. No mesmo estudo, 98% dos pacientes no grupo de HUMIRA® (adalimumabe) e 95% daqueles no grupo placebo atingiram níveis protetores de anticorpos contra antígenos do influenza. Um total de 52% dos indivíduos tratados com HUMIRA® (adalimumabe) e de 63% dos indivíduos em placebo alcançaram aumento de pelo menos 4 vezes em pelo menos 2 dos 3 antígenos do influenza.
Se possível, recomenda-se que os pacientes com artrite idiopática juvenil poliarticular estejam com todas as vacinas em dia de acordo com as recomendações locais, antes de iniciar o tratamento com HUMIRA® (adalimumabe).Os pacientes em tratamento com HUMIRA® (adalimumabe) podem receber vacinações simultâneas, com exceção das vacinas vivas. Não há dados disponíveis quanto à transmissão secundária de infecções por vacinas vivas em pacientes recebendo HUMIRA® (adalimumabe).
Insuficiência cardíaca congestiva: HUMIRA® (adalimumabe) não foi formalmente estudado em pacientes com insuficiência cardíaca congestiva (ICC). Entretanto, em estudos clínicos com outro antagonista de TNF, uma taxa mais elevada de eventos adversos sérios relacionados a ICC foi relatada, incluindo piora da ICC e novo episódio de ICC. Casos de piora da ICC também foram relatados em pacientes recebendo HUMIRA® (adalimumabe). Médicos devem ter cautela quando escolherem a terapia com HUMIRA® (adalimumabe) para pacientes que têm insuficiência cardíaca, e monitorá-los cuidadosamente.
Processos autoimunes: o tratamento com HUMIRA® (adalimumabe) pode resultar na formação de anticorpos autoimunes.
O impacto de um longo tratamento com HUMIRA® (adalimumabe) no desenvolvimento de doenças auto-imunes é desconhecido.
Se um paciente desenvolver sintomas que sugiram síndrome Lúpus símile durante o tratamento com HUMIRA® (adalimumabe), o tratamento deve ser descontinuado (ver Reações Adversas).
- Cuidados e advertências para populações especiais:
Uso em idosos: a frequência de infecções graves entre pacientes com mais de 65 anos de idade tratados com HUMIRA® (adalimumabe) foi maior do que para os sujeitos com menos de 65 anos de idade. Do número total de sujeitos no estudo clínico de HUMIRA® (adalimumabe),10,8% tinham 65 anos de idade ou mais, enquanto cerca de 2,3% tinham 75 anos ou mais. Não foram observadas diferenças em termos de eficácia entre essa população e a de indivíduos mais jovens. Não é necessário ajuste de dose para esta população. Devido a uma maior incidência de infecções na população idosa geral, deve-se ter cautela quando do tratamento de pacientes idosos.
Uso pediátrico: HUMIRA® (adalimumabe) não foi estudado em crianças com menos de 4 anos de idade e existem dados limitados referentes ao tratamento com HUMIRA® (adalimumabe) em crianças com peso menor que 15 kg. A segurança e eficácia do medicamento em pacientes pediátricos não foi estabelecida para outras indicações além da artrite idiopática juvenil poliarticular.
Uso na gravidez: foi realizado um estudo de toxicidade embrio-fetal perinatal em macacos Cynomolgus com doses de até 100 mg/kg (que implica em AUC 266 vezes maior que a produzida em humanos pela dose semanal de 40 mg SC com metotrexato, ou 373 vezes com dose de 40 mg SC sem metotrexato). Os resultados não revelaram evidências de danos fetais decorrentes do adalimumabe. Não existem, porém, ensaios adequados e bem controlados em mulheres grávidas. Considerando que os estudos de reprodução e desenvolvimento em animais nem sempre podem prever a resposta humana, este medicamento só deve ser usado durante a gravidez quando, na opinião do médico, os benefícios potenciais claramente justificarem os possíveis riscos ao feto. Mulheres em idade reprodutiva devem ser advertidas a não engravidar durante o tratamento com HUMIRA® (adalimumabe).
Categoria de risco: B
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Trabalho de parto ou parto: não existem efeitos conhecidos de HUMIRA® (adalimumabe) sobre o trabalho de parto.
Uso na lactação: não se sabe se o adalimumabe é excretado no leite humano ou se é absorvido sistemicamente após ingestão. Considerando que vários medicamentos e imunoglobulinas são excretados no leite humano, e devido ao potencial de reações adversas do adalimumabe nas lactentes, recomenda-se decidir entre descontinuar o tratamento com HUMIRA® (adalimumabe) ou interromper o aleitamento, levando em conta a importância do medicamento para a mãe.
Insuficiência renal e hepática: não há dados disponíveis sobre o metabolismo do medicamento em pacientes com insuficiência renal ou hepática.
Dados de segurança pré-clínicos: os dados pré-clínicos não revelaram risco especial para humanos, com base em estudos de toxicidade de dose única, toxicidade de dose repetida, e genotoxicidade.
Carcinogenicidade, mutagenicidade e alterações na fertilidade: não foram realizados estudos experimentais de longo prazo para avaliar o potencial carcinogênico ou os efeitos do adalimumabe sobre a fertilidade. Não foram observados efeitos clastogênicos ou mutagênicos do adalimumabe nos testes em micronúcleos de camundongos in vivo, ou no teste de AMES com Salmonella e Escherichia coli.
Interações medicamentosas.
Metotrexato: quando HUMIRA® (adalimumabe) foi administrado a 21 pacientes sob terapia estável com metotrexato, não houve alteração estatisticamente significante no perfil da concentração plasmática de metotrexato. Em contraste, após dose única e dose múltipla, metotrexato reduziu a depuração aparente de adalimumabe para 29% e 44%, respectivamente. No entanto, os dados não sugerem a necessidade de ajuste de doses de nenhum dos dois medicamentos. Outras: não foram realizados estudos formais de farmacocinética entre HUMIRA® (adalimumabe) e outras substâncias. O uso concomitante de HUMIRA® (adalimumabe) e anacinra ou abatacepte não é recomendado. Vacinas vivas não devem ser administradas concomitantemente a HUMIRA® (adalimumabe). Nos estudos clínicos, não foram observadas interações quando HUMIRA® (adalimumabe) foi administrado concomitantemente a DMCDs (sulfassalazina, hidroxicloroquina, leflunomida e ouro parenteral), glicocorticóides, salicilatos, antiinflamatórios não esteroidais ou analgésicos.
Interação com testes laboratoriais: não são conhecidas interferências
entre HUMIRA® (adalimumabe) e testes laboratoriais.
Cuidados de armazenamento.
HUMIRA® (adalimumabe) deve ser mantido em sua embalagem original e armazenado entre 2 e 8°C (na geladeira). Não congelar.
Prazo de validade: se armazenado nas condições indicadas, o medicamento se manterá próprio para consumo pelo prazo de validade de 24 meses, a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após aberto, este medicamento deve ser utilizado imediatamente. A parte da solução não utilizada e todo o material utilizado para a injeção devem ser adequadamente descartados.
Características físicas e organolépticas:
HUMIRA® (adalimumabe) é fornecido sob a forma de solução estéril, livre de conservantes, para administração subcutânea. A solução de HUMIRA® (adalimumabe) é límpida e incolor, com um pH de 5,2.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Posologia e modo de usar.
HUMIRA® (adalimumabe) deve ser utilizado sob a orientação e supervisão de um médico ou profissional habilitado.
Os pacientes podem se autoaplicar se o médico considerar apropriado e sob orientação médica, conforme necessário, após treinamento adequado do paciente na técnica de injeção subcutânea. Os locais da autoaplicação localizam-se na coxa ou no abdômen. Os locais de injeção devem ser alternados a cada aplicação. As novas injeções nunca devem ser administradas em áreas onde a pele estiver sensível, ferida, avermelhada ou áspera.
Soluções injetáveis devem ser inspecionadas visualmente para verificar a presença de partículas ou alterações de coloração antes de serem administradas, sempre que o recipiente permitir. Se partículas ou descoloração forem observadas, o produto não deve ser utilizado. HUMIRA® (adalimumabe) não contém conservantes e, portanto, o material não utilizado que permanecer na seringa deve ser adequadamente descartado. HUMIRA® (adalimumabe) não deve ser misturado na mesma seringa com qualquer outro medicamento.
NOTA: a capa branca da agulha contém borracha (látex), a qual não deve ser manuseada por pessoas sensíveis à substância.
Instruções para preparo, manuseio e aplicação de HUMIRA® (adalimumabe) seringa pronta para uso:
1) Lave cuidadosamente suas mãos.
2) Coloque os seguintes itens sobre uma superfície limpa:
- uma seringa pronta para uso de HUMIRA® (adalimumabe) 40 mg solução injetável
- lenço umedecido em álcoo
3) Verifique o prazo de validade da seringa. Não use o produto se este estiver vencido.
4) Escolha o local da injeção: coxa ou abdômen.
5) Cada nova injeção deve ser dada ao menos a 3 cm de distância do local da última injeção.
6) Não aplique o medicamento em área onde a pele estiver avermelhada, lesionada ou áspera.
Isto pode indicar uma infecção.
7) Com o lenço umedecido em álcool, limpe o local da injeção fazendo movimentos circulares.
8) Após a limpeza, não toque na área até a injeção.
9) Remova a tampa da agulha da seringa, sendo cuidadoso para não tocar na agulha ou deixar que ela toque em qualquer superfície.
10) Com uma das mãos, levante gentilmente a área da pele limpa e segure firmemente.
11) Com a outra mão, segure a seringa a um ângulo de 45° em relação à pele.
12)Com um movimento curto e rápido, insira a agulha na pele.
13) Solte a pele e injete a solução da seringa - isto pode levar de 2 a 5 segundos até o completo esvaziamento da seringa.
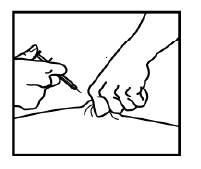
14) Quando a seringa estiver vazia, remova a agulha da pele, sendo cuidadoso para manter o mesmo ângulo com que a agulha foi inserida.
15) Usando uma gaze, pressione o local da injeção por 10 segundos. Um pequeno sangramento pode ocorrer. Não esfregue o local da injeção. Use um curativo adesivo, se você quiser.
16) Nunca reutilize a seringa. Nunca recoloque o protetor na agulha.
17) Após a injeção, descarte imediatamente a seringa conforme as instruções de seu médico, enfermeiro ou farmacêutico.
POSOLOGIA
HUMIRA® (adalimumabe) é um medicamento de uso crônico e a duração do tratamento será de acordo com cada paciente. O limite máximo diário de administração de HUMIRA® (adalimumabe) não foi determinado em humanos.
Artrite Reumatóide
A dose recomendada de HUMIRA® (adalimumabe) para pacientes adultos é de 40 mg de solução injetável, administrados em dose única por via subcutânea, a cada 14 dias.
O tratamento com metotrexato, glicocorticóides, salicilatos, antiinflamatórios não esteroidais, analgésicos ou outras drogas antirreumáticas modificadoras do curso da doença (DMCDs) pode ser mantido durante o tratamento com HUMIRA® (adalimumabe).
Alguns pacientes, não tratados concomitantemente com metotrexato, podem obter benefício adicional com o aumento da frequência da administração de HUMIRA® (adalimumabe) para 40 mg de solução injetável uma vez por semana.
Os dados clínicos disponíveis para artrite reumatóide sugerem que a resposta clínica normalmente é alcançada dentro de 12 semanas de tratamento. A continuação da terapia deve ser cuidadosamente reconsiderada se um paciente não responder ao tratamento dentro deste período.
Artrite Psoriásica
A dose recomendada de HUMIRA® (adalimumabe) para pacientes adultos
é de 40 mg de solução injetável, administrados em dose única por via subcutânea, a cada 14 dias.
O tratamento com metotrexato, glicocorticóides, salicilatos, antiinflamatórios não esteroidais, analgésicos ou outras drogas antirreumáticas modificadoras do curso da doença (DMCDs) pode ser mantido durante o tratamento com HUMIRA® (adalimumabe).
Espondilite Anquilosante
A dose recomendada de HUMIRA® (adalimumabe) para pacientes adultos é de 40 mg de solução injetável, administrados em dose única por via subcutânea, a cada 14 dias.
O tratamento com metotrexato, glicocorticóides, salicilatos, antiinflamatórios não esteroidais, analgésicos ou outros fármacos antirreumáticos modificadores do curso da doença (DMCDs) pode ser mantido durante o tratamento com HUMIRA® (adalimumabe).
Doença de Crohn
A dose recomendada de HUMIRA® (adalimumabe) para pacientes adultos com doença de Crohn é:
Início do tratamento - Semana 0: 160 mg por via subcutânea (a dose pode ser administrada em quatro injeções em um dia ou duas injeções por dia por dois dias consecutivos);
Semana 2: 80 mg por via subcutânea (a dose deve ser administrada em duas injeções no mesmo dia);
Manutenção do tratamento: a partir da Semana 4, 40 mg a cada 14 dias por via subcutânea.
O tratamento com corticosteróides, aminosalicilatos e/ou agentes imunomoduladores (6-mercaptopurina e azatioprina) pode ser mantido durante o tratamento com HUMIRA® (adalimumabe).
Alguns pacientes podem necessitar de um aumento na frequência da dose de manutenção de HUMIRA® (adalimumabe) para 40 mg de solução injetável por semana.
Os pacientes que não responderem ao tratamento até a Semana 4 podem continuar com a manutenção do tratamento até a Semana 12. Se não houver resposta neste período, a continuação da terapia deve ser cuidadosamente reconsiderada.
Durante a manutenção do tratamento, corticosteróides podem ser reduzidos em conformidade às diretrizes de prática clínica.
Psoríase
A posologia recomendada de HUMIRA® (adalimumabe) para pacientes adultos é de uma dose inicial de 80 mg por via subcutânea (duas injeções), seguida de doses de 40 mg de solução injetável por via subcutânea administradas em semanas alternadas, começando na semana seguinte à dose inicial.
Caso o paciente não apresente resposta dentro de 16 semanas de tratamento, a terapia deve ser cuidadosamente reconsiderada.
Artrite idiopática juvenil poliarticular
No estudo clínico utilizado como suporte para esta indicação foi realizado usando uma dose por área de superfície do corpo, durante toda a fase controlada. Na parte aberta do estudo, a dosagem foi alterada para uma dose fixa de acordo com o corte do peso corporal.
A dose recomendada de HUMIRA® (adalimumabe) para pacientes com artrite idiopática juvenil poliarticular com idade superior a 13 anos é de 40 mg solução injetável, administrados em dose única por via subcutânea, a cada 14 dias.
Os dados disponíveis sugerem que a resposta clínica é geralmente alcançada com 12 semanas de tratamento. A continuação do tratamento deve ser cuidadosamente reconsiderada em pacientes que não responderam dentro deste período de tempo.
Reações adversas.
Reações adversas nos estudos clínicos
HUMIRA® (adalimumabe) foi estudado em 7552 pacientes em estudos principais, abertos e controlados por até 60 meses ou mais.
Os dados apresentados a seguir baseiam-se em estudos controlados, envolvendo 5029 pacientes recebendo HUMIRA® (adalimumabe) e 3035 pacientes recebendo placebo ou comparador ativo durante o período de controle.
A proporção de pacientes que interrompeu o tratamento devido a reações adversas, durante a parte duplo-cega e controlada dos estudos clínicos de HUMIRA® (adalimumabe), foi de 5,8% para os pacientes tratados com HUMIRA® (adalimumabe), e de 5,9% para os pacientes controle.
Aproximadamente 14% dos pacientes podem esperar algum tipo de reação no local da injeção, considerado um dos mais comuns eventos adversos com adalimumabe em estudos clínicos controlados.
As reações adversas pelo menos possivelmente relacionadas ao tratamento com HUMIRA® (adalimumabe) em estudos clínicos são apresentadas a seguir por órgão de sistema e por frequência (muito comum ≥1/10; comum ≥1/100 a < 1/10; incomum ≥1/1.000 a < 1/100; raro 1/10.000 a 1/1.000).
- Infestações e Infecções
Reação muito comum (≥1/10): infecções no trato respiratório (incluindo infecção do trato respiratório inferior e superior, pneumonia, sinusite, faringite, nasofaringite e pneumonia por herpes viral).
Reação comum (≥1/100 e < 1/10): infecções sistêmicas (incluindo sepsis, candidíase e influenza), infecções intestinais (incluindo gastroenterite viral), infecções de pele e tecidos moles (incluindo paroníquia, celulite, impetigo, fasciíte necrosante e herpes zoster), infecções de ouvido, infecções orais (incluindo herpes simples, herpes oral e infecção dentária), infecções do trato reprodutivo (incluindo infecção vulvo vaginal micótica), infecção do trato urinário (incluindo pielonefrite), infecções fúngicas.
Reaçã