HEMCIBRA
ROCHE
emicizumabe
Profilaxia em pacientes com hemofilia A.
Apresentações.
Solução injetável de 30 mg/mL: caixa com 1 frasco-ampola de dose única de 30 mg (1 mL).
Solução injetável de 150 mg/mL: caixa com 1 frasco-ampola de dose única de 60 mg (0,4 mL), 105 mg (0,7 mL) ou 150 mg (1 mL).
VIA SUBCUTÂNEA
USO ADULTO E PEDIÁTRICO
Composição.
Hemcibra® solução injetável de 30 mg
Princípio ativo: emicizumabe (anticorpo monoclonal IgG4 modificado recombinante humanizado) 30 mg (30 mg/mL). Excipientes: histidina, ácido aspártico, arginina, poloxâmer e água para injetáveis
Hemcibra® solução injetável de 60 mg
Princípio ativo: emicizumabe (anticorpo monoclonal IgG4 modificado recombinante humanizado) 60 mg (150 mg/mL). Excipientes: histidina, ácido aspártico, arginina, poloxâmer e água para injetáveis
Hemcibra® solução injetável de 105 mg
Princípio ativo: emicizumabe (anticorpo monoclonal IgG4 modificado recombinante humanizado) 105 mg (150 mg/mL). Excipientes: histidina, ácido aspártico, arginina, poloxâmer e água para injetáveis
Hemcibra® solução injetável de 150 mg
Princípio ativo: emicizumabe (anticorpo monoclonal IgG4 modificado recombinante humanizado) 150 mg (150 mg/mL). Excipientes: histidina, ácido aspártico, arginina, poloxâmer e água para injetáveis
Informações técnicas.
1. INDICAÇÕES
Hemcibra® é indicado para profilaxia de rotina, para prevenir sangramento ou reduzir a frequência de episódios de sangramento em adultos e crianças com hemofilia A (deficiência congênita do fator VIII) com ou sem inibidores do fator VIII (FVIII).
Hemcibra® pode ser utilizado por todas as faixas etárias.
2. RESULTADOS DE EFICÁCIA
A eficácia do Hemcibra® para profilaxia de rotina em pacientes com hemofilia A, com ou sem inibidores, foi avaliada em quatro estudos clínicos (três estudos com adultos e adolescentes [HAVEN 3, HAVEN 1 E HAVEN 4] e um estudo pediátrico [HAVEN 2].
Estudos clínicos em pacientes adultos e adolescentes
HAVEN 31
O HAVEN 3 foi um estudo clínico de fase III, aberto, multicêntrico e randomizado com 152 pacientes adultos e adolescentes (≥ 12 anos e ≥ 40 kg) do sexo masculino com hemofilia A, sem inibidores do FVIII, que receberam tratamento episódico ("sob demanda") ou profilático com FVIII. Os pacientes receberam 3 mg/kg de Hemcibra® por via subcutânea, uma vez por semana, durante as primeiras quatro semanas e, em seguida, receberam 1,5 mg/kg uma vez por semana (braços A e D) ou 3 mg/kg a cada duas semanas (braço B) ou não receberam profilaxia (braço C). Os pacientes do braço C podiam passar a receber Hemcibra® (3 mg/kg a cada duas semanas) após completarem pelo menos 24 semanas sem profilaxia. Nos braços A e B, o aumento da dose para 3 mg/kg por semana foi permitido após 24 semanas para pacientes que apresentavam dois ou mais sangramentos qualificados (ou seja, sangramentos espontâneos e clinicamente significativos que ocorriam no estado de equilíbrio). Os pacientes no braço D podiam aumentar a dose após o segundo sangramento qualificado. Na ocasião da análise, cinco pacientes haviam aumentado sua dose de manutenção.
Oitenta e nove pacientes tratados previamente com FVIII episódico ("sob demanda") foram randomizados em uma razão de 2:2:1 para receber Hemcibra® uma vez por semana (braço A; N = 36) ou a cada duas semanas (braço B; N = 35) ou sem profilaxia (braço C; N = 18), com estratificação por taxa prévia de sangramento de 24 semanas ( < 9 ou ≥ 9). Sessenta e três pacientes tratados previamente com FVIII profilático foram incluídos no braço D para receber Hemcibra® (1,5 mg/kg uma vez por semana).
O objetivo primário do estudo foi avaliar, em pacientes tratados previamente com FVIII episódico, a eficácia da profilaxia com Hemcibra® uma vez por semana (braço A) ou a cada duas semanas (braço B) em comparação a pacientes sem profilaxia (braço C) com base no número de sangramentos que demandaram tratamento com fatores de coagulação (vide Tabela 1). Outros objetivos do estudo incluíram a avaliação da comparação randomizada dos braços A ou B e do braço C em termos de eficácia da profilaxia com Hemcibra® na diminuição do número de sangramentos em geral, sangramentos espontâneos, sangramentos articulares e sangramentos em articulações alvo (vide Tabela 2), bem como a avaliação da qualidade de vida relacionada à saúde (QVRS) relatada pelos pacientes (vide Tabela 9). O tratamento preferido dos pacientes também foi avaliado através de uma pesquisa de preferência.
A eficácia da profilaxia com Hemcibra® também foi comparada ao tratamento profilático anterior com FVIII (braço D) em pacientes que haviam participado em um estudo não intervencional (ENI) antes do recrutamento (vide Tabela 3). Somente os pacientes do ENI foram incluídos nessa comparação porque os dados sobre sangramento e tratamento foram coletados com o mesmo nível de granularidade usado no estudo HAVEN 3.
HAVEN 12
O estudo HAVEN 1, estudo clínico randomizado, multicêntrico, aberto que incluiu 109 adolescentes e adultos do sexo masculino (idade ≥12 anos e ≥ 40kg) com hemofilia A com inibidores do fator VIII que tinham recebido previamente tratamento episódico ou profilático com agentes de bypass. No estudo, os pacientes receberam profilaxia semanal com Hemcibra® (braços A, C e D) - doses de 3 mg/kg, uma vez por semana durante quatro semanas, seguido por doses de 1,5 mg/kg, uma vez por semana subsequentemente - ou sem profilaxia (braço B). Os pacientes randomizados para o braço B puderam trocar para profilaxia com Hemcibra® após completarem pelo menos 24 semanas sem profilaxia.
Foi permitido aumento de dosagem para 3 mg/kg, uma vez por semana, depois de 24 semanas sob profilaxia com Hemcibra® para pacientes que apresentaram dois ou mais sangramentos qualificados, em caso de eficácia abaixo da ideal (isto é, ≥ dois sangramentos espontâneos e clinicamente significativos que ocorreram no estado de equilíbrio). Durante o estudo, cinco pacientes foram submetidos ao aumento de sua dose de manutenção para 3 mg/kg, uma vez por semana.
Cinquenta e três pacientes previamente tratados com agentes de bypass de forma episódica foram randomizados em uma razão de 2:1 para receberem profilaxia com Hemcibra® (braço A) ou sem profilaxia, (braço B), com estratificação pela taxa de sangramento nas últimas 24 semanas ( < 9 ou ≥ 9).
Quarenta e nove pacientes previamente tratados com agentes de bypass profiláticos foram incluídos no braço C para receberem profilaxia com Hemcibra®. Sete pacientes previamente tratados de forma episódica com agentes de bypass que tinham participado de estudo não intervencional (ENI) antes do recrutamento, mas não puderam ser incluídos no estudo HAVEN 1 antes do fechamento dos braços A e B, foram incluídos no braço D, para receberem profilaxia com Hemcibra®.
O objetivo primário do estudo foi avaliar, entre pacientes previamente tratados de forma episódica com agentes de bypass, o efeito do tratamento profilático semanal com Hemcibra®, em comparação com pacientes sem profilaxia (braço A versus braço B) sobre o número de sangramentos com necessidade de tratamento com fatores de coagulação ao longo do tempo (mínimo de 24 semanas ou data de descontinuação), vide Tabela 1. Os objetivos secundários da comparação randomizada dos braços A e B foram a eficácia da profilaxia com Hemcibra® semanal na redução do número de todos os sangramentos, dos sangramentos espontâneos, sangramentos articulares e sangramentos em articulações-alvo (vide Tabela 4), bem como avaliar a qualidade de vida relacionada à saúde dos pacientes (QVRS) e seu estado de saúde (vide Tabelas 10 e 12).
A eficácia da profilaxia com Hemcibra® semanal, em comparação com agentes de bypass profiláticos prévios também foi avaliado em pacientes que tinham participado do ENI antes da inclusão (braço C) (vide Tabela 5). Apenas pacientes do ENI foram incluídos nessa comparação, porque os dados de sangramento e tratamento foram coletados com o mesmo grau de granularidade que o usado no estudo HAVEN 1.
HAVEN 46
O Hemcibra® foi investigado em um estudo clínico de fase III, multicêntrico e de braço único em 41 pacientes adultos e adolescentes (≥ 12 anos e ≥ 40 kg) do sexo masculino com hemofilia A, com ou sem inibidores do FVIII, que receberam tratamento episódico ("sob demanda") ou profilático com FVIII ou agentes de bypass.5,6 Os pacientes receberam profilaxia com Hemcibra® - 3 mg/kg uma vez por semana, durante quatro semanas, seguida por 6 mg/kg a cada quatro semanas desse ponto em diante.
O objetivo primário do estudo foi avaliar a eficácia da profilaxia com Hemcibra® na manutenção do controle adequado de sangramentos administrada a cada quatro semanas com base nos sangramentos tratados. Outros objetivos eram avaliar a eficácia clínica da profilaxia com Hemcibra® nos sangramentos em geral, sangramentos espontâneos tratados, sangramentos articulares tratados e sangramentos tratados em articulações alvo (vide Tabela 7) bem como avaliar a QVRS dos pacientes (vide Tabela 11).5,6 O tratamento preferido dos pacientes também foi avaliado através de uma pesquisa de preferência.5,6
Resultados de Eficácia em Adultos e Adolescentes
Os resultados de eficácia da profilaxia com Hemcibra® em relação à taxa de sangramentos tratados são apresentados na Tabela 1.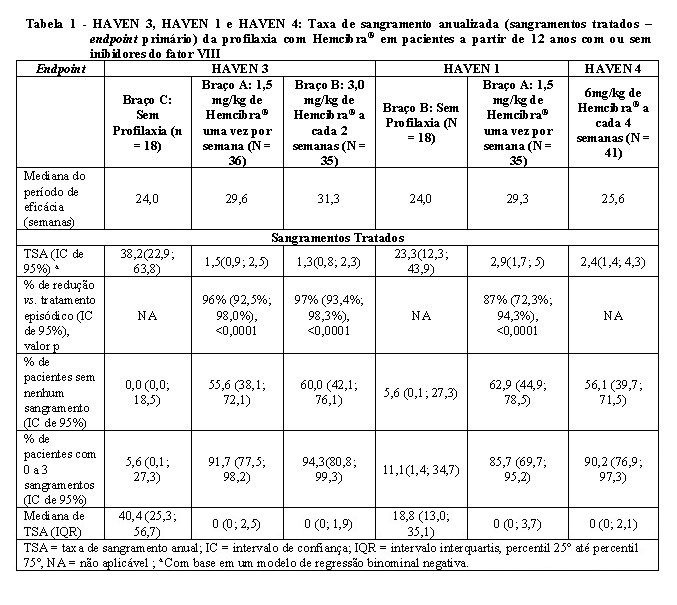
HAVEN 3
Os resultados de eficácia da profilaxia com Hemcibra® em comparação a nenhuma profilaxia em relação à taxa de sangramentos tratados, sangramentos em geral, sangramentos espontâneos tratados, sangramentos articulares tratados e sangramentos tratados em articulações alvo são apresentados a seguir, na Tabela 2.1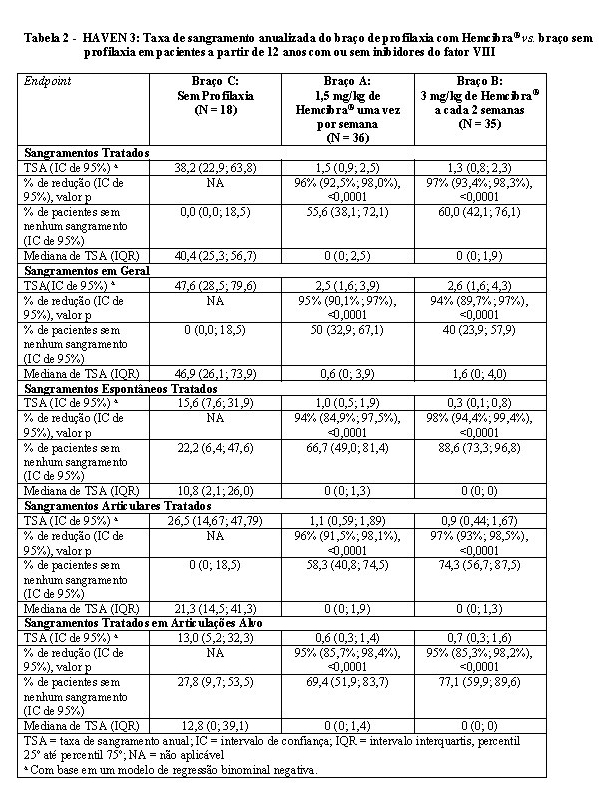
Na análise intrapaciente do estudo clínico HAVEN 3, a profilaxia com Hemcibra® resultou em uma redução (68%) estatisticamente significativa (p < 0,0001) na taxa de sangramentos tratados em comparação à profilaxia prévia com FVIII registrada no ENI antes da inclusão (vide Tabela 3).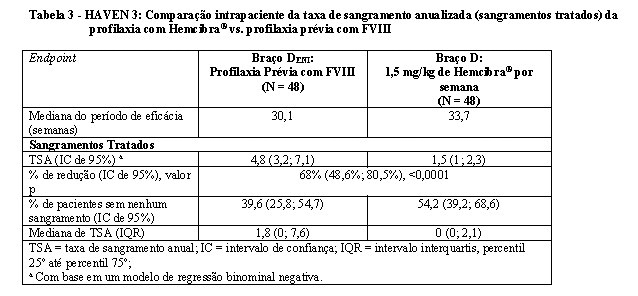
HAVEN 1
Os resultados de eficácia da profilaxia com Hemcibra® comparados com nenhuma terapia profilática, relacionados à taxa de sangramentos tratados, todos os sangramentos, sangramentos espontâneos tratados, sangramento articular tratado e sangramento das articulações alvo tratadas são mostrados na tabela 4.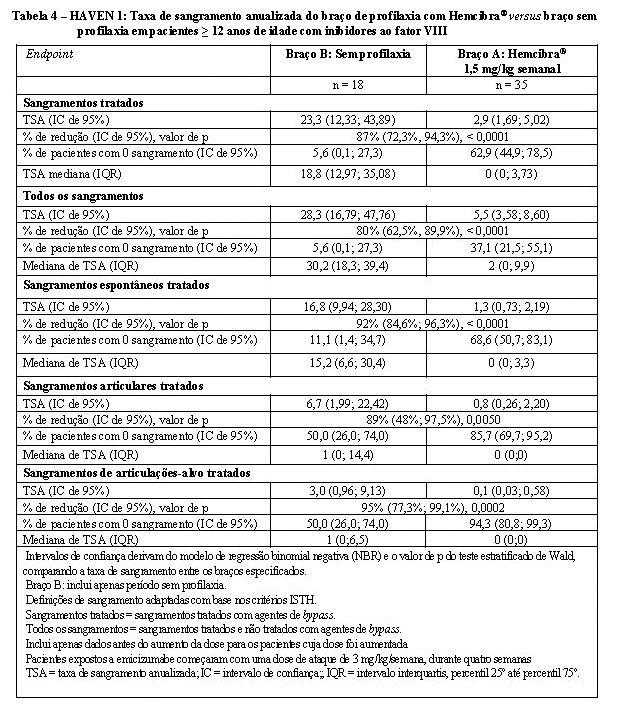
Análises adicionais foram realizadas para o estudo HAVEN 1 a fim de avaliar o controle a longo prazo dos sangramentos em pacientes tratados profilaticamente com o Hemcibra® usando o intervalo de tratamento de 12 semanas até a semana 72. Quando a TSA para hemorragias tratadas foi avaliada em intervalos de 12 semanas, as TSAs médias diminuíram ao longo do tempo e a melhoria durou até a semana 72, enquanto a mediana permaneceu consistentemente em zero (ver Tabela 5). Estes dados demonstram a eficácia a longo prazo da profilaxia com o Hemcibra®. As médias e medianas de TSA foram calculadas para hemorragias tratadas, conforme mostrado na Tabela 5.
Na análise intrapaciente do estudo clínico HAVEN 1, a profilaxia Hemcibra® resultou em uma redução estatisticamente significativa (p = 0,0003) (79%) na taxa de sangramento para sangramentos tratados em comparação com a profilaxia do agente de bypass anteriormente coletada no ENI antes do recrutamento (Tabela 6)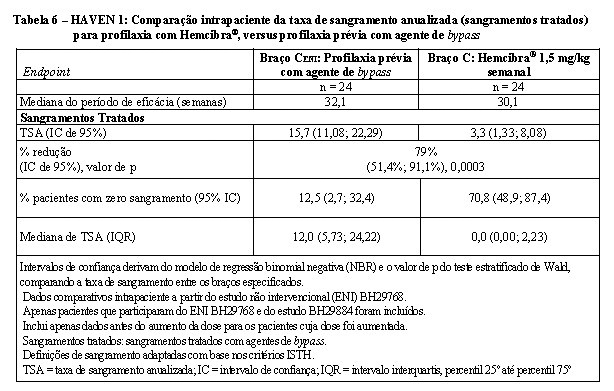
HAVEN 4 5,6
Os resultados de eficácia do estudo clínico HAVEN 4 são resumidos a seguir. Quarenta e um pacientes a partir de 12 anos de idade foram avaliados quanto à eficácia com um período de observação mediano de 25,6 semanas (faixa: 24,1 - 29,4 semanas). Os resultados de eficácia da profilaxia com Hemcibra® a cada quatro semanas em relação à taxa de sangramentos tratados, sangramentos em geral, sangramentos espontâneos tratados, sangramentos articulares tratados e sangramentos tratados em articulações alvo são apresentados na Tabela 7.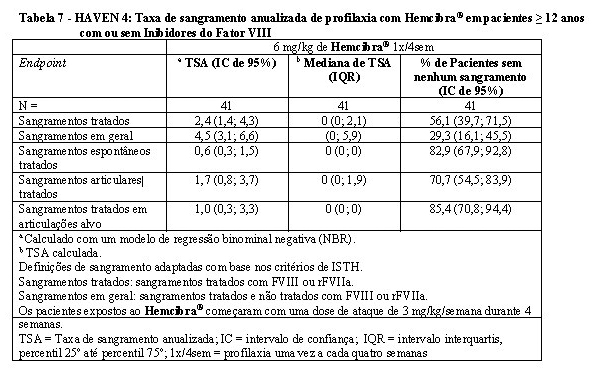
Medidas de Resultados Relacionados à Saúde em Adultos e Adolescentes
Os estudos clínicos HAVEN em adultos e adolescentes avaliaram os resultados relatados pelos pacientes com diversas medidas. O questionário de qualidade de vida específico para hemofilia (Haemophilia-Specific Quality of Life - Haem-A-QoL) para adultos (≥ 18 anos) e sua versão para adolescentes (Haemo-QoL-SF, 8 a < 18 anos) avaliaram a qualidade de vida relacionada à hemofilia nos pacientes. Nos questionários Haem-A-QoL e Haemo-QoL-SF, a pontuação de saúde física (ou seja, inchaços dolorosos, presença de dor articular, dor ao se mover, dificuldade para caminhar longas distâncias e necessidade de mais tempo para se preparar) e a pontuação total (resumo de todas as pontuações) foram definidas no protocolo como endpoint de interesse. Para medir a mudança no estado de saúde, a pontuação de utilidade do índice (Index Utility Score - IUS) e a escala visual analógica (EVA) do questionário EuroQoL de cinco dimensões e cinco níveis (EQ-5D-5L) foram examinadas.
Nos estudos HAVEN 3 e 4, para a avaliação referente ao tratamento preferido dos pacientes foi usada a pesquisa da preferência por emicizumabe (EmiPref).
Resultados Relacionados à Saúde no Estudo HAVEN 3
No estudo HAVEN 3, a qualidade de vida relacionada à saúde (QVRS) em pacientes a partir de 18 anos foi avaliada na semana 25 com base no questionário Haem-A-QoL para adultos. O questionário Haem-A-QoL é uma medida válida e confiável da qualidade de vida relacionada à saúde (vide Tabela 8).
Resultados relacionados à saúde no estudo HAVEN 12
No estudo HAVEN 1, a qualidade de vida relacionada à saúde (QVRS) em pacientes com 18 anos de idade ou mais foi avaliada na semana 25, com base no questionário para adultos Haemophilia-specific Quality of Life (Haem-A-QoL (vide Tabela 9).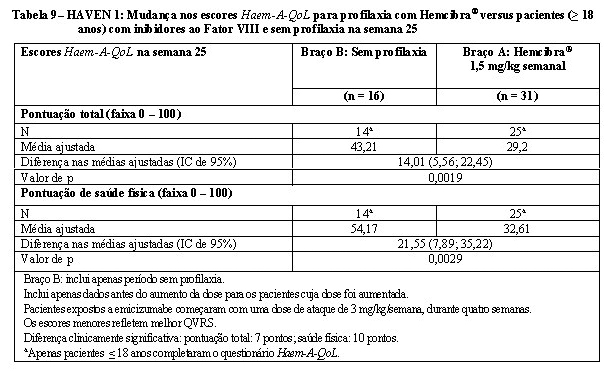
Resultados sobre estado de saúde do estudo HAVEN 12
No estudo HAVEN 1, o estado de saúde dos pacientes foi avaliado de acordo com o EuroQoL Five-Dimension-Five Levels Questionnaire (EQ-5D-5L), que é uma medida válida e confiável de estado de saúde (vide Tabela 10). 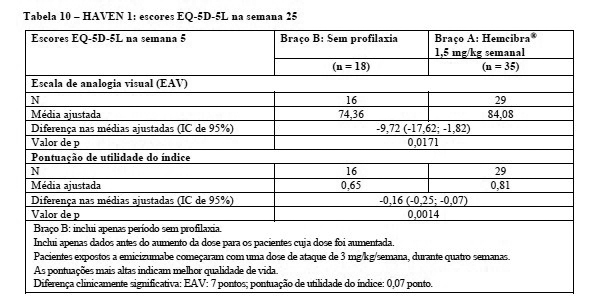
Resultados relacionados à saúde no estudo HAVEN 45,6
No estudo HAVEN 4, a qualidade de vida relacionada à saúde (QVRS) em pacientes com 18 anos de idade ou mais foi avaliada na semana 25, com base no questionário para adultos Haemophilia-specific Quality of Life (Haem-A-QoL (vide Tabela 11)).
Preferência dos pacientes nos estudos HAVEN 3 e 4
No estudo HAVEN 3 e 4, os pacientes que receberam Hemcibra® (uma vez por semana, a cada duas semanas e a cada 4 semanas) relataram se preferiam Hemcibra® subcutâneo, seu tratamento IV anterior ou se não tinham nenhuma preferência na semana 17. Dos pacientes no estudo HAVEN 3 que responderam o questionário de preferência, 89 de 95 (93,7%) relataram preferir Hemcibra® ao tratamento IV anterior, e especificamente 45 de 46 pacientes (97,8%) preferiam Hemcibra® ao seu tratamento profilático anterior com FVIII. No estudo HAVEN 4, todos os 41 pacientes (100%) responderam o questionário de preferência e relataram preferir Hemcibra® ao tratamento IV anterior. Nos estudos HAVEN 3 e 4, os dois motivos mencionados mais frequentemente pelos pacientes para justificar sua preferência pelo Hemcibra® foram a via de administração mais fácil e a menor frequência do tratamento.
Estudos Clínicos em Pacientes Pediátricos
HAVEN 2 (análise interina)3
A profilaxia semanal com Hemcibra® foi avaliada em um estudo clínico aberto, de braço único, multicêntrico que incluía pacientes pediátricos (idade < 12 anos ou de 12 a 17 anos com peso < 40 kg) com hemofilia A com inibidores do FVIII. Os pacientes receberam profilaxia com Hemcibra® na dose de 3 mg/kg, uma vez por semana, durante as primeiras quatro semanas, seguidos por 1,5 mg/kg, uma vez por semana subsequentemente.
O estudo avaliou a farmacocinética, a segurança e a eficácia, que incluía a eficácia da profilaxia semanal com Hemcibra®, em comparação com o tratamento episódico e o tratamento profilático com agentes de bypass em pacientes que tinham participado do ENI antes do recrutamento (comparação intrapaciente).
Estudo HAVEN 2: resultados de eficácia (análise interina)3
No momento da análise interina, a eficácia foi avaliada em 59 pacientes pediátricos com idade < 12 anos e recebendo profilaxia semanal com o Hemcibra® por pelo menos 12 semanas, incluindo 38 pacientes com idade entre 6 e < 12 anos; 17 pacientes com idade entre 2 e < 6 anos e quatro pacientes < 2 anos de idade.
A taxa de sangramento anualizada e a porcentagem de pacientes sem sangramentos foram calculadas para 59 pacientes (vide Tabela 12). A mediana do período de observação para estes pacientes foi de 29,6 semanas (intervalo: 18,4 - 63).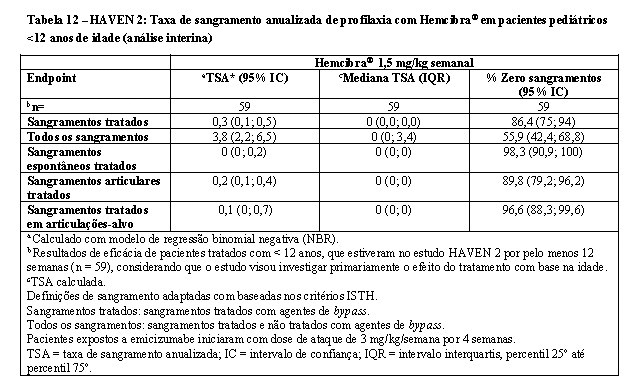
Na análise interina intrapaciente, a profilaxia semanal com Hemcibra® resultou em redução clinicamente significativa (98%) na taxa de sangramentos tratados em dezoito pacientes pediátricos que tiveram, pelo menos, 12 semanas de tratamento profilático com Hemcibra®, em comparação com sua taxa de sangramento coletada no ENI antes do recrutamento (vide Tabela 13).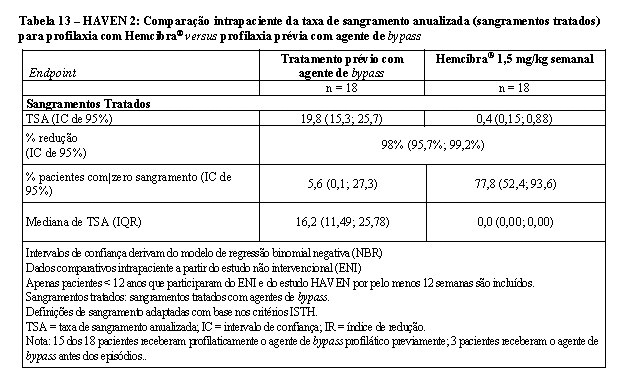
Resultados relacionados à saúde no estudo HAVEN 23
No HAVEN 2, o QVRS para pacientes entre ≥ 8 a < 12 anos foi avaliado na semana 25, com base no Haemo-QoL-SF questionnaire para crianças. O Haemo-QoL-SF é uma medida confiável e válida do QVRS (vide Tabela 14).
No HAVEN 2, o QVRS para pacientes < 12 anos também foi avaliado na semana 25, com base no questionário Adapted InhibQoL, completados pelos cuidadores, com aspectos relacionados a carga do cuidador. O Adapted InhibQoL é uma medida confiável e válida para avaliação da QVRS (vide Tabela 15).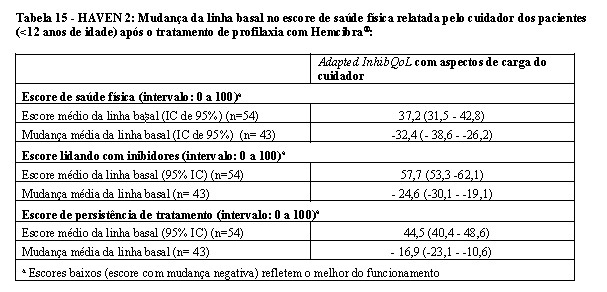
Cirurgias e procedimentos nos estudos clínicos HAVEN
A experiência com o uso de agentes bypass ou FVIII durante cirurgias e procedimentos em pacientes que recebem profilaxia com Hemcibra® é limitada (para maiores informações consultar o item 5. ADVERTÊNCIAS E PRECAUÇÕES - Hemcibra® e Concentrado de Complexo Protrombínico Ativado [CCPa]).
Referências bibliográficas
1. Clinical Study Report - HAVEN 3. Report No. 1082237. March 2018. (CDS version 3.0).
2. Clinical Study Report - BH29884 (HAVEN 1) - A multicenter, open-label phase III study to evaluate the efficacy, safety, and pharmacokinetics of prophylactic emicizumab versus no prophylaxis in hemophilia A patients with inhibitors to factor VIII. Report No. 1070071. May 2017.
3. Interim Clinical Study Report - BH29992 (HAVEN 2) - A single arm, multicenter, open-label, phase III study to evaluate the efficacy, safety and pharmacokinetics of once weekly subcutaneous administration of emicizumab in hemophilia A pediatric patients with inhibitors to factor VIII. Report No.1074617. May 2017.
4. Supplemental Results Report (BH29992) (CDS version 2.0).
5. Interim Clinical Study Report - BO39182 (HAVEN 4). A multicenter, open-label phase III study to evaluate the efficacy, safety, pharmacokinetics and pharmacodynamics of emicizumab given every 4 weeks (Q4W) in patients with hemophilia A. Report No. 1079217. March 2018. (CDS version 3.0).
6. Clinical Study Report - BO39182 (HAVEN 4) - A multicenter, open-label phase III study to evaluate the efficacy, safety, pharmacokinetics and pharmacodynamics of emicizumab given every 4 weeks (Q4W) in patients with hemophilia A. Report No. 1085099. May 2018. (CDS version 4.0).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Emicizumabe é um anticorpo tipo imunoglobulina G4 (IgG4) monoclonal humanizado com estrutura de dupla especificidade (anticorpo biespecífico), que liga o fator IXa ao fator X, produzido por tecnologia de DNA recombinante em células de ovário de hamster chinês (CHO).
Propriedades farmacodinâmicas
Mecanismo de ação
O emicizumabe liga o fator IX ativado ao fator X, restaurando a função faltante do fator VIII ativado, necessária para a hemostasia efetiva.
O emicizumabe não tem relação estrutural nem homologia sequencial com o fator VIII e, como tal, não induz nem reforça o desenvolvimento de inibidores diretos para o fator VIII.
Farmacodinâmica
A hemofilia A é um distúrbio hereditário da coagulação sanguínea ligado ao cromossomo X, decorrente de uma deficiência da função do fator VIII, que resulta em sangramento nas articulações, músculos ou órgãos internos, espontaneamente ou como resultado de trauma acidental ou cirúrgico. A terapia profilática com Hemcibra® encurta o tempo de tromboplastina parcial ativada (TTPA) e aumenta a atividade de fator VIII avaliada (usando um ensaio cromogênico com fatores de coagulação humanos). Esses dois marcadores farmacodinâmicos não refletem o verdadeiro efeito hemostático de emicizumabe in vivo (o TTPA é excessivamente encurtado, e a atividade de fator VIII pode ser superestimada), mas fornecem uma indicação relativa do efeito pró-coagulante de emicizumabe.
Propriedades farmacocinéticas
A farmacocinética do emicizumabe foi determinada através de uma análise não compartimental em indivíduos saudáveis e usando uma análise farmacocinética populacional em um banco de dados composto de 389 pacientes com hemofilia A
Absorção
Depois da administração subcutânea em pacientes com hemofilia A, a meia-vida de absorção foi de 1,6 dias.
Depois de múltiplas administrações subcutâneas de 3 mg/kg, uma vez por semana, durante as primeiras quatro semanas, em pacientes com hemofilia A, a média (± DP) de concentrações plasmáticas mínimas de emicizumabe aumentou até atingir 52,6 ± 13,6 mg/mL na semana 5. As concentrações plasmáticas mínimas médias sustentadas do emicizumabe em estado de equilíbrio foram de 51,1 mg/mL, 46,7 mg/mL e 38,3 mg/mL com as doses de manutenção recomendadas de 1,5 mg/kg uma vez por semana, 3 mg/kg a cada duas semanas ou 6 mg/kg a cada quatro semanas, respectivamente (vide Figura 1, Tabela 15).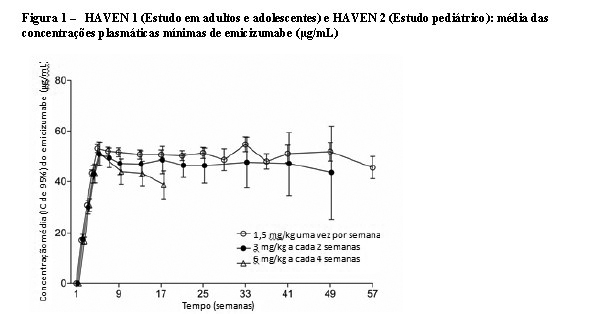
A Cmínima, a Cmáx e as razões de Cmáx/Cmínima médias (± DP) em estado de equilíbrio para as doses de manutenção recomendadas de 1,5 mg/kg uma vez por semana, 3 mg/kg a cada duas semanas ou 6 mg/kg a cada quatro semanas são apresentadas na Tabela 16.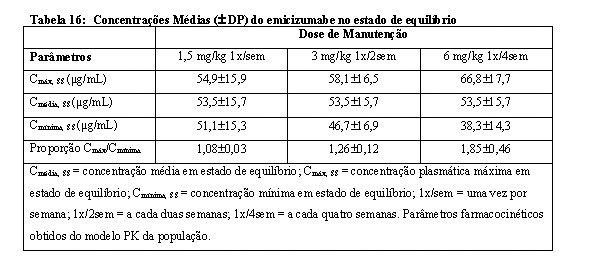
Foram observados perfis semelhantes de farmacocinética após a administração uma vez por semana (3 mg/kg/semana durante 4 semanas seguido por 1,5 mg/kg/semana) em adultos/adolescentes (a partir de 12 anos) e crianças (com menos de 12 anos) (vide Figura 2).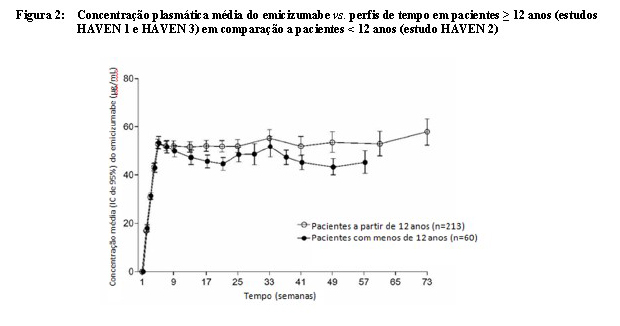
Em indivíduos saudáveis, a biodisponibilidade absoluta depois de administração subcutânea de 1 mg/kg foi entre 80,4% e 93,1%, dependendo do local da injeção. Perfis farmacocinéticos semelhantes foram observados depois da administração subcutânea no abdome, parte superior do braço e coxa. Emicizumabe pode ser administrado de forma intercambiável nessas regiões anatômicas (vide item 8. Posologia e Modo de Usar).
Distribuição
Depois de dose intravenosa única de 0,25 mg/kg de emicizumabe em indivíduos saudáveis, o volume de distribuição no estado de equilíbrio foi 106 mL/kg (isto é, 7,4 L para um adulto de 70 kg). Emicizumabe não se destina a uso intravenoso (vide item 8. Posologia e modo de usar).
O volume de distribuição aparente (V/F), estimado a partir da análise de farmacocinética populacional, em pacientes com hemofilia A, depois de múltiplas doses subcutâneas de emicizumabe, foi de 10,4 L.
Metabolismo
O metabolismo de emicizumabe não foi estudado. Os anticorpos IgG são catabolizados principalmente por proteólise lisossomal e depois eliminados ou reutilizados pelo organismo.
Eliminação
Depois da administração intravenosa de 0,25 mg/kg em indivíduos saudáveis, a depuração total de emicizumabe foi de 3,26 mL/kg/dia (isto é, 0,228 L/dia para um adulto de 70 kg), e a meia-vida terminal média foi de 26,7 dias.
Depois de injeção subcutânea única em indivíduos saudáveis, a meia-vida de eliminação foi de, aproximadamente, quatro a cinco semanas.
Depois de múltiplas injeções subcutâneas em pacientes com hemofilia A, a depuração aparente foi de 0,272 L/dia, e a meia-vida aparente de eliminação foi de 26,8 dias.
Linearidade da dose
Emicizumabe apresentou farmacocinética proporcional à dose em pacientes com hemofilia A em um intervalo de dose de 0,3 a 6 mg/kg, uma vez por semana, depois de administração subcutânea.
Farmacocinética em populações especiais
Insuficiência renal
Nenhum estudo dedicado ao efeito da insuficiência renal sobre a farmacocinética de emicizumabe foi conduzido.
A maioria dos pacientes com hemofilia A na análise farmacocinética da população apresentou função renal normal (N = 332; depuração de creatinina ≥ 90 mL/min) ou insuficiência renal leve (N = 27; depuração de creatinina de 60-89 mL/min). Apenas 2 pacientes apresentaram insuficiência renal moderada (depuração de creatinina de 30-59 mL/min).
Nenhum paciente apresentava insuficiência renal grave. A insuficiência renal leve ou moderada pareceu não ter impacto na farmacocinética do emicizumabe.
Insuficiência hepática
Nenhum estudo dedicado ao efeito da insuficiência hepática sobre a farmacocinética de emicizumabe foi conduzido. A maioria dos pacientes com hemofilia A na análise de farmacocinética populacional tinha função hepática normal (bilirrubina e AST ≤ LSN, N=300) ou insuficiência hepática leve (bilirrubina ≤ LSN e AST > LSN ou bilirrubina < 1,0 a 1,5 x LSN e qualquer AST, N= 51). Apenas 6 pacientes apresentaram insuficiência hepática moderada (1,5 x LSN < bilirrubina ≤ 3 x LSN e qualquer AST). Insuficiência hepática leve ou moderada não afetou a farmacocinética de emicizumabe (vide item 8. Posologia e modo de usar - Instruções de dosagem especiais). O comprometimento hepático foi definido pelos critérios do Instituto Nacional do Câncer (NCI) de disfunção hepática.
Pediatria
O efeito da idade sobre a farmacocinética de emicizumabe foi avaliado em uma análise de farmacocinética populacional que incluiu 5 lactentes (≥ 1 mês a < 2 anos), 55 crianças (≥ 2 anos a < 12 anos) e 50 adolescentes (12 a < 18 anos) com hemofilia A. A idade não afetou a farmacocinética de emicizumabe em pacientes pediátricos (vide item 8. Posologia e modo de usar - Instruções de dosagem especiais).
Geriatria
O efeito da idade sobre a farmacocinética de emicizumabe foi avaliado em uma análise de farmacocinética populacional que incluiu 13 pacientes com 65 anos ou mais (nenhum dos pacientes tinha mais que 77 anos de idade). A biodisponibilidade relativa diminuiu com o aumento da idade, mas não foram observadas diferenças clinicamente importantes na farmacocinética de emicizumabe entre pacientes < 65 anos e pacientes ≥65 anos.
Raça
As análises de farmacocinética populacional em pacientes com hemofilia A mostraram que a raça não afetou a farmacocinética de emicizumabe.
Segurança pré-clínica
Dados pré-clínicos não revelaram nenhum risco especial para humanos com base nos estudos de toxicidade com dose aguda e doses repetidas, que incluíam desfechos de segurança farmacológica e desfechos de toxicidade reprodutiva.
Carcinogenicidade
Não foram realizados estudos de carcinogenicidade para estabelecer o potencial de carcinogenicidade de emicizumabe.
Genotoxicidade
Não foram realizados estudos para estabelecer o potencial de mutagenicidade de emicizumabe.
Comprometimento da fertilidade
Emicizumabe não causou nenhuma alteração toxicológica nos órgãos reprodutivos de macacos cynomolgus machos ou fêmeas em doses de até 30 mg/kg/semana nos estudos de toxicidade geral subcutânea de até 26 semanas de duração e em doses de até 100 mg/kg/semana em um estudo de toxicidade geral intravenosa de quatro semanas.
Toxicidade reprodutiva
Não há dados disponíveis em relação aos possíveis efeitos colaterais de emicizumabe no desenvolvimento embriofetal.
Outros
Em um estudo in vitro de liberação de citocinas que utilizou sangue total de adultos saudáveis, os níveis de citocinas induzidos por emicizumabe foram comparáveis aos induzidos por anticorpos de referência de baixo risco.
Imunogenicidade
Os dados refletem o número de pacientes cujos resultados de exames foram considerados positivos para anticorpos anti-emicizumabe ao ser utilizado o ensaio enzimático por imunoabsorbância (ELISA). Os resultados de ensaios de imunogenicidade podem ser influenciados por diversos fatores, e isso inclui sensibilidade e especificidade do ensaio, manipulação da amostra, momento da coleta da amostra, medicamentos concomitantes e doença subjacente.
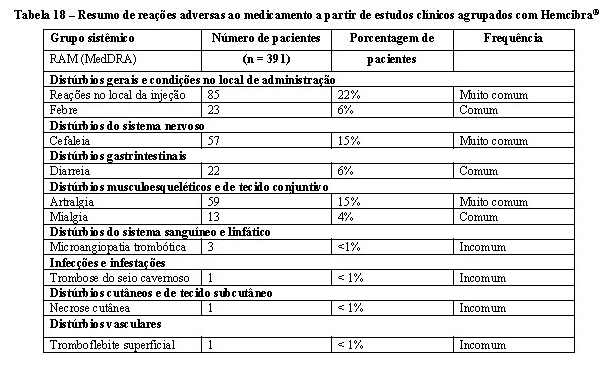
Por essas razões, a comparação da incidência de anticorpos anti-emicizumabe com a incidência de anticorpos contra outros produtos pode induzir a equívocos. A imunogenicidade do Hemcibra® foi avaliada utilizando um ensaio imunoenzimático (ELISA) ou um ensaio de eletroquimioluminescência (ECL). Quatro pacientes foram positivos para anticorpos anti-emicizumabe no estudo de determinação da dose (n = 18). Nos ensaios clínicos agrupados HAVEN, 3,5% (14/398) dos pacientes apresentaram resultado positivo para anticorpos anti-emicizumabe e < 1% (3/398) dos pacientes desenvolveram anticorpos anti-emicizumabe com potencial neutralizante (com base na farmacocinética decrescente). Um paciente do estudo HAVEN 2 que desenvolveu um anticorpo neutralizante anti-emicizumabe apresentou perda de eficácia após 5 semanas de tratamento. Não houve nenhum impacto clinicamente aparente da presença de anticorpos anti-emicizumabe na segurança do produto.
4. CONTRAINDICAÇÕES
Hemcibra® é contraindicado a pacientes com hipersensibilidade conhecida a emicizumabe ou qualquer um de seus excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Gerais
A fim de melhorar a rastreabilidade de medicamentos biológicos, a marca e o número do lote do produto administrado devem ser claramente registrados (ou declarados) no arquivo do paciente.
Oriente os pacientes / cuidadores a registrar o número do lote do produto sempre que Hemcibra® for aplicado fora do ambiente hospitalar.
Hemcibra® e Concentrado de Complexo Protrombínico Ativado (CCPa)
Microangiopatia trombótica associada à Hemcibra® e ao CCPa
Foram relatados casos de microangiopatia trombótica (MAT) em um estudo clínico que incluiu pacientes que recebiam a profilaxia com Hemcibra®, quando foram aplicadas doses cumulativas de concentrado de complexo protrombínico ativado (CCPa) superiores a 100 U/kg/24 horas (vide item 2. Resultados de eficácia). O tratamento para os eventos de MAT incluiu tratamento de suporte com ou sem plasmaferese e hemodiálise. Observou-se evidência de melhora da MAT dentro de uma semana após a descontinuação do CCPa e interrupção do tratamento com Hemcibra®. Essa melhora clínica rápida é diferente da evolução clínica habitual observada na síndrome hemolítico-urêmica atípica e MATs clássicas, como púrpura trombocitopênica trombótica (vide item 2. Resultados de eficácia).
Deve-se ter cautela ao tratar pacientes que correm alto risco de MAT (por exemplo, ter um histórico médico prévio ou história familiar de MAT), ou aqueles que estão recebendo medicações concomitantes conhecidas como um fator de risco para o desenvolvimento de MAT (por exemplo, ciclosporina, quinina, tacrolimus).
Pacientes que recebem profilaxia com Hemcibra® devem ser monitorados quanto ao desenvolvimento de MAT quando recebem CCPa. O médico deve descontinuar imediatamente o CCPa e interromper a terapia com Hemcibra® se ocorrerem sintomas clínicos e/ou achados laboratoriais compatíveis com MAT e tratar conforme indicação clínica. Os médicos e pacientes / cuidadores devem ponderar sobre os benefícios e riscos de reiniciar a profilaxia com Hemcibra® depois da completa resolução de MAT, caso a caso. Se houver indicação de um agente de bypass em um paciente que está fazendo uso de profilaxia com Hemcibra®, consulte a seguir as recomendações de dose para uso dos agentes de bypass.
Tromboembolismo associado à Hemcibra® e ao CCPa
Foram relatados eventos trombóticos em um estudo clínico com pacientes que recebiam profilaxia com Hemcibra® quando doses cumulativas superiores a 100 U/kg / 24 horas de CCPa foram aplicadas (vide item 2. Resultados de eficácia). Nenhum dos casos precisou de terapia de anticoagulação, diferentemente dos tratamentos habituais de eventos trombóticos. Foi observada evidência de melhora ou resolução após a descontinuação do CCPa (vide item 2. Resultados de eficácia).
Pacientes que recebem a profilaxia com Hemcibra® devem ser monitorados quanto ao desenvolvimento de tromboembolismo quando aplicam o CCPa. O médico deve descontinuar imediatamente o CCPa e interromper a terapia com Hemcibra® se ocorrerem sintomas clínicos, resultados de exames de imagem e/ou achados laboratoriais compatíveis com eventos trombóticos e tratar conforme indicação clínica. Os médicos e pacientes / cuidadores devem ponderar sobre os benefícios e riscos de reiniciar a profilaxia com Hemcibra® depois da completa resolução dos eventos trombóticos, caso a caso. Se houver indicação de um agente de bypass em um paciente em uso da profilaxia com Hemcibra®, consulte a seguir as recomendações de dose para uso dos agentes de bypass.
Orientação sobre o uso de agentes de bypass em pacientes em uso de profilaxia com Hemcibra®
O tratamento com agentes de bypass deve ser descontinuado no dia anterior ao início da terapia com Hemcibra®.
Os médicos devem discutir com todos os pacientes e/ou cuidadores a dose exata e o esquema dos agentes de bypass a serem utilizados, se necessário, enquanto fazem uso da profilaxia com Hemcibra®.
Hemcibra® aumenta o potencial de coagulação dos pacientes. A dose necessária do agente de bypass pode, portanto, ser menor que a utilizada sem a profilaxia com Hemcibra®. A dose e a duração do tratamento com agentes de bypass vai depender da localização e extensão do sangramento e da condição clínica do paciente. Evite o uso de CCPa, a menos que não existam outras opções / alternativas de tratamento disponíveis. Se houver indicação do CCPa a paciente em uso de profilaxia com Hemcibra®, a dose inicial não deve exceder 50 U/kg e o monitoramento laboratorial é recomendado (incluindo, mas não restringindo ao monitoramento da função renal, avaliações plaquetárias, e avaliação para trombose). Se a hemorragia não for controlada com a dose inicial de CCPa até 50 U/kg, devem ser administradas doses adicionais de CCPa sob orientação ou supervisão médica, tendo em consideração a monitorização laboratorial para o diagnóstico de MAT ou tromboembolismo e verificação de hemorragias antes da administração repetida. A dose total de CCPa não deve exceder 100 U/kg nas primeiras 24 horas de tratamento. Os médicos assistentes precisam ponderar cuidadosamente sobre o risco de MAT e tromboembolismo (TE) frente o risco de sangramento ao considerar um tratamento com CCPa acima da dose máxima de 100 U/kg nas primeiras 24 horas.
Nos estudos clínicos, não foram observados casos de microangiopatia trombótica ou eventos trombóticos com o uso de FVII humano recombinante ativado (rFVIIa) isoladamente em pacientes em uso de profilaxia com Hemcibra®.
A orientação para administração de agentes de bypass deve ser seguida durante, pelo menos, seis meses depois da descontinuação da profilaxia com Hemcibra® (vide item 3. Características farmacológicas - Propriedades farmacocinéticas, eliminação).
Interferência em exame laboratorial de coagulação
Hemcibra® restaura a atividade perdida do cofator tenase do fator VIII ativado (FVIIIa). Exames laboratoriais de coagulação baseados em coagulação intrínseca (por exemplo, TTPA) medem o tempo total de coagulação, e isso inclui o tempo necessário para ativação de FVIII para FVIIIa pela trombina. Esses testes baseados na via intrínseca fornecerão tempos de coagulação evidentemente encurtados com Hemcibra®, que não precisa de ativação pela trombina. O tempo de coagulação intrínseca excessivamente encurtado vai então afetar todos os ensaios de fator único baseados em TTPA, como o ensaio de atividade de FVIII em um estágio (vide Tabela 17). No entanto, ensaios de fator único que utilizam métodos cromogênicos ou de base imunológica não são afetados por Hemcibra® e podem ser usados para monitorar parâmetros de coagulação durante o tratamento, com considerações específicas para ensaios de atividade cromogênica de FVIII, como descrito a seguir. Testes cromogênicos de atividade de fator VIII podem ser produzidos com proteínas de coagulação humanas ou bovinas. Ensaios que contêm fatores de coagulação humanos são responsivos a Hemcibra®, mas podem superestimar o potencial hemostático clínico de Hemcibra®. De forma inversa, os ensaios que contêm fatores de coagulação bovinos são insensíveis a Hemcibra® (sem atividade medida) e podem ser usados para monitorar a atividade de fa