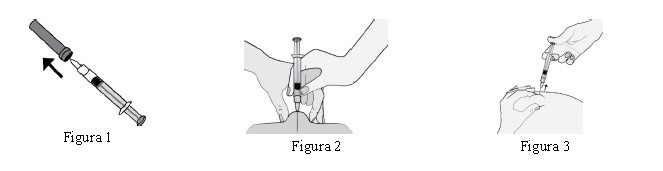
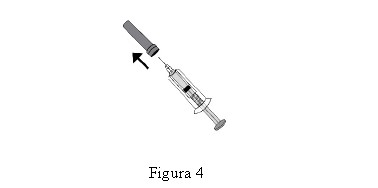
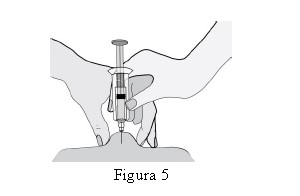
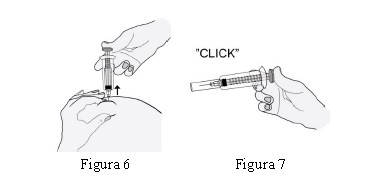
HEPTRIS
VIATRIS
enoxaparina sódica
Anticoagulante.
Apresentações.
- Solução injetável 20 mg/0,2 mL: embalagem com 2, 6 e 10 seringas preenchidas com e sem sistema de segurança.
- Solução injetável 40 mg/0,4 mL: embalagem com 2, 6 e 10 seringas preenchidas com e sem sistema de segurança.
- Solução injetável 60 mg/0,6 mL: embalagem com 2, 6 e 10 seringas preenchidas graduadas com e sem sistema de segurança.
- Solução injetável 80 mg/0,8 mL: embalagem com 2, 6 e 10 seringas preenchidas graduadas com e sem sistema de segurança.
- Solução injetável 100 mg/1,0 mL: embalagem com 2 seringas preenchidas graduadas com e sem sistema de segurança.
USO SUBCUTÂNEO OU INTRAVENOSO (a via de administração varia de acordo com a indicação do produto)
USO ADULTO
Composição.
Cada seringa preenchida de HEPTRIS® contém: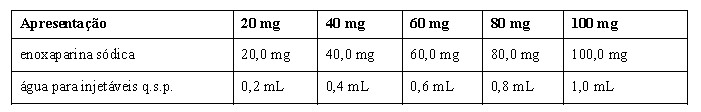
Informações técnicas.
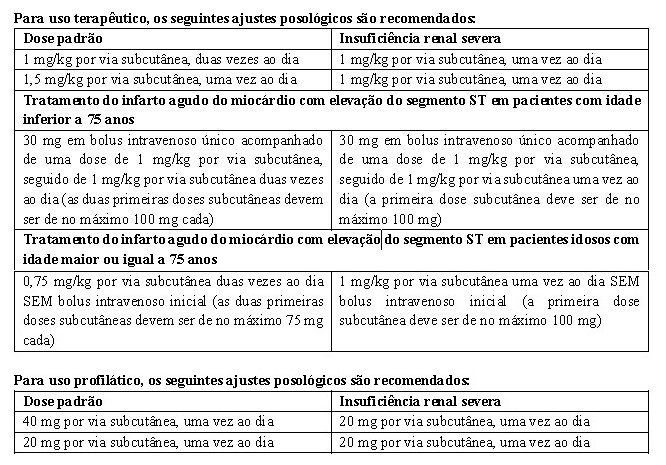
1. INDICAÇÕES
- Tratamento da trombose venosa profunda com ou sem embolismo pulmonar;
- Tratamento da angina instável e infarto do miocárdio sem elevação do segmento ST, administrado concomitantemente ao ácido acetilsalicílico;
- Tratamento de infarto agudo do miocárdio com elevação do segmento ST, incluindo pacientes a serem tratados clinicamente ou com subsequente intervenção coronariana percutânea;
- Profilaxia do tromboembolismo venoso, em particular aqueles associados à cirurgia ortopédica ou à cirurgia geral;
- Profilaxia do tromboembolismo venoso em pacientes acamados devido a doenças agudas incluindo insuficiência cardíaca, falência respiratória, infecções severas e doenças reumáticas;
- Prevenção da formação de trombo na circulação extracorpórea durante a hemodiálise.
2. RESULTADOS DE EFICÁCIA
HEPTRIS® é um medicamento biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre HEPTRIS® e o medicamento comparador Clexane.
Dados de CLEXANE
Cirurgia abdominal
Em um estudo duplo-cego em pacientes submetidos à cirurgia eletiva de tumores gastrointestinais, urológicos, ou do trato ginecológico, um total de 1116 pacientes foram incluídos e 1115 receberam profilaxia de TEV.
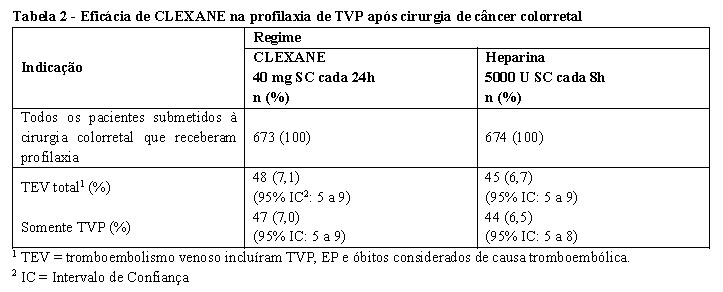
CLEXANE 40 mg SC, uma vez ao dia, começando 2h antes da cirurgia e continuado por um período de no
máximo 12 dias após a cirurgia, teve sua eficácia comparada a da heparina não fracionada (HNF) 5000 U SC a cada 8h na redução do risco de trombose venosa profunda (TVP). Os dados de eficácia são apresentados abaixo [ver tabela 1] (Bergqvist et al, 1997).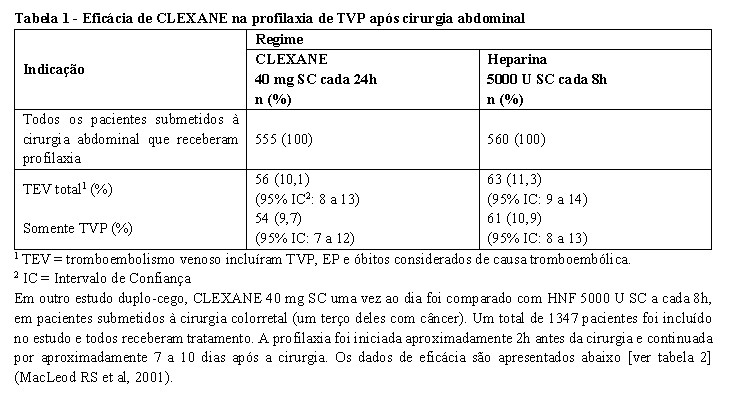
Cirurgia geral oncológica
Em estudo duplo cego randomizado multicêntrico conduzido com pacientes submetidos à cirurgia abdominal ou pélvica oncológica curativa, 332 pacientes receberam CLEXANE 40 mg uma vez ao dia, por 6 a 10 dias e após este período, foram randomizados para receber placebo ou manter a profilaxia com CLEXANE 40 mg uma vez ao dia por 19 a 21 dias adicionais, totalizando um período de tratamento de 25 a 31 dias. O desfecho primário de eficácia analisado foi a incidência de de tromboembolismo venoso diagnosticada à venografia entre os dias 25 e 31. Os resultados de eficácia são apresentados na tabela abaixo [ver tabela 3]. Todos os pacientes foram acompanhados por 3 meses. (Berqgvist, 2002).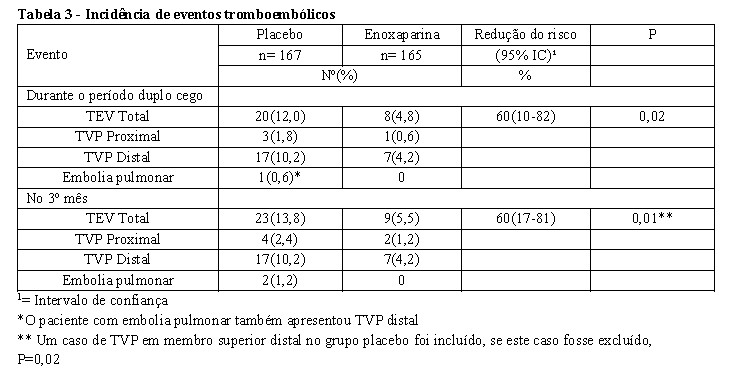
Artroplastia de quadril
Em um estudo duplo-cego, randomizado, CLEXANE 40 mg SC uma vez ao dia foi comparado com HNF 5000 U SC a cada 8h, após artroplastia total de quadril. A profilaxia foi iniciada 12h antes da cirurgia, no caso da enoxaparina, e 2h antes da cirurgia, no caso da heparina. Um total de 237 pacientes foram randomizados no estudo e receberam profilaxia. Os resultados de eficácia são mostrados na tabela abaixo [ver tabela 3] (Planes et al 1988).
Um estudo duplo-cego, multicêntrico, comparou três regimes de dose de CLEXANE em pacientes submetidos à artroplastia de quadril. Um total de 572 pacientes foram randomizados e 568 receberam a profilaxia proposta. Profilaxia com CLEXANE foi iniciada 2 dias após a cirurgia e continuou por 7 a 11 dias após a cirurgia. Os dados de eficácia são fornecidos abaixo [ver tabela 4] (Spiro et al, 1994).
Não houve diferença significativa entre os regimes de 30 mg cada 12h e 40 mg cada 24h.
Em um estudo de profilaxia estendida para pacientes submetidos à artroplastia de quadril, os pacientes receberam durante a internação, CLEXANE 40 mg SC iniciado 12h antes da cirurgia para prevenir TVP pós-operatória. Ao final do período perioperatório, todos os pacientes foram submetidos à venografia bilateral. Seguindo um desenho duplo-cego, todos os pacientes sem evidência de doença tromboembólica foram randomizados para um regime pós-alta de CLEXANE 40 mg (n = 90) por via SC, uma vez ao dia ou de placebo (n = 89) por 3 semanas. Nessa população de pacientes, a incidência de TVP durante a fase de profilaxia estendida foi significativamente mais baixa no grupo que recebeu CLEXANE comparado ao placebo. Os dados de eficácia são apresentados na tabela abaixo [ver tabela 5] (Planes et al 1996).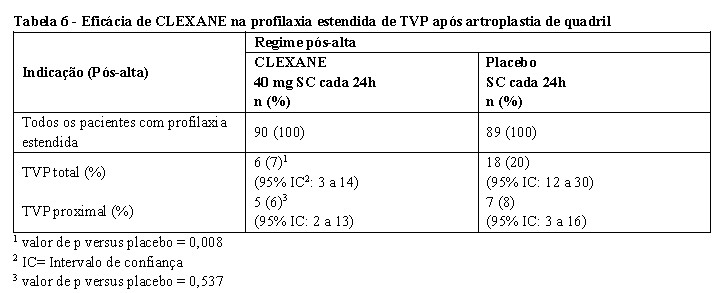
Em um segundo estudo, pacientes submetidos à artroplastia de quadril receberam durante a hospitalização CLEXANE 40 mg SC, iniciado 12h antes da cirurgia. Todos os pacientes foram examinados em busca de sinais e sintomas de doença tromboembólica. Pacientes sem qualquer sinal de TEV foram randomizados para um regime pós-alta com CLEXANE 40 mg SC uma vez ao dia (n = 131) ou placebo (n = 131) por 3 semanas. Um total de 262 pacientes foram randomizados nessa fase duplo-cega. De modo semelhante ao primeiro estudo, a incidência de TVP durante a profilaxia estendida foi significativamente menor com CLEXANE quando comparado ao placebo, com diferença estatisticamente significativa tanto na incidência TVP total (CLEXANE [16%] versus placebo 45 [34%]; p = 0,001) quanto na de TVP proximal (CLEXANE 8 [6%] versus placebo 28 [21%]; p = < 0,001) (Bergqvist et al, 1996).
Artroplastia de joelho
Um total de 132 pacientes foram randomizados no estudo e 131 receberam profilaxia. Após hemostasia, profilaxia foi iniciada 12 a 24h após a cirurgia e continuada por até 15 dias. A incidência de TVP total e proximal após cirurgia foi significativamente mais baixa no grupo que recebeu CLEXANE comparado ao placebo. Os dados de eficácia são mostrados abaixo [ver tabela 6] (Leclerc et al, 1992).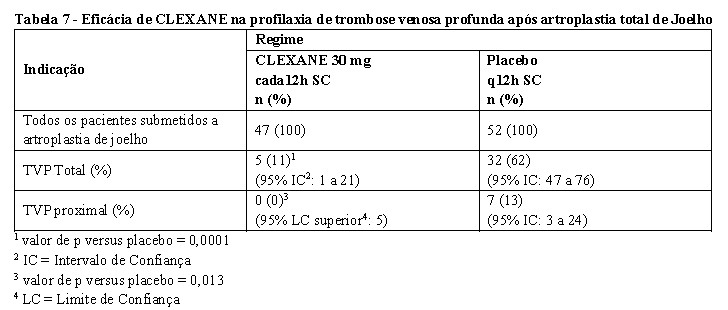
Ainda em artroplastia eletiva de joelho, outro estudo clínico aberto, de grupos paralelos, randomizado, comparou
CLEXANE 30 mg SC a cada 12h com heparina 5000 U SC a cada 8h. Um total de 453 pacientes foram randomizados e todos receberam profilaxia conforme o grupo designado. A profilaxia iniciou-se após a cirurgia e continuou até 14 dias. A incidência de trombose venosa profunda foi significativamente menor com CLEXANE, comparada com heparina (Cowell et al, 1995).
Profilaxia de tromboembolismo em pacientes clínicos com mobilidade reduzida durante doença aguda
Em um estudo multicêntrico, duplo-cego, de grupos paralelos, CLEXANE 20 mg ou 40 mg SC uma vez ao dia foi comparado com placebo na profilaxia de TVP em pacientes clínicos com mobilidade restrita durante uma doença aguda (definida como distância percorrida < 10 metros em tempo ≤ 3 dias). Esse estudo incluiu pacientes com insuficiência cardíaca (NYHA Classe funcional III ou IV); insuficiência respiratória aguda ou insuficiência respiratória crônica complicada (sem necessidade de suporte ventilatório): infecção aguda (exceto choque séptico) ou doença reumatológica aguda. Um total de 1102 pacientes foi incluído no estudo, e 1073 pacientes receberam profilaxia. A terapia foi continuada por até 14 dias (média de duração de 7 dias). Quando administrado numa dose de 40 mg SC 1x/dia, CLEXANE reduziu significativamente a incidência de TVP comparado ao placebo. Dados de eficácia são mostrados abaixo [ver tabela 7] (Samama et al, 1999).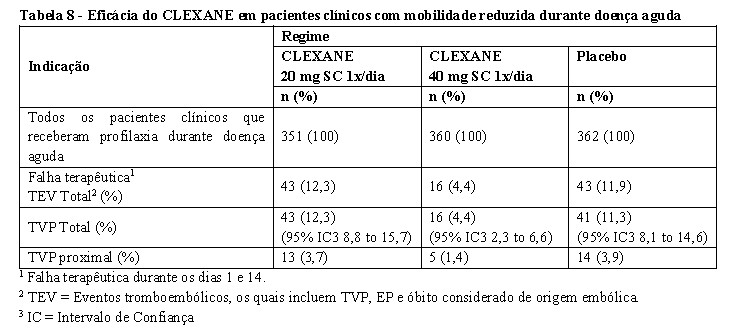
O tratamento profilático com CLEXANE 40 mg SC ao dia reduziu em 63% o risco de TEV. Em aproximadamente 3 meses após a inclusão, a incidência de tromboembolismo permaneceu significativamente mais baixa no grupo que recebeu CLEXANE 40 mg versus o grupo placebo.
Tratamento de trombose venosa profunda (TVP) com ou sem embolia pulmonar (EP)
Em um estudo multicêntrico, de grupos paralelos, 900 pacientes com TVP aguda de membro inferior associada ou não à embolia pulmonar foram randomizados para tratamento hospitalar com CLEXANE 1,5 mg/kg SC 1x/dia, CLEXANE 1 mg/kg SC cada 12h ou heparina em bolus (5000 UI) seguido de infusão contínua (administrada até atingir um TTPa de 55 a 85 segundos). Todos os pacientes receberam tratamento. Todos os pacientes também receberam varfarina sódica (dose ajustada de acordo com o TP para atingir um RNI (relação normatizada internacional) entre 2 e 3), a partir de 72h do início da terapia com CLEXANE ou HNF. CLEXANE ou HNF foram administrados por no mínimo 5 dias e até que o RNI desejado fosse atingido. Ambos os regimes de CLEXANE foram equivalentes à terapia com HNF em reduzir o risco de TEV recorrente. Os dados de eficácia são mostrados abaixo [ver tabela 8] (Merli et al, 2001).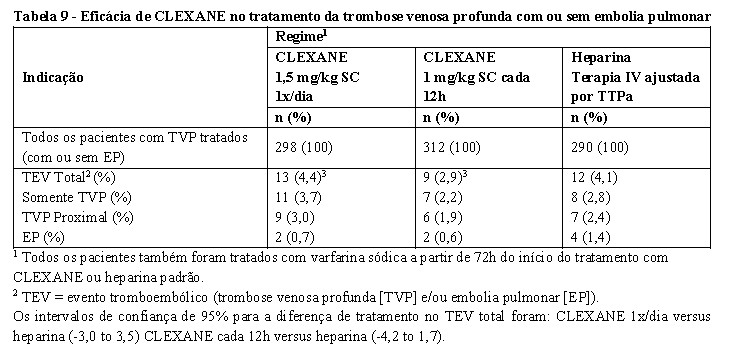
Hemodiálise
Em um estudo, pacientes com insuficiência renal terminal (n=36) fazendo hemodiálise 3 vezes por semana, receberam CLEXANE ou HNF e depois foram trocados para o outro tratamento, a cada 12 semanas. Enoxaparina 1 mg/kg foi administrada na forma de bolus dentro da linha arterial antes da diálise. Doses subsequentes de enoxaparina eram reduzidas para 0,2 mg/kg caso ocorresse sangramento. HNF 50 UI/kg foi administrada na forma de bolus dentro da linha arterial, seguida por uma dose de manutenção de 1000 UI/hora. As linhas e filtros de diálise ficaram significativamente mais limpas (sem coágulos) com CLEXANE em comparação com HNF (p < 0,001) (Saltissi et al, 1999).
Tratamento de angina instável e infarto do miocárdio (IM) sem elevação do segmento ST (Cohen et al, 1997)
Em um grande estudo multicêntrico, 3171 pacientes incluídos na fase aguda de angina instável ou IM sem elevação do segmento ST foram randomizados para receber, em associação com ácido acetilsalicílico (100 a 325 mg, uma vez ao dia), 1 mg/kg de enoxaparina sódica em injeção subcutânea a cada 12 horas, ou HNF por administração IV, ajustada com base no tempo de tromboplastina parcial ativada (TTPa). Os pacientes foram tratados em ambiente hospitalar por um período mínimo de 2 e máximo de 8 dias, até estabilização clínica, procedimentos de revascularização ou alta hospitalar. Os pacientes foram acompanhados por 30 dias. A enoxaparina sódica, em comparação à HNF, diminuiu significativamente a incidência de angina recorrente, IM e óbito, com redução do risco relativo de 16,2% no 14° dia, sustentado durante o período de 30 dias. Além disto, um número menor de pacientes do grupo tratado com enoxaparina sódica foi submetido à revascularização por angioplastia coronariana transluminal percutânea (ACTP) ou por enxerto de ponte arterial coronariana (15,8% de redução do risco relativo no 30° dia).
Tratamento do IM com elevação do segmento ST (Antman et al, 2006)
Em um grande estudo multicêntrico, 20479 pacientes diagnosticados com IM com elevação do segmento ST, elegíveis para receber terapia fibrinolítica, foram randomizados para receber: 1) enoxaparina sódica em bolus IV único de 30 mg acompanhado de 1 mg/kg por via SC, seguido de doses SC de 1 mg/kg a cada 12 horas; ou, 2) HNF por administração IV, ajustada com base no tempo de TTPa por 48 horas. Todos os pacientes também foram tratados com ácido acetilsalicílico por um período mínimo de 30 dias. A estratégia posológica de enoxaparina foi ajustada para pacientes acometidos por insuficiência renal severa e para pacientes idosos com idade maior ou igual a 75 anos. As injeções SC de enoxaparina foram administradas por um período máximo de 8 dias ou até que o paciente recebesse alta do hospital (considerando o que ocorresse primeiro).
Em um subgrupo deste mesmo estudo, 4716 pacientes foram submetidos à intervenção coronariana percutânea (ICP) recebendo suporte antitrombótico com fármaco do estudo de modo cego. Portanto, para pacientes que utilizaram a enoxaparina, a ICP foi realizada com enoxaparina (sem troca) utilizando-se o regime estabelecido em estudos prévios, ou seja, caso a última dose SC tenha sido administrada há menos de 8 horas antes de o balão ser inflado, não se administra dose adicional e caso a última dose subcutânea tenha sido administrada há mais de 8 horas antes de o balão ser inflado, administra-se uma dose adicional de 0,3 mg/kg através de bolus intravenoso.
A enoxaparina sódica quando comparada com a HNF reduziu significativamente a incidência do desfecho primário, uma combinação de morte por qualquer causa ou reinfarto do miocárdio nos primeiros 30 dias após a randomização [9,9% no grupo tratado com enoxaparina, comparado a 12,0% no grupo tratado com heparina não fracionada] com uma redução relativa do risco igual a 17% (P < 0,001).
Os benefícios do tratamento com enoxaparina, evidenciados por uma série de resultados de eficácia, surgiram em 48 horas, tempo no qual houve uma redução de 35% do risco relativo de reinfarto do miocárdio, quando comparado com o tratamento com HNF (P < 0,001).
O efeito benéfico da enoxaparina no desfecho primário foi consistente entre os subgrupos principais do estudo, incluindo idade, sexo, local do infarto, histórico de diabetes, histórico de infarto do miocárdio anterior, tipo do fibrinolítico administrado e tempo para tratamento com o fármaco em estudo.
Houve um benefício significativo do tratamento com enoxaparina, quando comparado com o tratamento com HNF, em pacientes submetidos à ICP dentro de 30 dias após a randomização (23% de redução do risco relativo) ou em pacientes tratados com terapia medicamentosa (15% de redução do risco relativo, P = 0,27 para interação).
A incidência do desfecho composto de morte, reinfarto do miocárdio ou hemorragia intracraniana (uma medida do benefício clínico líquido), considerando-se os 30 primeiros dias, foi significativamente menor (p < 0,0001) no grupo tratado com enoxaparina (10,1%) quando comparado com o grupo tratado com HNF (12,2%), representando uma redução de 17% do risco relativo em favor do tratamento com CLEXANE.
O efeito benéfico da enoxaparina no desfecho primário, observado durante os primeiros 30 dias, foi mantido por um período de acompanhamento de 12 meses.
REFERÊNCIAS BIBLIOGRÁFICAS
Antman EM et al. for the ExTRACT-TIMI 25 Investigators. Enoxaparin versus UFH with Fibrinolysis for STElevation Myocardial Infarction. N Engl J Med 2006;354:1477-88.
Bergqvist D et al. Low-molecular-weight heparin (enoxaparin) as prophylaxis against venous thromboembolism after total hip replacement. N Engl J Med. 1996;335(10):696-700.
Bergqvist et al. for the ENOXACAN Study Group. Efficacy and safety of enoxaparin versus unfractionated heparin for prevention of deep vein thrombosis in elective cancer surgery: A double-blind randomized multicentre trial with venographic assessment. Br J Surg. 1997;84:1099-1103.
Bergqvist D et al; ENOXACAN II Investigators. Duration of prophylaxis against venous thromboembolism with enoxaparin after surgery for cancer. N Engl J Med. 2002 Mar 28;346(13):975-80.
Cohen M el al. A comparison of low-molecular-weight heparin with UFH for unstable coronary artery disease. N Engl J Med. 1997;337:447-452.
Colwell CW et al for the Enoxaparin Clinical Trial Group. Efficacy and safety of enoxaparin versus unfractionated heparin for prevention of deep venous thrombosis after elective knee arthroplasty. Clin Orthop. 1995; 321:19-27.
Leclerc JR et al. Prevention of deep vein thrombosis after major knee surgery. A randomized, double-blind trial comparing a low molecular weight heparin fragment (enoxaparin) to placebo. Thromb Haemost. 1992; 67:417- 423.
MacLeod RS et al. Subcutaneous heparin versus low-molecular-weight heparin as thromboprophylaxis in patients undergoing colorectal surgery: results of the Canadian Colorectal DVT Prophylaxis Trial: a randomized, double-blind trial. Ann Surg 2001; 233: 438-444.
Merli G et al. Subcutaneous enoxaparin once or twice daily compared with intravenous UFH for treatment of venous thromboembolic disease. Ann Intern Med 2001;134,191-202.
Planès A et al. Prevention of postoperative venous thrombosis: A randomized trial comparing unfractionated heparin with low molecular weight heparin in patients undergoing total hip replacement. Thromb Haemost. 1988; 60:407-410.
Planes A et al. Risk of deep-venous thrombosis after hospital discharge in patients having undergone total hip replacement: double-blind randomised comparison of enoxaparin versus placebo. Lancet. 1996; 348:224-8.
Saltissi D, Morgan C, Westhuyzen J, et al: Comparison of low-molecular-weight heparin (enoxaparin sodium) and standard unfractionated heparin for haemodialysis anticoagulation. Nephrol Dial Transplant 1999; 14:2698-
2703.
Samama MM et al. Comparison of enoxaparin with placebo for the prevention of venous thromboembolism in acutely ill medical patients. N Engl J Med 1999; 341: 793-800.
Spiro T et al. Efficacy and Safety of Enoxaparin to Prevent Deep Venous Thrombosis after Hip Replacement Surgery Ann Intern Med 1994; 121;2; 81-89.
Clinical Overview "Enoxaparin And History Of Heparin-Induced Thrombocytopenia" M. Berthon, PharmD (09-Feb-2017 / GPE-CL-2017-00111)
Dados comparativos de CLEXANE X HEPTRIS®
Foi realizado um estudo randomizado, multicêntrico, paralelo e aberto para a comprovação da não-inferioridade de HEPTRIS® referente à incidência de eventos de sangramento durante o período de tratamento, comparação da segurança de HEPTRIS® versus Clexane e da eficácia dos medicamentos na prevenção de tromboembolismo venoso em pacientes cirúrgicos. Foi planejada a avaliação de 300 indivíduos para recebimento de HEPTRIS® 40 mg/0,4mL ou Clexane 40 mg/0,4mL na proporção 1:1. Foram analisados 299 indivíduos: HEPTRIS® 40 mg/0,4mL (n=153) com Clexane 40 mg/0,4mL (n=146). Os indivíduos foram tratados por 14 dias, com uma injeção diária de enoxaparina sódica 40 mg/0,4mL administrada via subcutânea. O objetivo primário foi avaliar a incidência de pacientes com eventos de sangramento até 24 horas após a descontinuação do tratamento. Os objetivos secundários do estudo estavam relacionados à segurança/eficácia. Para o objetivo primário, em ambas as análises de população ITT e PP, a não-inferioridade de HEPTRIS® em relação ao Clexane foi comprovada. Os resultados de ambas as análises estão em conformidade e confirmam mutuamente a não-inferioridade de HEPTRIS® em relação ao Clexane. Para o objetivo de segurança analisado neste estudo, não foi detectada uma diferença significativa entre a incidência deste endpoint particular no grupo de referência de Clexane e HEPTRIS® (enoxaparina sódica 40mg/mL). Além disso, os resultados obtidos na análise com base na população ITT estão em conformidade com os resultados obtidos na análise com base na população PP e levam à conclusão de que não há diferenças significativas entre HEPTRIS® e Clexane no que diz respeito à incidência de qualquer endpoint de segurança. Os desfechos de eficácia foram avaliados não tendo sido detectada nenhuma diferença significativa entre HEPTRIS® e Clexane. Além disso, os resultados obtidos na análise com base na população ITT estão de acordo com os resultados obtidos na análise com base na população PP e levam à conclusão de que não há diferenças significativas entre ambos.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
O princípio ativo de HEPTRIS® é a enoxaparina sódica. Trata-se de uma heparina de baixo peso molecular com peso médio de 4.500 dáltons. A enoxaparina sódica é um sal de sódio. A distribuição do peso molecular é:
< 2000 dáltons ≤ 20%
2000 a 8000 dáltons ≥ 68%
> 8000 dáltons ≤ 18%
A enoxaparina sódica é obtida pela despolimerização alcalina do éster benzil heparina derivado da mucosa intestinal suína. Sua estrutura é caracterizada por um grupo ácido 2-O-sulfo-4-enepiranosurônico no final não redutor da cadeia e um 2-N, 6-O-dissulfo-D-glicosamina no final redutor da cadeia. Aproximadamente 20% (variando entre 15% e 25%) da estrutura da enoxaparina contêm um derivado 1,6 anidro no final redutor da cadeia polissacarídica.
Em um sistema purificado in vitro, a enoxaparina sódica apresenta alta atividade anti-Xa (aproximadamente 100 UI/mg) e baixa atividade anti-IIa ou antitrombina (aproximadamente 28 UI/mg). Estas atividades anticoagulantes são mediadas por antitrombina III (ATIII) resultando em atividade antitrombótica em humanos.
Além da sua atividade anti-Xa/IIa, as propriedades antitrombótica e anti-inflamatória da enoxaparina foram identificadas em indivíduos saudáveis e em pacientes, bem como em modelos não clínicos.
Estes incluem inibição ATIII-dependente de outros fatores de coagulação, como fator VIIa, indução da liberação do inibidor da Via do Fator Tecidual endógeno, assim como uma liberação reduzida de fator de von Willebrand do endotélio vascular para a circulação sanguínea. Estes fatores são conhecidos por contribuir para o efeito antitrombótico global da enoxaparina.
Propriedades farmacocinéticas
Características gerais
Os parâmetros farmacocinéticos da enoxaparina sódica foram estudados principalmente com relação ao tempo da atividade plasmática anti-Xa e também com relação à atividade anti-IIa, nos intervalos de dose recomendados após administrações subcutâneas únicas e repetidas e após administração intravenosa única.
A determinação quantitativa das atividades farmacocinéticas anti-Xa e anti-IIa foi realizada por métodos amidolíticos validados com substratos específicos e com a enoxaparina padrão calibrada contra o padrão internacional para heparinas de baixo peso molecular (NIBSC).
Biodisponibilidade e Absorção
A biodisponibilidade absoluta da enoxaparina sódica após administração subcutânea, baseada na atividade anti-Xa, é próxima de 100%. Os volumes de injeção e concentração de doses no intervalo de 100-200 mg/mL não afetam os parâmetros farmacocinéticos em voluntários saudáveis.
A máxima atividade anti-Xa plasmática média é observada 3 a 5 horas após administração subcutânea alcançando, aproximadamente, 0,2; 0,4; 1,0 e 1,3 anti-Xa UI/mL após administração subcutânea de doses únicas de 20 mg, 40 mg, 1 mg/Kg e 1,5 mg/Kg, respectivamente.
Um bolus intravenoso de 30 mg seguido imediatamente por uma dose subcutânea de 1 mg/kg a cada 12 horas forneceu um pico inicial de níveis de fator anti-Xa igual a 1,16 UI/mL (n = 16) e uma exposição média correspondente a 88% dos níveis do estado de equilíbrio. O estado de equilíbrio é alcançado no segundo dia de tratamento.
A farmacocinética da enoxaparina parece ser linear nos intervalos de dose recomendados. A variabilidade intra e interpacientes é baixa. Após repetidas administrações subcutâneas não ocorre acumulação. Após repetidas administrações subcutâneas de 40 mg, uma vez ao dia, e de 1,5 mg/kg uma vez ao dia, em voluntários saudáveis, o estado de equilíbrio é alcançado no 2° dia, com uma taxa de exposição média aproximadamente 15% maior do que após a administração de dose única. O nível de atividade da enoxaparina no estado de equilíbrio é bem previsível pela farmacocinética de dose única. Após administrações subcutâneas repetidas de 1 mg/kg, num regime de 2 vezes ao dia, o estado de equilíbrio é alcançado entre o 3° e o 4° dia, com uma exposição média aproximadamente 65% maior do que após administração de dose única, e as concentrações máxima e mínima médias de aproximadamente 1,2 e 0,52 UI/mL, respectivamente. Baseada na farmacocinética da enoxaparina sódica, esta diferença no estado de equilíbrio é esperada e está dentro do intervalo terapêutico.
A atividade plasmática anti-IIa após a administração subcutânea é aproximadamente 10 vezes menor do que a atividade anti-Xa. A máxima atividade anti-IIa média é observada aproximadamente 3 - 4 horas após administração subcutânea e alcança 0,13 UI/mL e 0,19 UI/mL após administração repetida de 1 mg/kg, duas vezes ao dia e de 1,5 mg/kg, uma vez ao dia, respectivamente.
Dados comparativos PK/PD de CLEXANE e HEPTRIS®
Foi realizado um estudo randomizado, aberto, dose única, PK/PD comparativo cross-over de 2 vias (n=20) entre os medicamentos HEPTRIS® 40 mg/0,4mL versus Clexane 40 mg/0,4mL após administração subcutânea em indivíduos saudáveis. O objetivo do estudo foi avaliar as propriedades PK / PD e investigar a bioequivalência de HEPTRIS® com Clexane, bem como comparar a segurança e tolerabilidade dessas formulações. Nenhum indivíduo experimentou qualquer evento adverso e nenhum evento adverso sério (SAE) foi observado.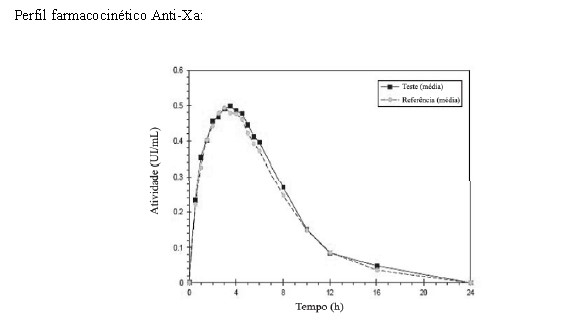
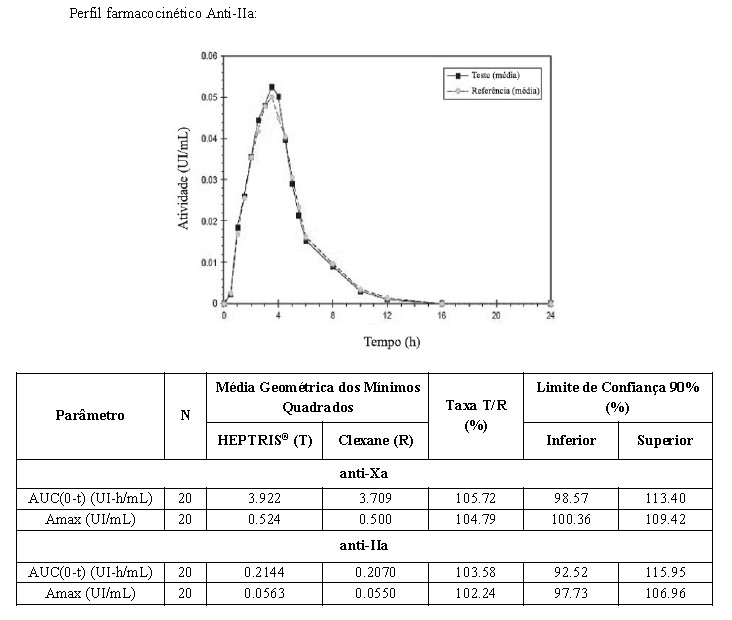
De acordo com o protocolo do estudo, os parâmetros AUC (0-t) e Amax para a atividade anti-Xa e anti-IIa foram usados para avaliar a bioequivalência. Os resultados confirmam que o intervalo de confiança 90% para as taxas T/R das razões das médias dos mínimos quadrados geométricos dos parâmetros avaliados estavam dentro da bioequivalência faixa de aceitação de 80,00 a 125,00%. A bioequivalência entre HEPTRIS® e Clexane foi confirmada neste estudo.
Distribuição
O volume de distribuição da atividade anti-Xa da enoxaparina sódica é de aproximadamente 5 L e é próximo do volume sanguíneo.
Metabolismo
A enoxaparina é metabolizada principalmente no fígado por dessulfatação e/ou despolimerização formando moléculas de peso menor, que apresentam potência biológica muito reduzida.
Eliminação
A enoxaparina sódica é um fármaco de baixa depuração, com média de clearance plasmático anti-Xa de 0,74 L/h após infusão intravenosa de 1,5 mg/kg em 6 horas.
A eliminação parece ser monofásica, com meia-vida de aproximadamente 5 4 horas após uma dose subcutânea única, e até aproximadamente 7 horas após doses repetidas.
O clearance renal dos fragmentos ativos representa aproximadamente 10% da dose administrada e a excreção renal total dos fragmentos ativos e não-ativos é de 40% da dose.
Características em Populações Especiais
Idosos: com base nos resultados da análise farmacocinética populacional, o perfil cinético de enoxaparina sódica não é diferente em voluntários idosos comparados a voluntários jovens quando a função renal é normal. Entretanto, como é conhecido que a função renal diminui com o aumento da idade, pacientes idosos podem apresentar retardo na eliminação de enoxaparina sódica (vide item "5. Advertências e Precauções - Populações Especiais", "8. Posologia e modo de usar - Populações Especiais" e o item a seguir "Insuficiência renal").
Insuficiência renal: observou-se uma relação linear entre o clearance plasmático de anti-Xa e o clearance de creatinina no estado de equilíbrio, o que indica um decréscimo do clearance de enoxaparina sódica em pacientes com função renal reduzida. A exposição anti-Xa representada pela AUC (área sob a curva), no estado de equilíbrio, é levemente aumentada na insuficiência renal leve (clearance de creatinina 50 - 80 mL/min) e moderada (clearance de creatinina 30 - 50 mL/min) após repetidas doses subcutâneas de 40 mg, uma vez ao dia. Em pacientes com insuficiência renal severa (clearance de creatinina < 30 mL/min), a AUC no estado de equilíbrio é significativamente aumentada em média em 65% após repetidas doses únicas diárias subcutâneas de 40 mg (vide itens "5. Advertências e Precauções - Populações especiais" e "8. Posologia e modo de usar - Populações especiais").
Peso: após repetidas doses subcutâneas de 1,5 mg/kg, uma vez ao dia, a AUC média de atividade anti-Xa é levemente maior no estado de equilíbrio em voluntários saudáveis obesos (IMC 30 - 48 Kg/m2) em comparação aos voluntários controle não-obesos, embora a atividade máxima observada não tenha aumentado. Há menor clearance ajustado ao peso em voluntários obesos tratados com doses subcutâneas.
Quando se administram doses não ajustadas ao peso, a exposição da atividade anti-Xa é 52% maior em mulheres de peso baixo ( < 45 Kg) e 27% maior em homens de peso baixo ( < 57 Kg), após uma dose subcutânea única de 40 mg, quando comparada aos voluntários controle com peso normal (vide item "5. Advertências e Precauções - Populações especiais").
Hemodiálise: em um único estudo, a taxa de eliminação apresentou-se semelhante, porém a AUC foi duas vezes maior que na população controle, após uma dose intravenosa única de 0,25 ou 0,50 mg/kg.
Interações farmacocinéticas
Não foram observadas interações farmacocinéticas entre a enoxaparina e trombolíticos quando administrados concomitantemente.
Dados de Segurança Pré-Clínicos
Não foram realizados estudos de longa duração em animais para avaliar o potencial carcinogênico da enoxaparina.
A enoxaparina não se mostrou mutagênica em testes in vitro, incluindo o teste Ames, o teste de mutação de células de linfoma em camundongos, o teste de aberração cromossômica linfocítica em humanos e os testes in vivo de aberração cromossômica na medula óssea de ratos.
Demonstrou-se que a enoxaparina não tem nenhum efeito na fertilidade ou no desempenho reprodutivo de ratos machos e fêmeas em doses subcutâneas de até 20 mg/kg/dia. Estudos teratológicos foram conduzidos em ratas e coelhas prenhes em doses subcutâneas de enoxaparina de até 30 mg/kg/dia. Não houve nenhuma evidência de efeitos teratogênicos ou fetotoxicidade devido à enoxaparina.
Além dos efeitos anticoagulantes da enoxaparina, não houve evidência de efeitos adversos em doses de 15 mg/kg/dia em 13 semanas de estudos de toxicidade subcutânea, ambos em ratos e cães e em doses de 10 mg/kg/dia em 26 semanas de estudos de toxicidade subcutânea e intravenosa ambos em ratos e macacos.
4. CONTRAINDICAÇÕES
- Hipersensibilidade à enoxaparina sódica, à heparina e seus derivados, inclusive outras heparinas de baixo peso molecular;
- História de trombocitopenia induzida pela heparina mediada por imunidade (TIH) nos últimos 100 dias ou na presença de anticorpos circulantes;
- Hemorragias ativas de grande porte e condições com alto risco de desenvolvimento de hemorragia, incluindo acidente vascular cerebral hemorrágico recente, úlcera gastrointestinal, presença de neoplasia maligna com alto risco de sangramento cerebral recente, cirurgia espinhal ou oftálmica, varizes esofágicas conhecidas ou suspeitas, malformações arteriovenosas, aneurismas vasculares ou anomalias vasculares intra-espinhais ou intracerebrais importantes;
- Anestesia raquidiana ou epidural ou anestesia loco-regional quando a enoxaparina sódica é usada para tratamento nas 24 horas anteriores.
5. ADVERTÊNCIAS E PRECAUÇÕES
Não administrar HEPTRIS® por via intramuscular.
Hemorragia
Assim como com outros anticoagulantes, pode ocorrer sangramento em qualquer local (vide item "9. Reações Adversas"). Se ocorrer sangramento, a origem da hemorragia deve ser investigada e tratamento apropriado deve ser instituído.
HEPTRIS®, assim como qualquer outra terapia anticoagulante, deve ser utilizado com cautela em condições com alto risco de hemorragia, tais como:
- alterações na hemostasia;
- histórico de úlcera péptica;
- acidente vascular cerebral isquêmico recente;
- hipertensão arterial severa não controlada;
- retinopatia diabética;
- neurocirurgia ou cirurgia oftálmica recente;
- uso concomitante de medicamentos que afetem a hemostasia (vide item "6. Interações Medicamentosas").
Monitoramento da contagem plaquetária
O risco de trombocitopenia induzida por heparina (reação mediada por anticorpos) também existe com heparinas de baixo peso molecular. Pode ocorrer trombocitopenia, geralmente entre o 5° e 21° dia após o início do tratamento com HEPTRIS®. Portanto, recomenda-se a realização de contagem plaquetária antes do início e regularmente durante o tratamento. Na prática, em caso de confirmação de diminuição significativa da contagem plaquetária (30 a 50% do valor inicial), o tratamento com HEPTRIS® deve ser imediatamente interrompido e substituído por outra terapia.
Advertências Gerais
As diferentes classes de heparinas de baixo peso molecular (HBPM), por exemplo, enoxaparina, dalteparina, bemiparina e nadroparina não devem ser intercambiáveis (unidade por unidade), pois existem diferenças entre elas quanto ao processo de fabricação, peso molecular, atividade anti-Xa específica, unidade e dosagem. Isto ocasiona diferenças em suas atividades farmacocinéticas e biológicas associadas (por exemplo, a atividade antitrombina e a interação plaquetária). Portanto, é necessário obedecer às instruções de uso de cada medicamento.
Anestesia espinhal/peridural
Foram relatados casos de hematoma intraespinhal com o uso concomitante de HEPTRIS® e anestesia espinhal/peridural, resultando em paralisia prolongada ou permanente. Estes eventos são raros com a administração de doses iguais ou inferiores a 40 mg/dia de HEPTRIS®. O risco destes eventos pode ser aumentado com administração de doses maiores de HEPTRIS®, uso de cateter epidural no pós-operatório ou em caso de administração concomitante de medicamentos que alteram a hemostasia, tais como anti-inflamatórios não esteroidais (vide item "6. Interações Medicamentosas"). O risco parece também ser aumentado por traumatismo ou punções espinhais repetidas ou em pacientes com histórico de cirurgia ou deformidade espinhal.
Para reduzir o risco potencial de sangramento associado ao uso concomitante de HEPTRIS® e anestesia/analgesia peridural ou espinhal, deve-se considerar o perfil farmacocinético do fármaco (vide item "3. Características Farmacológicas - Propriedades Farmacocinéticas"). A introdução e remoção do cateter devem ser realizadas quando o efeito anticoagulante de HEPTRIS® estiver baixo, no entanto, o momento exato para chegar a um efeito anticoagulante suficientemente baixo em cada paciente não é conhecido.
A introdução ou remoção do cateter deve ser postergada por pelo menos 12 horas após a administração de doses baixas de HEPTRIS® (20 mg uma vez ao dia, 30 mg uma ou duas vezes ao dia, ou 40 mg uma vez ao dia) e, pelo menos, 24 horas após a administração de doses mais elevadas de HEPTRIS® (0,75 mg/kg, duas vezes ao dia, 1 mg/kg duas vezes ao dia, ou 1,5 mg/kg uma vez ao dia). Níveis de anti-Xa ainda são detectáveis neste momento, e estes atrasos não são uma garantia de que um hematoma neuroaxial (espinhal) será evitado. Pacientes recebendo a dose de 0,75 mg/kg duas vezes ao dia, ou a dose de 1 mg/kg duas vezes ao dia não devem receber a segunda dose de enoxaparina no regime de duas vezes ao dia para permitir um atraso maior antes da colocação ou remoção do cateter. Da mesma forma, apesar de uma recomendação específica para o intervalo da dose subsequente de enoxaparina após a remoção do cateter não poder ser feita, considerar adiar esta dose seguinte por pelo menos quatro horas, com base numa avaliação do risco-benefício considerando tanto o risco de trombose como o risco de sangramento no contexto do procedimento e fatores de risco do paciente. Para pacientes com clearance de creatinina < 30mL/minuto, são necessárias considerações adicionais porque a eliminação de enoxaparina é mais prolongada; considerar a duplicação do tempo de remoção de um cateter, pelo menos, 24 horas para a menor dose prescrita de enoxaparina (30 mg uma vez ao dia) e, pelo menos, 48 horas para a dose mais elevada (1mg/kg/dia).
Caso o médico decida administrar anticoagulantes durante o uso de anestesia peridural/espinhal ou punção lombar, deve-se empregar o monitoramento frequente para detectar qualquer sinal ou sintoma de lesão neurológica, tais como, dor na linha média da região dorsal, deficiências sensoriais e motoras (entorpecimento ou fraqueza dos membros inferiores), alterações intestinais e/ou urinárias. Os pacientes devem ser instruídos a informar imediatamente a seu médico caso apresentem qualquer sintoma ou sinal descrito acima. Em caso de suspeita de sinais ou sintomas de hematoma intraespinhal, devem ser efetuados o diagnóstico e tratamento, incluindo descompressão da medula espinhal, com urgência.
Trombocitopenia induzida pela heparina
A utilização de enoxaparina sódica em pacientes com história de HIT mediada por imunidade nos últimos 100 dias ou na presença de anticorpos circulantes está contraindicada. Os anticorpos circulantes podem persistir vários anos.
A enoxaparina sódica deve ser usada com extrema cautela em pacientes com história (mais de 100 dias) de trombocitopenia induzida por heparina sem anticorpos circulantes. A decisão de utilizar enoxaparina sódica neste caso, deve ser feita apenas após uma cuidadosa avaliação do risco benefício e após terem sido considerados tratamentos alternativos sem heparina.
Procedimentos de revascularização coronariana percutânea
Para minimizar o risco de sangramento após a instrumentação vascular durante o tratamento da angina instável, infarto do miocárdio sem elevação do segmento ST e infarto agudo do miocárdio com elevação do segmento ST, devem-se respeitar precisamente os intervalos entre as doses recomendadas de HEPTRIS®. É importante estabelecer a hemostasia no local da punção após a intervenção coronariana percutânea. Caso tenha sido utilizado um dispositivo de fechamento, a bainha de acesso vascular pode ser removida imediatamente. Caso tenha sido utilizado um método de compressão manual, a bainha deve ser removida 6 horas após a última administração intravenosa ou subcutânea de HEPTRIS®. Se o tratamento com HEPTRIS® continuar, a próxima dose programada não deve ser administrada antes de 6 a 8 horas após a remoção da bainha. Deve-se ter atenção especial ao local do procedimento para detecção de sinais de sangramento ou formação de hematoma.
Gravidez e lactação
Estudos em animais não demonstraram qualquer evidência de fetotoxicidade ou teratogenicidade. Em ratas prenhes, a passagem de 35 S-enoxaparina sódica através da placenta para o feto é mínima.
Em humanos, não existe evidência da passagem da enoxaparina sódica através da placenta durante o segundo trimestre da gravidez. Ainda não existem informações disponíveis a este respeito durante o primeiro e terceiro trimestres da gravidez.
Como não foram realizados estudos adequados e bem controlados em gestantes, e uma vez que os estudos realizados em animais nem sempre são bons indicativos da resposta humana, deve-se utilizar HEPTRIS® durante a gravidez somente se o médico considerar como estritamente necessário.
Categoria de risco na gravidez: B. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Em ratas lactant