HARVONI
GILEAD
sofosbuvir + ledipasvir
Antiviral contra o vírus da hepatite C.
Apresentações.
Harvoni é apresentado em frascos contendo 28 comprimidos revestidos. Cada comprimido revestido contém 90 mg de ledipasvir e 400 mg de sofosbuvir.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 12 ANOS
Composição.
Cada comprimido revestido contém 90 mg de ledipasvir e 400 mg de sofosbuvir. Excipientes: copovidona, lactose monoidratada, celulose microcristalina, croscarmelose sódica, dióxido de silício coloidal, estearato de magnésio, álcool polivinílico, dióxido de titânio, macrogol e talco.
Informações técnicas.
1. INDICAÇÕES
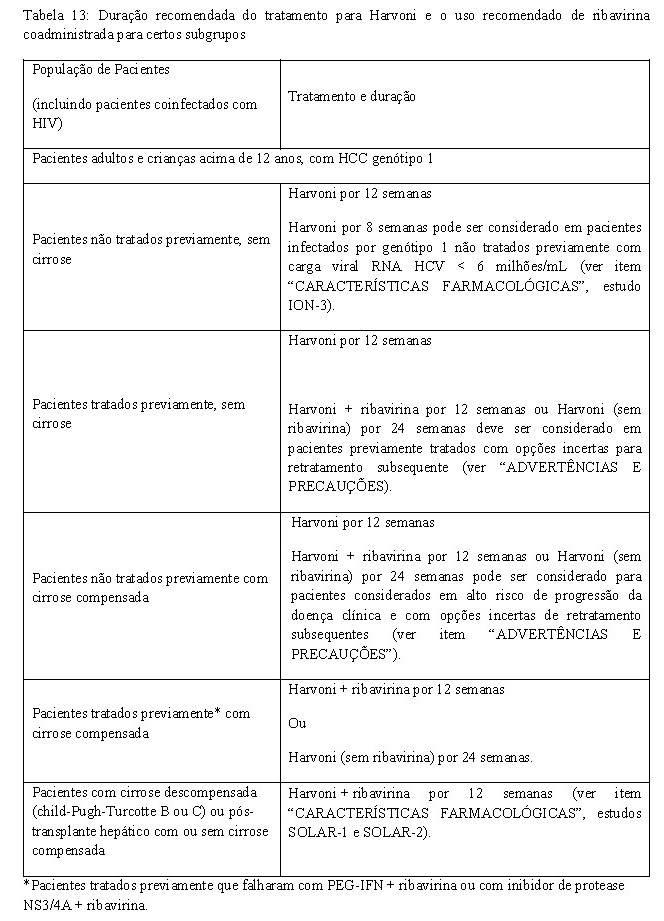
Harvoni é indicado, em combinação ou não com ribavirina, para o tratamento da Hepatite C Crônica (HCC) genótipo 1 em adultos e crianças acima de 12 anos (ver POSOLOGIA E MODO DE USAR, ADVERTÊNCIAS E PRECAUÇÕES e CARACTERÍSTICAS FARMACOLÓGICAS).
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
A eficácia de Harvoni (ledipasvir [LDV]/sofosbuvir [SOF]) foi avaliada em três estudos de Fase 3 em regime aberto com dados disponíveis para um total de 1.950 pacientes com HCC genótipo 1. Os três estudos de Fase 3 incluíram: um estudo conduzido em pacientes não cirróticos virgens de tratamento (ION-3); um estudo em pacientes cirróticos e não cirróticos virgens de tratamento (ION-1); e um estudo em pacientes cirróticos e não cirróticos que falharam em terapia anterior com um regime baseado em interferon, incluindo regimes contendo um inibidor de protease do VHC (ION-2). Os pacientes nesses estudos tinham doença hepática compensada. Todos os três estudos de Fase 3 avaliaram a eficácia de ledipasvir/sofosbuvir em associação ou não com ribavirina.
A duração do tratamento foi fixada em cada estudo. Os valores plasmáticos do RNA do VHC foram medidos durante os estudos clínicos usando o teste VHC COBAS TaqMan (versão 2.0), para uso com o "High Pure System". O ensaio teve um limite inferior de quantificação (LIQ) de 25 UI/mL. A RVS foi o desfecho primário para determinar a taxa de cura do VHC que foi definida como RNA de VHC menor que o LIQ 12 semanas após o término do tratamento.
- Adultos virgens de tratamento sem cirrose - ION-3 (estudo 0108) - Genótipo 1
O estudo de fase 3 ION-3 avaliou 8 semanas de tratamento com ledipasvir/sofosbuvir com ou sem ribavirina e 12 semanas de tratamento com ledipasvir/sofosbuvir em pacientes virgens de tratamento não cirróticos com HCC genótipo 1. Os pacientes foram randomizados em uma razão 1:1:1 para um dos três grupos de tratamento e estratificados por genótipo do VHC (1a versus 1b).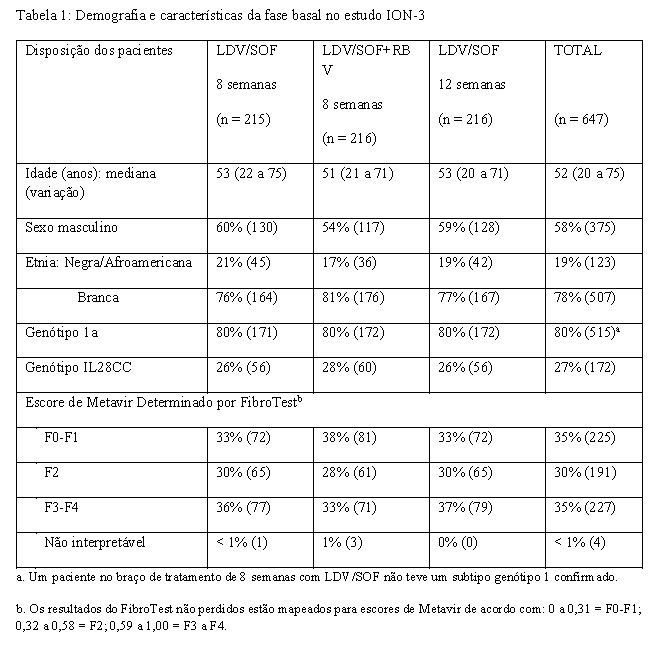
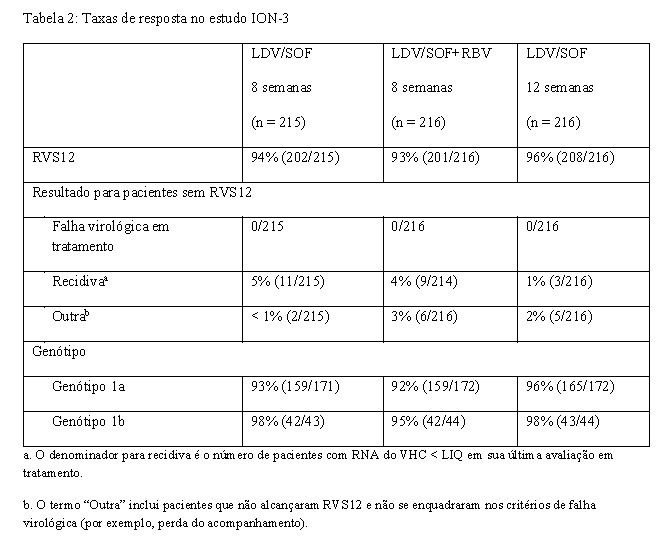
O tratamento de 8 semanas com ledipasvir/sofosbuvir sem ribavirina foi não-inferior ao tratamento de 8 semanas com ledipasvir/sofosbuvir com ribavirina (diferença de tratamento de 0,9%; intervalo de confiança de 95%: -3,9% a 5,7%) e ao tratamento de 12 semanas de ledipasvir/sofosbuvir (diferença de tratamento de -2,3%; intervalo de confiança de 97,5%: -7,2% a 3,6%). Dentre os pacientes com um RNA do VHC na fase basal < 6 milhões UI/mL, a RVS foi de 97% (119/123) com o tratamento de 8 semanas com ledipasvir/sofosbuvir e de 96% (126/131) com o tratamento de 12 semanas com ledipasvir/sofosbuvir.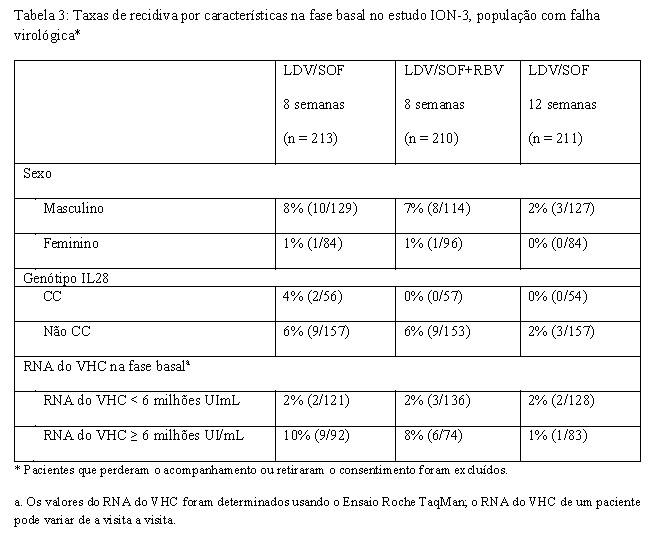
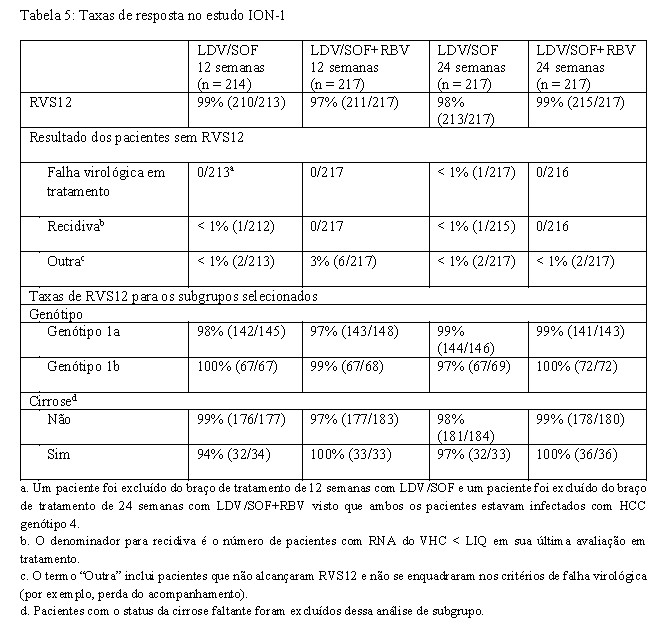
- Adultos virgens de tratamento com ou sem cirrose - ION-1 (estudo 0102) - Genótipo 1
O ION-1 foi um estudo de fase 3, randomizado, em regime aberto que avaliou 12 e 24 semanas de tratamento com ledipasvir/sofosbuvir com ou sem ribavirina em 865 pacientes virgens de tratamento com HCC genótipo 1 incluindo aqueles com cirrose (randomizados 1:1:1:1). A randomização foi estratificada para a presença ou ausência de cirrose e pelo genótipo do VHC (1a versus 1b).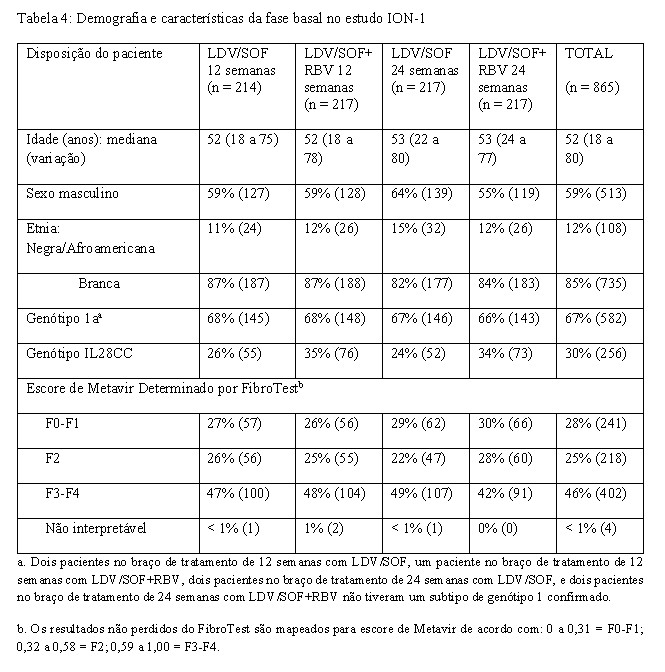
- Adultos previamente tratados com ou sem cirrose - ION-2 (estudo 0109) - Genótipo 1
O ION-2 foi um estudo de fase 3, randomizado, em regime aberto que avaliou 12 e 24 semanas de tratamento com ledipasvir/sofosbuvir com ou sem ribavirina (randomizados 1:1:1:1) em pacientes infectados pelo VHC genótipo 1 com ou sem cirrose que falharam antes da terapia com um regime baseado em interferon, incluindo regimes contendo um inibidor de protease do VHC. A randomização foi estratificada pela presença ou ausência de cirrose, genótipo do VHC (1a versus 1b) e resposta a terapia prévia para o VHC (recidiva/reativação versus ausência de resposta).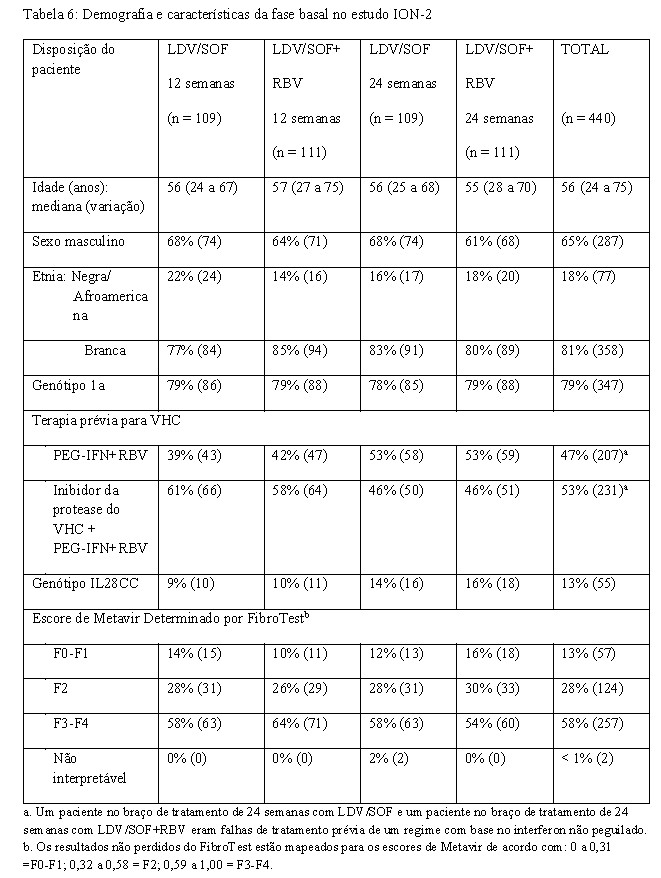
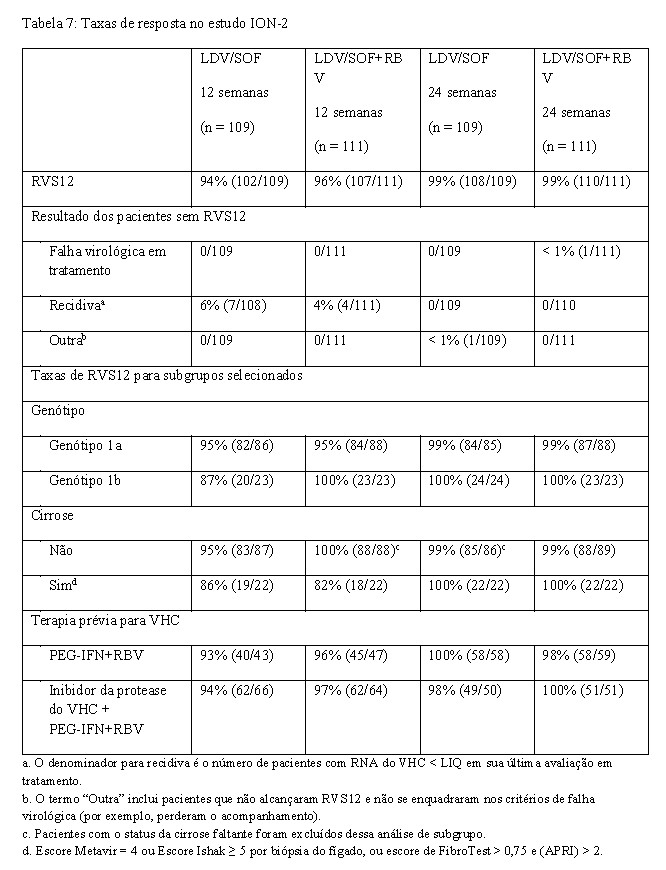
A Tabela 8 apresenta taxas de recidiva com os regimes de 12 semanas (com ou sem ribavirina) para subgrupos selecionados (ver também "Efeito das variantes associadas à resistência ao VHC na fase basal sobre o resultado do tratamento"). Em pacientes não cirróticos, recidivas ocorreram apenas na presença de RAVs na NS5A na fase basal, e durante terapia com ledipasvir/sofosbuvir sem ribavirina. Em pacientes cirróticos, as recidivas ocorreram com ambos os regimes, e na ausência e na presença de RAVs na NS5A na fase basal.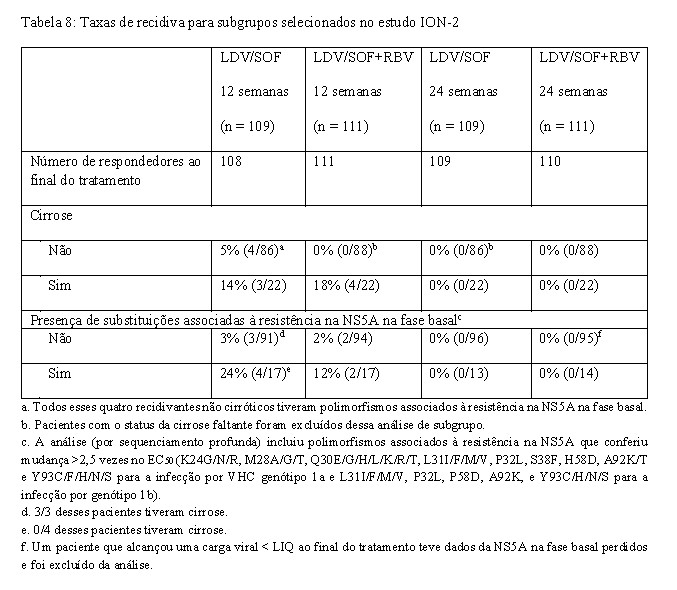
- Adultos cirróticos previamente tratados- SIRIUS - Genótipo 1
O estudo de fase 2 SIRIUS incluiu pacientes com cirrose compensada que falharam ao primeiro tratamento com interferon peguilado (PEG-IFN) + ribavirina e que, falharam também ao tratamento que consistiu em interferon peguilado + ribavirina + um inibidor da protease NS3/4A. A cirrose foi determinada por biópsia hepática, FibroScan ( > 12,5 kPa) ou FibroTest > 0,75 e por um índice da razão aspartato aminotransferase (AST):plaquetas (APRI) de > 2.
O estudo (duplo cego, controlado com placebo) avaliou 24 semanas de tratamento com ledipasvir/sofosbuvir (com placebo da ribavirina) versus 12 semanas de tratamento com ledipasvir/sofosbuvir com ribavirina. Os pacientes deste último braço de tratamento receberam placebo (para ledipasvir/sofosbuvir e ribavirina) durante as primeiras 12 semanas, seguido de tratamento ativo cego durante as 12 semanas seguintes. Os pacientes foram estratificados em função do genótipo do VHC (1a versus 1b) e da resposta ao tratamento anterior (se obtido um RNA do VHC < LIQ).
As características basais e demográficas foram balanceadas nos dois grupos de tratamento. A idade mediana era de 56 anos (entre 23 a 77); 74% dos pacientes eram do sexo masculino; 97% eram de etnia branca: 63% tinham infecção pelo VHC de genótipo 1a; 94% tinham alelos IL28B não CC (CT ou TT).
Dos 155 pacientes incluídos, 1 paciente descontinuou o tratamento enquanto estava recebendo tratamento com placebo. Dos 154 pacientes remanescentes, um total de 149 alcançou RVS12 nos dois grupos de tratamento: 96% (74/77) dos pacientes no grupo de ledipasvir/sofosbuvir com ribavirina durante 12 semanas e 97% (75/77) dos pacientes no grupo ledipasvir/sofosbuvir durante 24 semanas. Os 5 pacientes que não alcançaram uma RVS12 recidivaram após terem resposta de fim de tratamento (ver a subitem "Resistência" - do item "CARACTERÍSTICAS FARMACOLÓGICAS").
- Adultos previamente tratados que não responderam a sofosbuvir + ribavirina ± PEG-IFN
Os resultados de dois estudos clínicos sugerem que ledipasvir/sofosbuvir podem ser eficazes em pacientes que não responderam anteriormente ao tratamento com sofosbuvir + ribavirina ± PEG-IFN. No estudo de fase 2 1118, 44 pacientes com infecção do genótipo 1, incluindo 12 pacientes cirróticos, que não responderam anteriormente ao tratamento com sofosbuvir + ribavirina + PEG-IFN ou com sofosbuvir + ribavirina, foram tratados com ledipasvir/sofosbuvir + ribavirina durante 12 semanas; a RVS foi de 100% (44/44). No estudo de fase 3 ION-4 foram incluídos 13 pacientes com VHC de genótipo 1 coinfectados por HIV-1, incluindo 1 paciente cirrótico, que não responderam a um regime com sofosbuvir + ribavirina; a RVS foi de 100% (13/13) após 12 semanas de tratamento com ledipasvir/sofosbuvir.
- Adultos coinfectados por VHC/HIV - ION-4
ION-4 consistiu num estudo clínico de fase 3 aberto que avaliou a segurança e eficácia de 12 semanas de tratamento com ledipasvir/sofosbuvir sem ribavirina em pacientes virgens de tratamento e em pacientes com exposição anterior ao tratamento para o VHC, com HCC de genótipo 1 ou 4, que estavam coinfectados com o HIV-1. Os pacientes com exposição anterior ao tratamento falharam ao tratamento prévio com PEG-IFN + ribavirina ± um inibidor da protease do VHC ou com sofosbuvir + ribavirina ± PEG-IFN. Os pacientes estavam em tratamento antirretroviral estável para o HIV-1 que incluía emtricitabina/ fumarato de tenofovir desoproxila, administrados com efavirenz, rilpivirina ou raltegravir.
A idade mediana era de 52 anos (intervalo: 26 a 72); 82% dos pacientes eram do sexo masculino; 61% eram de etnia branca; 34% eram de etnia negra; 75% tinham infecção pelo VHC de genótipo 1a; 2% tinham infecção pelo VHC de genótipo 4; 76% tinham alelos IL28B não CC (CT ou TT); e 20% tinham cirrose compensada. Cinquenta e cinco por cento (55%) dos pacientes tinham tido exposição anterior a tratamento.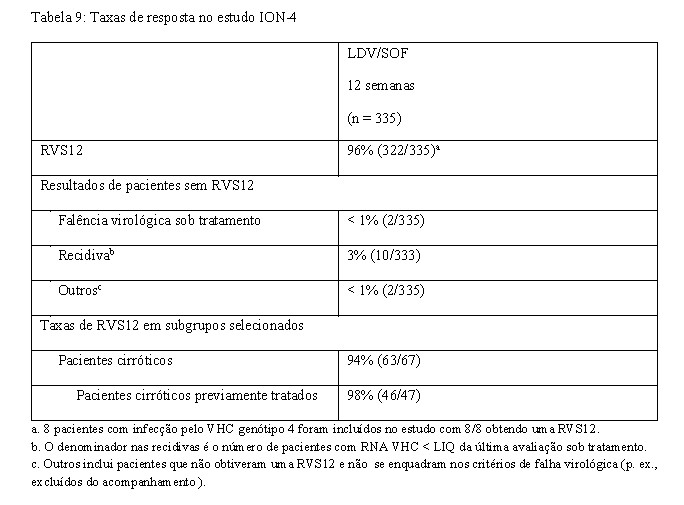
- Adultos coinfectados com VHC/HIV - ERADICATE
O ERADICATE foi um estudo de fase 2 em regime aberto para avaliar 12 semanas de tratamento com ledipasvir/sofosbuvir em 50 pacientes com HCC genótipo 1 coinfectados com HIV. Todos os pacientes eram virgens de tratamento para terapia para o VHC sem cirrose, 26% (13/50) dos pacientes eram virgens para antirretrovirais para HIV e 74% (37/50) dos pacientes estavam recebendo terapia antirretroviral para HIV concomitante. No momento da análise interina 40 pacientes haviam alcançado 12 semanas pós-tratamento e a RVS12 foi de 98% (39/40).
- Pacientes aguardando transplante de fígado e pós-transplante de fígado - SOLAR-1 e SOLAR-2
O SOLAR-1e SOLAR-2 são dois estudos de fase 2 em regime aberto, multicêntricos avaliando 12 e 24 semanas de tratamento com ledipasvir/sofosbuvir em combinação com ribavirina em pacientes com HCC genótipo 1 e 4 que passaram por transplante de fígado ou que possuem doença hepática descompensada. Ambos os estudos são idênticos quanto ao desenho. Os pacientes foram recrutados em um dos sete grupos com base à condição de transplante hepático e a gravidade da comprometimento hepática (veja Tabela 10). Pacientes com CPT > 12 foram excluídos. Dentre cada grupo, os pacientes foram randomizados em uma relação 1:1 para receber ledipasvir/sofosbuvir + ribavirina por 12 ou 24 semanas.
As características basais e demográficas foram balanceadas entre os grupos de tratamento. Dos 670 pacientes tratados, a idade média era de 59 anos (variação: 21 a 81 anos); 77% dos pacientes eram do sexo masculino; 91% eram brancos; o índice médio de massa corporal era 28Kg/m2 (variação de 18 a 49 Kg/m2); 94% e 6% tinham infecção por HCV genótipo 1 e 4, respectivamente; 78% dos pacientes falharam um tratamento de HCV anterior. Entre os pacientes que tinham cirrose descompensada (pre- ou pós-transplante), 64% e 36% eram CPT classe B e C no momento da seleção, respectivamente, 24% tinham um MELD (Modelo para Doença Hepática em Estágio Terminal) maior do que 15.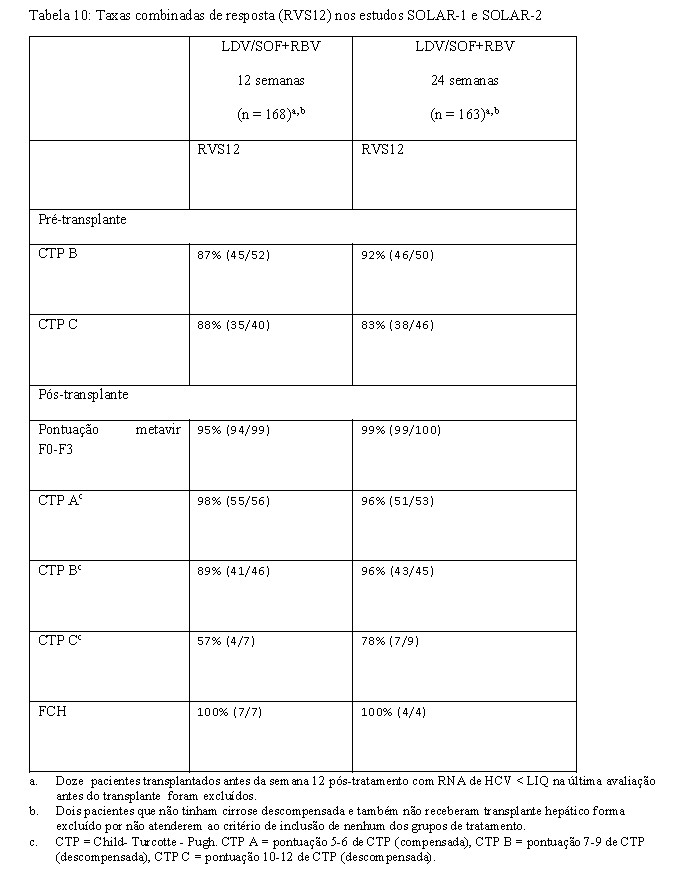
Quarenta pacientes com hepatite C genótipo 4 foram incluídos nos estudos SOLAR-1 e SOLAR-2, as RVS12 foram 92% (11/12) e 100% (10/10) nos pacientes pós-transplante sem cirrose descompensada e 60% (6/10) e 75% (6/8) nos pacientes com cirrose descompensada (pré e pós-transplante hepático) tratados por 12 ou 24 semanas, respectivamente. Dos 7 pacientes que falharam em alcançar a RVS12, 3 apresentaram recidiva, todos tiveram cirrose descompensada e foram tratados com ledipasvir/sofosbuvir + ribavirina por 12 semanas.
Alterações na pontuação MELD e CPT a partir do basal até a semana 12 do pós-tratamento foram analisadas para todos os pacientes com cirrose descompensada (pré ou pós-transplante) que atingiram a RVS12 e de quem os dados estavam disponíveis (n = 123) para análise do efeito da RVS12 na função hepática.
Alterações na pontuação MELD: dentre os pacientes que atingiram a RVS12 com 12 semanas de tratamento com ledipasvir/sofosbuvir + ribavirina, 57% (70/123) e 19% (23/123) tiveram melhora ou nenhuma mudança na pontuação MELD a partir do basal até a semana 12 pós-tratamento, respectivamente; dos 32 pacientes cuja pontuação MELD estava ≥ 15 no basal, 59% (19/32) tinham pontuação MELD < 15 na semana 12 pós-tratamento. A melhora na pontuação MELD observada deveu-se largamente a melhoras na bilirrubina total.
Alterações na pontuação e classe CPT: dentre aqueles que atingiram a RVS12 com 12 semanas de tratamento com ledipasvir/sofosbuvir com ribavirina, 60% (74/123) e 34% (42/123) tiveram melhora ou nenhuma alteração na pontuação CPT do basal ao pós-tratamento com 12 semanas, respectivamente; dos 32 pacientes que tiveram cirrose CPT C no basal, 53% (17/32) tinham cirrose CPT B na semana 12 pós-tratamento; dos 88 pacientes que tinha cirrose CPT B no basal, 25% (22/88) tinham cirrose CPT A na semana 12 pós-tratamento. A melhora na pontuação CPT observada deveu-se largamente a melhoras na bilirrubina total e na albumina.
- Pacientes com Compromentimento renal
O estudo 0154 foi um ensaio clínico aberto que avaliou 12 semanas de tratamento com Harvoni em 18 indivíduos infectados pelo VHC genótipo 1 com comprometimento renal grave sem diálise. No início do estudo, dois indivíduos (11%) apresentaram cirrose e a TFGe média foi de 24,9 mL/min (variação: 9,0-39,6). A taxa de RVS foi de 100% (18/18).
O estudo 4063 foi um ensaio clínico de três braços, aberto, que avaliou 8, 12 e 24 semanas de tratamento com Harvoni em um total de 95 indivíduos com genótipo 1 (72%), 2 (22%), 4 (2%), 5 (1%) ou 6 (2%) com Hepatite C crônica e Doença Renal em Estágio Final (DREF) necessitando hemodiálise: 45 indivíduos virgens de tratamento infectados pelo VHC genótipo 1 e sem cirrose receberam Harvoni por 8 semanas; 31 indivíduos previamente tratados infectados pelo VHC com genótipo 1 e indivíduos virgens de tratamento ou previamente tratados com infecção pelos genótipos 2, 5 e 6 sem cirrose receberam Harvoni por 12 semanas; e 19 indivíduos infectados pelo genótipo 1, 2 e 4 com cirrose compensada receberam Harvoni por 24 semanas. Do total de 95 indivíduos, no início do estudo, 20% dos indivíduos tinham cirrose, 22% foram previamente tratados, 21% tinham recebido transplante de rim, 92% estavam em hemodiálise e 8% estavam em diálise peritoneal; a duração média da diálise foi de 11,5 anos (variação: 0,2 a 43,0 anos). As taxas de RVS para os grupos de tratamento com Harvoni de 8, 12 e 24 semanas foram de 93% (42/45), 100% (31/31) e 79% (15/19), respectivamente. Dos 7 indivíduos que não atingiram RVS12, nenhum apresentou falha virológica ou recidivou.
- População pediátrica
A eficácia do ledipasvir/sofosbuvir em adolescentes infectados pelo VHC com 12 a < 18 anos de idade foi avaliada num ensaio clínico aberto de Fase 2 que incluiu 100 pacientes com HCC de genótipo 1 (Estudo 1116). O estudo contou com um total de 80 pacientes (80%) sem exposição anterior ao tratamento e 20 pacientes (20%) anteriormente tratados. Todos os pacientes incluídos no estudo foram tratados com ledipasvir/sofosbuvir durante 12 semanas.
As características iniciais e demográficas foram equilibradas nos dois grupos de pacientes. A mediana da idade dos 100 pacientes tratados foi de 15 anos (intervalo: 12 a 17); 63% dos pacientes eram do sexo feminino; 90% eram caucasianos, 7% eram de raça negra e 2% eram asiáticos; 13% eram de origem hispânica/latina; o peso médio era de 61,3 kg (intervalo: 33,0 a 126,0 kg); 55% tinham níveis iniciais de RNA do VHC iguais ou superiores a 800.000 UI/ml; 81% tinham infecção pelo VHC de genótipo 1; 76% tinham alelos IL28B não CC (CT ou TT) e 1% tinham cirrose conhecida. A maior parte dos pacientes (84%) tinha sido infectada por transmissão vertical.
A taxa de RVS12 foi de 98% globalmente (98% [78/80] em pacientes sem exposição anterior ao tratamento e 100% [20/20] em pacientes anteriormente tratados). Um total de 2 em 100 pacientes (2%), ambos sem exposição anterior ao tratamento, não obtiveram uma RVS12 (devido à perda de seguimento). Nenhum paciente teve falha virológica (ver item POSOLOGIA E MODO DE USAR para informações sobre utilização pediátrica).
- Referências bibliográficas
Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 2014a; 370 (16):1483-93.
Afdhal N, Zeuzem S, Kwo P, al. e. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection [Supplementary Appendix]. N Engl J Med 2014b:1-15.
Balistreri WF, Murray KF, Rosenthal P, et al. The Safety and Effectiveness of Ledipasvir2Sofosbuvir in Adolescents 12-17 Years Old With Hepatitis C Virus Genotype 1 Infection Hepatology. 2017;66(2):371-378.
Bourliere M, Bronowicki JP, De Ledinghen V, Hezode C, Zoulim F, Mathurin P, et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect Dis 2015; 15: 397-404
Charlton M, Everson GT, Flamm SL, Kumar P, Landis C, Brown Jr RS, et al. Ledipasvir and Sofosbuvir Plus Ribavirin for Treatment of HCV Infection in Patients with Advanced Liver Disease. Gastroenterology 2015:649-59.
Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med 2014; 370 (20):1879-88.
Manns M, Samuel D, Gane EJ, Mutimer D, et al. Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis. 2016 Jun;16(6):685-97.
Naggie S, Cooper C, Saag M, Workowski K, Ruane P, Towner WJ, et al. Ledipasvir and Sofosbuvir for HCV in Patients Coinfected with HIV-1. N Engl J Med 2015:705-13.
Osinusi A, Townsend K, Kohli A, Nelson A, Seamon C, Meissner EG, et al. Virologic Response Following Combined Ledipasvir and Sofosbuvir Administration in Patients With HCV Genotype 1 and HIV Co-infection. JAMA 2015; 313 (12):1232-9.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
- Mecanismo de ação
Ledipasvir é um inibidor do VHC voltado à proteína NS5A do VHC, que é essencial tanto para a replicação do RNA quanto para a montagem dos virions do VHC. A confirmação bioquímica da inibição da NS5A pelo ledipasvir não é possível atualmente visto que a NS5A não tem função enzimática. Estudos de seleção de resistência in vitro e de resistência cruzada indicam que ledipasvir tem como alvo a NS5A como seu modo de ação.
Sofosbuvir é um inibidor pan-genotípico da RNA polimerase dependente de RNA NS5B do VHC, que é essencial para a replicação viral. Sofosbuvir é um pró-fármaco de nucleotídeos que sofre metabolismo intracelular para formar o análogo de trifosfato uridina farmacologicamente ativo (GS 461203), que pode ser incorporado ao RNA do VHC pela polimerase NS5B e atua como um terminador da cadeia. O GS-461203 (o metabólito ativo do sofosbuvir) não é nem um inibidor das polimerases de DNA e RNA humanas nem um inibidor de RNA polimerase mitocondrial.
- Atividade antiviral
Os valores de EC50 de ledipasvir e sofosbuvir contra replicons completos ou replicons quiméricos que codificam as sequências da NS5A e NS5B de isolados clínicos são detalhados na Tabela 11. A presença de 40% de soro humano não teve efeito sobre a atividade anti-VHC de sofosbuvir, mas reduziu a atividade anti-VHC de ledipasvir em 12 vezes contra replicons de VHC de genótipo 1a.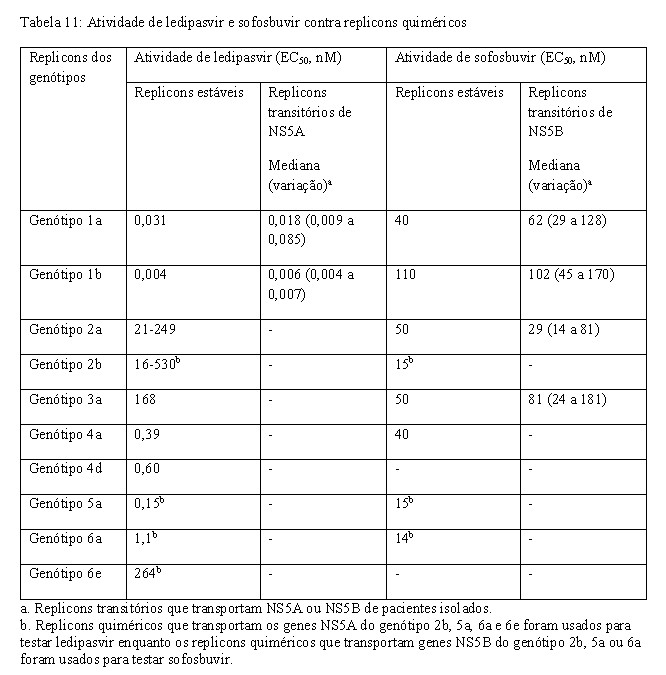
-Resistência
Em cultura de células
Replicons de VHC com suscetibilidade reduzida a ledipasvir foram selecionados em cultura de células para o genótipo 1a e 1b. Suscetibilidade reduzida a ledipasvir foi associada com a substituição primária da NS5A Y93H tanto no genótipo 1a quanto no 1b. Adicionalmente uma substituição Q30E desenvolveu-se nos replicons de genótipo 1a. Mutagênese sítio-dirigida das variantes associadas à resistência (RAVs) na NS5A mostrou que as substituições conferindo uma mudança > 100 e ≤ 1.000 vezes na suscetibilidade a ledipasvir são Q30H/R, L31I/M/V, P32L e Y93T no genótipo 1a e P58D e Y93S no genótipo 1b; e as substituições conferindo uma mudança > 1.000 vezes são M28A/G, Q30E/G/K, H58D, Y93C/H/N/S no genótipo 1a e A92K e Y93H no genótipo 1b.
Replicons de VHC com suscetibilidade reduzida a sofosbuvir foram selecionados em cultura de células para múltiplos genótipos incluindo 1b, 2a, 2b, 3a, 4a, 5a e 6a. A suscetibilidade reduzida a sofosbuvir foi associada com a substituição primária da NS5B S282T em todos os replicons dos genótipos examinados. Mutagênese sítio-dirigida à substituição S282T em replicons de 8 genótipos conferiu suscetibilidade 2 a 18 vezes reduzida a sofosbuvir e reduziu a capacidade de replicação viral em 89% a 99% em comparação com o tipo selvagem correspondente.
Em estudos clínicos - Genótipo 1
Em uma análise agrupada de pacientes que receberam ledipasvir/sofosbuvir em estudos de Fase 3 (ION-3, ION-1 and ION-2), 37 pacientes (29 com genótipo 1a e 8 com genótipo 1b) foram qualificados para análise de resistência devido a falha virológica ou descontinuação precoce da medicação de estudo e que tinham RNA de VHC > 1.000 IU/mL. Os dados de sequenciamento de NS5A e NS5B pós-fase basal (limite do ensaio de 1%) estavam disponíveis para 37/37 e 36/37 pacientes, respectivamente.
As RAVs na NS5A foram observadas em isolados pós-basais de 29/37 pacientes (22/29 com genótipo 1a e 7/8 com genótipo 1b) não alcançaram resposta virológica sustentada (RVS). Dos 29 pacientes com genótipo 1a que se qualificaram para os testes de resistência, 22/29 (76%) pacientes eram portadores de uma ou mais RAVs na NS5A nas posições K24, M28, Q30, L31, S38 e Y93 na falha, enquanto os restantes 7/29 pacientes não tiveram RAVs na NS5A detectadas na falha. As variantes mais comuns foram Q30R, Y93H e L31M. Dos 8 pacientes com genótipo 1b que se qualificaram para os testes de resistência, 7/8 (88%) eram portadores de uma ou mais RAVs na NS5A nas posições L31 e Y93 na falha, enquanto 1/8 pacientes não tiveram RAVs na NS5A na falha. A variante mais comum foi Y93H. Entre os 8 pacientes que não apresentaram RAVs na NS5A na falha, 7 pacientes receberam 8 semanas de tratamento (n = 3 com ledipasvir/sofosbuvir; n = 4 com ledipasvir/sofosbuvir + ribavirina) e 1 paciente recebeu ledipasvir/sofosbuvir por 12 semanas. Nas análises fenotípicas, isolados pós-basais de pacientes que eram portadores de RAVs na NS5A na falha mostraram suscetibilidade reduzida de 20 até pelo menos 243 vezes (a dose mais elevada testada) a ledipasvir. A mutagênese sítio-dirigida da substituição Y93H tanto no genótipo 1a quando no 1b bem como das substituições Q30R e L31M no genótipo 1a conferiram suscetibilidade reduzida a ledipasvir (mudança na EC50 variando de 544 vezes a 1.677 vezes).
A substituição S282T na NS5B associada à resistência a sofosbuvir não foi detectada em nenhum isolado de falha virológica dos estudos de Fase 3. Contudo, a substituição S282T na NS5B em combinação com as substituições L31M, Y93H e Q30L na NS5A foram detectadas em um paciente na falha após 8 semanas de tratamento com ledipasvir/sofosbuvir a partir de um estudo de Fase 2 (LONESTAR). Esse paciente foi subsequentemente retratado com ledipasvir/sofosbuvir + ribavirina por 24 semanas e alcançou RVS após o retratamento.
No estudo SIRIUS (veja item "RESULTADOS DE EFICÁCIA") 5 pacientes com infecção pelo VHC genótipo 1 recidivaram após tratamento com ledipasvir/sofosbuvir com ou sem ribavirina. Observaram-se RAVs da NS5A da recidiva em 5/5 pacientes (por genótipo 1a: Q30R/H + L31M/V [1] e Q30R [=1]; para genótipo 1b: Y93H [n=3]).
No estudo SOLAR-1 (veja item "RESULTADOS DE EFICÁCIA"), 13 pacientes com infecção do genótipo 1 recidivaram após tratamento com ledipasvir/sofosbuvir com ribavirina. Observaram-se RAVs da NS5A da recidiva em 11/13 pacientes (com o genótipo 1a: Q30R isolada [n = 2], Y93C [n = 1], Y93H/C [n = 2], Q30R + H58D [n = 1], M28T + Q30H [n = 1]; com o genótipo 1b: Y93H [n = 3], Y93H/C [n = 1]).
- Efeito das variantes associadas à resistência ao VHC na fase basal sobre o resultado do tratamento
Genótipo 1:
Análises foram conduzidas para explorar a associação entre as RAVs na NS5A pré-existentes na fase basal e o resultado do tratamento. Na análise agrupada dos estudos de Fase 3, 16% dos pacientes apresentavam RAVs na NS5A na fase basal identificadas por população ou sequenciamento independentemente do subtipo. As RAVs na NS5A na fase basal estavam super- representada em pacientes que experimentaram recaídas nos estudos de Fase 3 (ver "RESULTADOS DE EFICÁCIA").
Após 12 semanas de tratamento com ledipasvir/sofosbuvir (sem ribavirina) em pacientes anteriormente tratados (braço 1 do estudo ION-2), 4/4 pacientes com RAVs da NS5A basal que conferiram uma alteração ≤ 100 vezes relativamente ao ledipasvir atingiram uma RVS. No mesmo braço de tratamento, em pacientes com VARs da NS5A basais que conferiram uma alteração > 100 vezes superior, ocorreu recidiva em 4/13 (31%), em comparação com 3/95 (3%) em pacientes sem RVAs basal ou sem RVAs conferindo uma alteração ≤ 100 vezes.
Após 12 semanas de tratamento com ledipasvir/sofosbuvir com ribavirina em pacientes já tratados anteriormente com cirrose descompensada (SIRIUS, n=77), 8/8 pacientes com RAVs da NS5A basal que obtendo > 100 reduzindo a suscetibilidade para o ledipasvir alcançar SVR12.
O grupo de RAVs na NS5A que conferiu mudança > 100 vezes e que foi observado em pacientes englobou as seguintes substituições no genótipo 1a (M28A, Q30H/R/E, L31M/V/I, H58D, Y93H/N/C) ou no genótipo 1b (Y93H). A proporção destas RAVs na NS5A na fase basal observada com sequenciamento profundo variou de muito baixa (limite para o ensaio = 1%) até alta (parte principal da população plasmática).
A substituição S282T associada à resistência ao sofosbuvir não foi detectada na sequência da NS5B na fase basal de nenhum paciente nos estudos de Fase 3 por população ou sequenciamento profundo. A RVS foi alcançada em todos os 24 pacientes (n = 20 com L159F+C316N; n = 1 com L159F e n = 3 com N142T) que tinham variantes associadas à resistência aos inibidores nucleotídeos da NS5B na fase basal.
Após o tratamento com ledipasvir/sofosbuvir com ribavirina durante 12 semanas em pacientes pós-transplante hepático com doença hepática compensada (SOLAR-1), nenhum (n = 8) dos pacientes com RAVs da NS5A no início do estudo, que conferiram uma alteração > 100 vezes superior ao ledipasvir, recidivou. Após o tratamento com ledipasvir/sofosbuvir com ribavirina durante 12 semanas em pacientes com doença descompensada (independentemente da relação com o transplante hepático), 3/7 pacientes com RAVs da NS5A no início do estudo, que conferiram uma sensibilidade > 100 vezes inferior ao ledipasvir, recidivaram, em comparação com 4/68 pacientes sem RAVs no início do estudo ou com RAVs que conferiram uma sensibilidade ≤ 100 vezes inferior ao ledipasvir.
- Resistência cruzada
Ledipasvir apresentou uma atividade completa contra a substituição S282T na NS5B associada à resistência a sofosbuvir, enquanto todas as substituições na NS5A associadas à resistência ao ledipasvir foram completamente sensíveis ao sofosbuvir. Tanto sofosbuvir quanto ledipasvir foram completamente ativos contra substituições associadas a resistência a outras classes de antivirais de ação direta com diferentes mecanismos de ação, como inibidores não nucleosídeos da NS5B e inibidores da protease NS3. As substituições na NS5A que conferem resistência ao ledipasvir podem reduzir a atividade antiviral de outros inibidores da NS5A.
Propriedades farmacocinéticas
- Absorção
Após administração oral de ledipasvir/sofosbuvir a pacientes infectados com VHC, o pico da concentração plasmática mediana de ledipasvir foi observado 4,0 horas após a administração. O sofosbuvir foi rapidamente absorvido e o pico da concentração plasmática mediana foi observado aproximadamente 1 hora após a administração. O pico da concentração plasmática mediana de GS-331007 foi observado 4 horas após a administração.
Com base na análise farmacocinética de população em pacientes com infecção por VHC, a média geométrica da ASC0-24 no estado de equilíbrio dinâmico de ledipasvir (n = 2.113), sofosbuvir (n = 1.542), e GS-331007 (n = 2.113) foram 7.290, 1.320 e 12.000 ng•h/mL, respectivamente. As Cmax no estado de equilíbrio dinâmico para ledipasvir, sofosbuvir e GS-331007 foram 323, 618 e 707 ng/mL, respectivamente. As ASC0-24 e Cmax de sofosbuvir e GS-331007 foram semelhantes em indivíduos adultos saudáveis e em pacientes com infecção por VHC. Em relação aos indivíduos saudáveis (n = 191), a ASC0-24 e a Cmax de ledipasvir foram 24% menores e 32% menores, respectivamente, em pacientes infectados por VHC. A ASC de ledipasvir é proporcional à dose no intervalo de doses de 3 a 100 mg. As ASCs de sofosbuvir e GS-331007 são quase proporcionais à dose no intervalo de doses de 200 mg a 400 mg.
Efeitos do alimento
Em relação às condições de jejum, a administração de uma dose única de ledipasvir/sofosbuvir com uma refeição moderadamente gordurosa ou rica em gorduras aumentou a ASC0-inf de sofosbuvir em aproximadamente 2 vezes, mas não afetou significantemente a Cmax de sofosbuvir. As exposições a GS-331007 e ledipasvir não foram alteradas na presença de qualquer tipo de refeição. Harvoni pode ser administrado independentemente dos alimentos.
- Distribuição
Ledipasvir é > 99,8% ligado às proteínas do plasma humano. Após uma dose única de 90 mg de ledipasvir marcado com [14C] em indivíduos sadios, a razão sangue:plasma de radioatividade do [14C] variou entre 0,51 e 0,66.
Sofosbuvir é aproximadamente 61 a 65% ligado às proteínas do plasma humano e a ligação é independente da concentração de droga em um intervalo de 1 mg/mL a 20 mg/mL. A ligação à proteína de GS 331007 foi mínima no plasma humano. Após uma única dose de 400 mg de sofosbuvir marcado com [14C] em indivíduos sadios, a razão sangue:plasma de radioatividade do [14C] foi de aproximadamente 0,7.
- Biotransformação
Não se observou, in vitro, um metabolismo detectável de ledipasvir por CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4 humanas. Foram observadas evidências de metabolismo oxidativo lento por um mecanismo desconhecido. Após uma dose única de 90 mg de [14C]-ledipasvir, a exposição sistêmica foi quase exclusivamente devida à droga original ( > 98%). Ledipasvir inalterado também é a principal forma presente nas fezes.
Sofosbuvir é altamente metabolizado no fígado para formar o análogo do nucleosídeo farmacologicamente ativo trifosfato GS 461203. O metabólito ativo não é observado. A via de ativação metabólica envolve hidrólise sequencial da porção de éster carboxílico catalisada pela catepsina A humana ou carboxilesterase 1 e clivagem fosforamidata pela proteína com tríade de histidina de ligação à nucleotídeo 1 seguida por fosforilação pela via da biossíntese do nucleótido pirimídico. A desfosforilação resulta na formação do metabolito nucleosídeo GS 331007 que não pode ser eficientemente refosforilado e não tem atividade anti-VHC invitro. Na associação ledipasvir/sofosbuvir, o GS-331007 é responsável por aproximadamente 85% da exposição sistêmica total.
- Eliminação
Após uma única dose oral de 90 mg de [14C]-ledipasvir, a recuperação total média da radioatividade [14C] nas fezes e na urina foi de 87%, com a maior parte da dose radioativa recuperada a partir das fezes (86%). Ledipasvir inalterado excretado nas fezes foi responsável por uma média de 70% da dose administrada e o metabólito oxidativo M19 foi responsável por 2,2% da dose. Esses dados sugerem que a excreção biliar de ledipasvir inalterado é uma importante via de eliminação com excreção renal sendo uma via menor (aproximadamente 1%). A meia-vida terminal mediana de ledipasvir em voluntários sadios após administração de ledipasvir/sofosbuvir em jejum foi de 47 horas.
Após dose oral única de 400 mg de [14C]-sofosbuvir, a recuperação total média da dose foi superior a 92%, compreendendo aproximadamente 80%, 14%, e 2,5% recuperados na urina, fezes, e no ar expirado, respectivamente. A maior parte da dose de sofosbuvir recuperada na urina foi GS-331007 (78%) enquanto 3,5% foi recuperada como sofosbuvir. Esses dados indicam que a depuração (clearance) renal é a principal via de eliminação para GS-331007 com uma grande parte secretada ativamente. As meias-vidas terminais medianas de sofosbuvir e GS-331007 após administração de ledipasvir/sofosbuvir foram de 0,5 e 27 horas, respectivamente.
O ledipasvir e sofosbuvir não são substratos dos transportadores de captação hepática, transportador do cátion orgânico (OCT) 1, polipeptídeo de transporte do ânion orgânico (OATP) 1B1 ou OATP1B3. O GS-331007 não é um substrato para os transportadores renais incluindo transportador de ânion orgânico (OAT) 1 ou OAT3 ou OCT2.
- Potencial de ledipasvir/sofosbuvir afetar outros medicamentos in vitro
Nas concentrações alcançadas na clínica, ledipasvir não é um inibidor de transportadores hepáticos incluindo o OATP 1B1 ou 1B3, BSEP, OCT1, OCT2, OAT1, OAT3, extrusão de múltiplos fármacos e compostos tóxicos (MATE) 1, proteína associada à resistência a múltiplos fármacos (MRP) 2 ou MRP4. O sofosbuvir e GS-331007 não são inibidores dos transportadores de fármaos P-gp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3, OCT1 e GS-331007 não é um inibidor de OAT1, OCT2 e MATE1.
O sofosbuvir e GS-331007 não são inibidores ou indutores das enzimas do CYP ou da enzima uridina difosfato glicuronosiltransferase (UGT) 1A1.
- Farmacocinética em populações especiais
Etnia e sexo
Não foram identificadas diferenças farmacocinéticas clinicamente relevantes devido à etnia para ledipasvir, sofosbuvir ou GS-331007. Não foram identificadas diferenças farmacocinéticas clinicamente relevantes devido ao sexo para sofosbuvir ou GS-331007.
A ASC e a Cmax do ledipasvir foram respetivamente 77% e 58% mais elevadas em mulheres do que nos homens; contudo, a relação entre sexo e exposições a ledipasvir não foi considerada clinicamente relevante.
Idosos
Uma análise farmacocinética de população em pacientes infectados por VHC mostrou que dentro da faixa etária analisada (18 a 80 anos), a idade não teve um efeito clinicamente relevante sobre a exposição a ledipasvir, sofosbuvir ou GS-331007. Os estudos clínicos com ledipasvir/sofosbuvir incluíram 235 pacientes (8,6% do número total de pacientes) com idade igual ou superior a 65 anos.
Comprometimento renal
A farmacocinética de ledipasvir foi estudada com uma dose única de 90 mg de ledipasvir em pacientes negativos para VHC com comprometimento renal grave (eTFG < 30 mL/min pelo Cockcroft-Gault, mediana [variação] CrCl 22 [17-29] mL/min). Não foram observadas diferenças clinicamente relevantes na farmacocinética de ledipasvir entre indivíduos sadios e pacientes com comprometimento renal grave.
A farmacocinética de sofosbuvir foi estudada em pacientes negativos para VHC com comprometimento renal leve (eTFG ≥ 50 e < 80 mL/min/1,73 m2), moderado (eTFG ≥ 30 e < 50 mL/min/1,73 m2), grave (eTFG < 30 mL/min/1,73 m2) e pacientes com Doença Renal em Estágio Final (DREF) necessitando hemodiálise após uma dose única de 400 mg de sofosbuvir. Em relação aos pacientes com função renal normal (eTFG > 80 mL/min/1,73 m2), a ASC0-inf de sofosbuvir foi 61%, 107% e 171% maior no comprometimento renal leve, moderado e grave, enquanto a ASC0-inf de GS-331007 foi 55%, 88% e 451% maior, respectivamente. Em pacientes com DREF, em relação aos pacientes com função renal normal, a ASC0-inf de sofosbuvir foi 28% maior quando sofosbuvir foi administrado 1 hora antes da hemodiálise em comparação com 60% maior quando sofosbuvir foi administrado 1 hora após a hemodiálise. A ASC0-inf de GS-331007 em pacientes com DREF que receberam sofosbuvir 1 hora antes ou 1 hora após a hemodiálise foi pelo menos 10 vezes e 20 vezes maior, respectivamente. GS-331007 é removido eficientemente pela hemodiálise com um coeficiente de extração de aproximadamente 53%. Após uma dose única de 400 mg de sofosbuvir, uma hemodiálise de 4 horas removeu 18% da dose de sofosbuvir administrado.
Em indivíduos infectados pelo VHC com comprometimento renal grave tratados com Harvoni por 12 semanas (N = 18), a farmacocinética do ledipasvir, sofosbuvir e GS-331007 foi consistente com a observada em indivíduos negativos para o VHC com comprometimento renal grave. A ASC0-24 no estado estacionário do sofosbuvir e GS-331007 foi aproximadamente 2 vezes e 6 vezes superior, respectivamente, do que a observada em indivíduos infectados pelo VHC com função renal normal.
A farmacocinética do ledipasvir, sofosbuvir e GS-331007 foi estudada em indivíduos infectados pelo HCV com DREF necessitando hemodiálise tratados com Harvoni por 8, 12 ou 24 semanas. A ASCtau no estado estacionário de ledipasvir, sofosbuvir e GS-331007 aumentou 61%, 89% e 2180%, respectivamente, em comparação com indivíduos sem comprometimento renal nos ensaios de fase 2/3 do ledipasvir/sofosbuvir. Não foram observadas relações de exposição-segurança em indivíduos infectados pelo VHC com DREF que requerem hemodiálise tratados com Harvoni.
Não é necessário ajuste da dose de Harvoni para pacientes com comprometimento renal, incluindo DREF que necessitam de diálise.
Comprometimento hepático
A farmacocinética de ledipasvir foi estudada com uma dose única de 90 mg de ledipasvir em pacientes negativos para VHC com comprometimento hepático grave (CTP classe C). A exposição plasmática de ledipasvir (ASCinf) foi semelhante em pacientes com comprometimento hepático grave e os pacientes controle com função hepática normal. Uma análise farmacocinética de população em pacientes infectados por VHC indicou que a cirrose não teve efeito clinicamente relevante sobre a exposição ao ledipasvir.
A farmacocinética do sofosbuvir foi estudada após administração por 7 dias de 400 mg de sofosbuvir em pacientes infectados por VHC com comprometimento hepático moderado e grave (CTP classe B e C). Em relação aos pacientes com função hepática normal, a ASC0-24 de sofosbuvir foi 126% e 143% maior no comprometimento hepático moderado e grave, enquanto a ASC0-24 de GS-331007 foi 18% e 9% maior, respectivamente. Uma análise farmacocinética de população em pacientes infectados por VHC indicou que a cirrose não teve efeito clinicamente relevante sobre a exposição a sofosbuvir e GS-331007.
Peso corpóreo
O peso corpóreo não teve um efeito significativo sobre a exposição a sofosbuvir de acordo com uma análise farmacocinética de população. A exposição a ledipasvir diminui com o aumento do peso corpóreo, mas o efeito não é considerado como sendo clinicamente relevante.
População pediátrica
A exposição ao ledipasvir, ao sofosbuvir e ao GS-331007 em adolescentes com 12 a < 18 anos de idade foram semelhantes àquelas observadas em adultos em estudos de Fase 2/3, após administração de ledipasvir/sofosbuvir (90 mg/400 mg). A farmacocinética do ledipasvir, do sofosbuvir e do GS-331007 não foi estabelecida em pacientes pediátricos com < 12 anos de idade (ver POSOLOGIA E MODO DE USAR).- Dados de segurança pré-clínica
Ledipasvir<