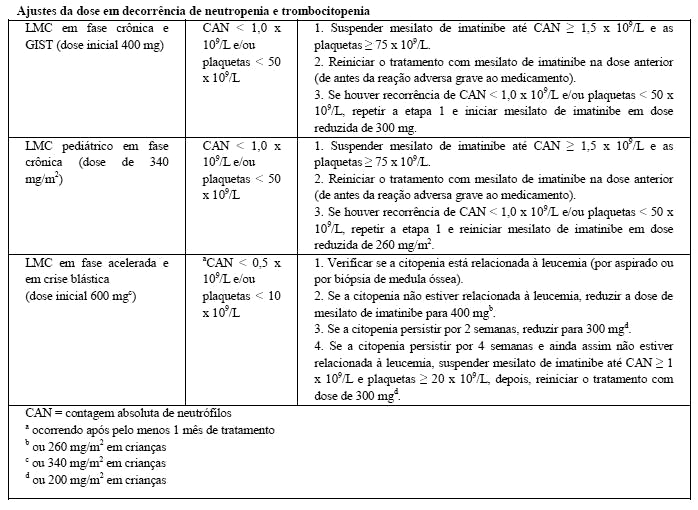
GLIMATIN
EMS
imatinibe
Antineoplásico.
Apresentações.
Comprimidos revestidos 100mg e 400mg.
Embalagens contendo 60 ou 100 (embalagem hospitalar) comprimidos revestidos de 100 mg.
Embalagens contendo 30 ou 100 (embalagem hospitalar) comprimidos revestidos de 400 mg.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 1 ANO (vide indicações)
Composição.
Cada comprimido revestido de 100 mg contém: mesilato de imatinibe* 119,500 mg. Excipiente** q.s.p. 1 com rev. * Cada comprimido revestido contém 119,500 mg de mesilato de imatinibe que equivalem a 100,000 mg de imatinibe. ** manitol, celulose microcristalina, crospovidona, povidona, estearil fumarato de sódio, hipromelose + macrogol, dióxido de titânio, óxido de ferro amarelo.
Cada comprimido revestido de 400 mg contém: mesilato de imatinibe* 478,000 mg. Excipiente** q.s.p. 1 com rev. * Cada comprimido revestido contém 478,000 mg de mesilato de imatinibe que equivalem a 400,000 mg de imatinibe. ** manitol, celulose microcristalina, crospovidona, povidona, estearil fumarato de sódio, hipromelose + macrogol, dióxido de titânio, óxido de ferro amarelo.
Informações técnicas.
1. INDICAÇÕES
O GLIMATIN é indicado para:
- pacientes adultos e pediátricos (acima de 2 anos) com Leucemia Mieloide Crônica (LMC) cromossomo Philadelphia positivo (Ph+) recém-diagnosticada e sem tratamento anterior;
- pacientes adultos com LMC cromossomo Philadelphia positivo em crise blástica, fase acelerada ou fase crônica após falha ou intolerância à terapia com alfainterferona;
- tratamento de pacientes adultos e pediátricos (acima de 1 ano) com Leucemia Linfoblástica Aguda (LLA Ph+) cromossomo Philadelphia positivo, recentemente diagnosticada, integrados com quimioterapia;
- tratamento de pacientes adultos com tumores estromais gastrintestinais (GIST), não ressecáveis e/ou metastáticos; - tratamento adjuvante de pacientes adultos após ressecção de GIST primário.
2. RESULTADOS DE EFICÁCIA
- Estudos Clínicos em LMC
A eficácia de GLIMATIN
baseia-se nas taxas globais de resposta hematológica e citogenética e na sobrevida livre de progressão.
Foram conduzidos três grandes estudos internacionais, abertos, não controlados, de fase II incluindo pacientes com leucemia mieloide crônica (LMC) cromossomo Philadelphia positivo (Ph+) na doença em fase avançada, blástica ou acelerada, outras leucemias Ph+ ou em LMC em fase crônica com falha à terapêutica anterior com alfa-interferona (IFN)1,2,3.
Foi conduzido um estudo extenso, aberto, multicêntrico, internacional, randomizado, de fase III em pacientes com LMC Ph+ recentemente diagnosticada4. Adicionalmente, crianças foram tratadas em dois estudos clínicos fase I e um estudo clínico de fase II aberto, multicêntrico de braço único.
Em todos os estudos clínicos, 38-40% dos pacientes tinham idade superior ou igual a 60 anos e 10-12% dos pacientes tinham 70 anos ou mais.
Fase crônica, recentemente diagnosticada4,5,6,7: este estudo de fase III comparou o tratamento com GLIMATIN em monoterapia ou uma combinação de alfa-interferona (IFN) mais Citosina Arabinosídeo (Ara-C). Foi permitido aos pacientes não apresentando resposta (sem resposta hematológica completa - RHC aos 6 meses, leucócitos em aumento, sem resposta citogenética Maior (RCM) aos 24 meses), ou com perda da resposta (perda de RHC ou de RCM) ou intolerância severa ao tratamento, serem transferidos para o braço de tratamento alternativo. No braço recebendo GLIMATIN, os pacientes foram tratados com 400 mg, diariamente. No braço recebendo IFN, os pacientes foram tratados com uma dose alvo de IFN de 5 MUI/m2/dia por via subcutânea em combinação com Ara-C 20 mg/m2/dia por via subcutânea por 10 dias/mês.
Um total de 1.106 pacientes foram randomizados em 177 centros de 16 países, 553 para cada braço. As características basais foram bem balanceadas entre os dois braços. A idade mediana foi 51 anos (faixa entre 18-70 anos), 21,9% dos pacientes com idade ≥ 60 anos. Cinquenta e nove por cento dos pacientes eram do sexo masculino e 41% do sexo feminino; 89,9% caucasianos e 4,7% de raça negra. Por ocasião da data de corte desta análise (7 anos após o recrutamento do último paciente), a duração média do tratamento de primeira linha foi 82 meses e 8 meses nos braços de GLIMATIN e IFN, respectivamente. A duração média do tratamento de segunda linha com GLIMATIN foi de 64 meses. Sessenta por cento dos pacientes randomizados para mesilato de imatinibe ainda estão recebendo tratamento de primeira linha. Nestes pacientes, a dose média de mesilato de imatinibe foi de 403 ± 57 mg. Em geral, em pacientes recebendo mesilato de imatinibe como primeira linha de tratamento, a dose média diária administrada foi de 406 ± 76 mg. Como consequência de uma taxa mais elevada tanto de descontinuações como de transferências para o braço alternativo de tratamento, somente 2% dos pacientes randomizados para IFN ainda estão recebendo tratamento de primeira linha. No braço de IFN, a retirada do consentimento (14%) foi a razão mais frequente para descontinuação da terapia de primeira linha, e a razão mais frequente para a transferência para o braço de mesilato de imatinibe foi intolerância grave ao tratamento (26%) e progressão da doença (14%)8,9. O objetivo primário de avaliação de eficácia do estudo foi a sobrevida livre de progressão. Define-se como progressão da doença, a ocorrência de qualquer um dos seguintes eventos: progressão para a fase acelerada ou crise blástica (FA/CB), óbito, perda de RHC, ou RCM ou, em pacientes que não conseguiram atingir RHC, o aumento de leucócitos, apesar do tratamento terapêutico apropriado. Resposta citogenética Maior, resposta hematológica, resposta molecular (avaliação da doença residual mínima), tempo até a fase acelerada ou crise blástica e sobrevida foram os principais objetivos secundários de avaliação de eficácia. Os dados de resposta são apresentados na Tabela 16,7,10.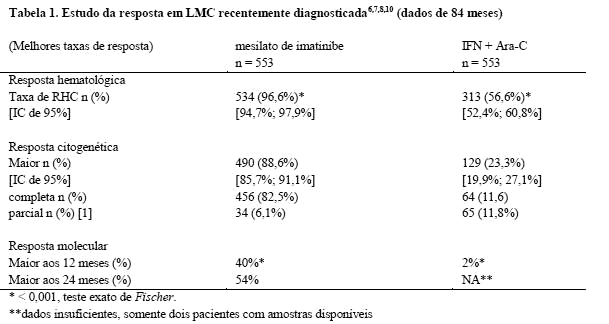
Critérios de resposta hematológica (todas as respostas devem ser confirmadas após ≥ 4 semanas): leucócitos < 10 x 109/L, plaquetas < 450 x 109/L, mielócito + metamielócito < 5% no sangue, ausência de blastos e promielócitos no sangue, basófilos < 20%, ausência de comprometimento extramedular.
Critérios de resposta citogenética: completa (0% metáfases Ph+), parcial (1-35%), Menor (36- 65%) ou mínima (66-95%). A reposta citogenética Maior (0-35%) combina ambas as respostas completa e parcial [1].
Critérios de resposta molecular Maior: no sangue periférico, redução de ≥ 3 logaritmos na quantidade de transcritos de Bcr-Abl (medidos por ensaio de PCR por transcriptase reversa quantitativo em tempo real) comparado em relação a um valor basal padronizado.
As taxas de resposta hematológica completa (RHC), resposta citogenética Maior (RCM) e resposta citogenética completa (RCC) no tratamento de primeira linha foram estimadas usando o método de Kaplan-Meier, para os quais as não-respostas foram censuradas na data do último exame. Usando este método, as taxas de resposta acumulativa estimada para o tratamento de primeira linha com GLIMATIN, são mostradas na Tabela 2.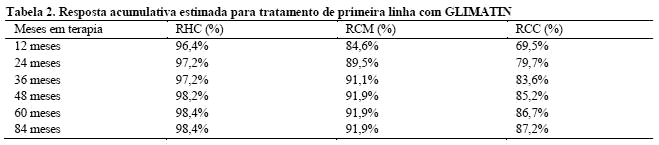
Para análise dos resultados de longa duração, os pacientes randomizados para receber GLIMATIN foram comparados aos pacientes randomizados para receber IFN. Os pacientes transferidos antes da progressão não foram censurados no momento da transferência, e os eventos que ocorreram nestes pacientes após a transferência foram atribuídos ao tratamento randomizado inicial.
Em sete anos de acompanhamento, havia 93 (16,8%) eventos com progressão no braço de mesilato de imatinibe: 37 (6,7%) envolvendo progressão para FA/CB, 31 (5,6%) envolvendo perda de RCM, 15 (2,7%) envolvendo perda de RHC ou aumento em leucócitos e 10 (1,8%) envolvendo óbitos não relacionados a LMC. Em contraste, houve 165 (29,8%) eventos no braço recebendo IFN + Ara-C dos quais 130 ocorreram durante o tratamento de primeira linha com IFN + Ara-C.
A taxa estimada para sobrevida livre de progressão aos 84 meses é 81,2% com IC de 95% (78, 85) no braço de mesilato de imatinibe e 60,6% (56, 5) no braço controle (p < 0,001) (Figura 1). As taxas anuais de progressão para GLIMATIN foram 3,3% no primeiro ano após início do estudo, 7,5% no segundo ano e 4,8%, 1,7%, 0,8%, 0,3% e 2,0% no terceiro, quarto, quinto, sexto e sétimo ano, respectivamente.
A taxa estimada de pacientes livres de progressão para a fase acelerada ou crise blástica aos 84 meses foi significativamente mais alta no braço de GLIMATIN comparado ao braço de IFN 6,7,10,11 (92,5%12,13 versus 85,1%, p < 0,00114,15) (Figura 2). A taxa anual de progressão diminuiu com o tempo de terapia: taxas anuais de progressão da doença para fase acelerada ou crise blástica foram 1,5%, 2,8%, 1,6%, 0,9%, 0,5%, 0% e 0,4% do primeiro ao sétimo ano, respectivamente.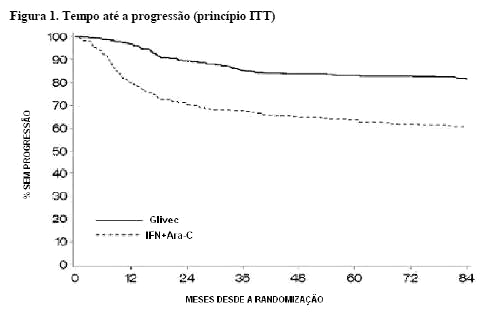
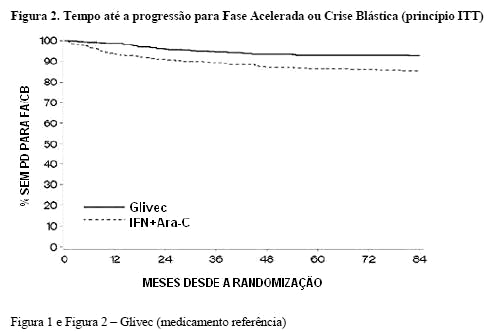
Um total de 71 (12,8%) e 85 (15,4%) pacientes foram a óbito nos grupos de GLIMATIN e IFN + Ara-C, respectivamente. Aos 84 meses, a sobrevida global estimada é 86,4% (83, 90) vs 83,3% (80, 87) nos grupos randomizados de mesilato de imatinibe e IFN + Ara-C, respectivamente (p = 0,073, teste log rank). Este objetivo de tempo para o evento foi fortemente afetado pela alta taxa de transferência do grupo IFN + Ara-C para o grupo de GLIMATIN. Além disso, um grande número de pacientes recebeu transplante de medula óssea (TMO) após a descontinuação do tratamento de estudo no grupo IFN + Ara-C (n = 66, 38 após transferência para mesilato de imatinibe) comparado com o grupo de mesilato de imatinibe (n = 50, 8 após transferência para IFN) na atualização de 84 meses. Quando censurados os 48 óbitos que ocorreram após o transplante de medula óssea, as taxas de sobrevida aos 84 meses foram 89,6 vs 88,1 (p=0,200, teste log rank). Apenas 31 óbitos (antes do TMO) do grupo de pacientes do GLIMATIN (5,6%) foram atribuídos a LMC, comparado à 40 pacientes do grupo IFN + Ara-C (7,2%). Quando considerado apenas os óbitos relacionados à LMC e desconsiderando quaisquer óbitos após TMO ou devido a quaisquer outras razões, as taxas estimadas de sobrevida aos 84 meses foram 93,6% vs 91,1% (p = 0,1, teste log rank). O efeito do tratamento com GLIMATIN na sobrevida da fase crônica, em LMC recentemente diagnosticada, foi examinado em uma análise retrospectiva dos dados de mesilato de imatinibe 4 reportados acima com os dados primários em um outro estudo de fase III usando IFN + Ara-C (n = 325) em um esquema terapêutico idêntico16. Nesta publicação, a superioridade de mesilato de imatinibe sobre IFN + Ara-C em relação a sobrevida global foi comprovada (p < 0,001); dentro de 42 meses, 47 (8,5%) pacientes de mesilato de imatinibe e 63 (19,4%) pacientes de IFN + Ara-C foram a óbito.
O grau de resposta citogenética afetou claramente nos resultados de longa duração em pacientes sendo tratados com GLIMATIN. Visto que uma estimativa de 96% dos pacientes com RCC e 93% dos pacientes com RCP (resposta citogenética parcial) aos 12 meses estavam livres de progressão para FA/CB aos 84 meses. Somente 81% dos pacientes sem RCM aos 12 meses estavam livres de progressão para LMC avançada aos 84 meses (p < 0,001 total, p = 0,25 entre RCC e RCP). Com base no marco de 18 meses, as estimativas foram 99%, 90% e 83% respectivamente, incluindo também uma diferença estatisticamente significante entre RCC e RCP (p < 0,001)17.
O monitoramento molecular representou uma informação prognóstica adicional importante. Para pacientes com RCC e redução de transcritos Bcr-Abl de pelo menos 3 logaritmos aos 12 meses, a probabilidade de permanecer livre de progressão de doença aos 60 meses foi numericamente maior quando comparados a pacientes que tiveram RCC e uma redução menor do que 3 log (95% vs 89%, p = 0,068), e significantemente maior quando comparados ao pacientes que não alcançaram RCC aos 12 meses (70%, p < 0,001). Considerando somente a progressão para FA/CB, as taxas estimadas sem evento foram 100%, 95% e 88%, respectivamente (p < 0,001 total, p = 0,007 entre RCC com e sem RMM - reposta molecular Maior). Considerando o marco de 18 meses, as taxas estimadas sem FA/CB aos 60 meses foram 100% para pacientes com RCC e RMM, 98% para pacientes com RCC, mas sem RMM e apenas 87% para pacientes sem RCC (p < 0,001 total, p = 0,105 entre RCC com e sem RMM)6,10.
Neste estudo, foram permitidos escalonamentos de dose de 400 mg ao dia para 600 mg ao dia, e depois, de 600 mg ao dia para 800 mg ao dia. Após 42 meses de acompanhamento, 11 pacientes que atingiram uma resposta hematológica completa aos 3 meses e uma resposta citogenética Maior aos 12 meses, enquanto em uso de dose de 400 mg ao dia, tiveram perda confirmada (dentro de 4 semanas) de suas respostas citogenéticas. Entre estes 11 pacientes, 4 pacientes foram escalonados até 800 mg ao dia, 2 dos quais recuperaram resposta citogenética (1 parcial e 1 completa, o último também atingiu resposta molecular), enquanto que dos 7 pacientes nos quais a dose não foi escalonada, somente um recuperou resposta citogenética completa. A porcentagem de algumas reações adversas ao medicamento foi mais alta nos 40 pacientes nos quais a dose foi escalonada para 800 mg ao dia comparada com a população de pacientes antes do aumento da dose (n = 551). As reações adversas ao medicamento mais frequentes incluíram hemorragias gastrintestinais, conjuntivite e elevação de transaminases ou bilirrubina. Outras reações adversas ao medicamento foram reportadas com frequência menor ou igual18.
A Qualidade de Vida (QdV) foi medida utilizando-se o instrumento validado FACT-BRM. Todos os domínios foram avaliados e revelaram escores significativamente mais elevados para o braço de GLIMATIN comparado ao braço de IFN. Os dados de QdV mostraram que os pacientes mantêm seu bem-estar enquanto estão sendo tratados com GLIMATIN 19.
Fase crônica, pós-falha à interferona: 532 pacientes foram tratados com uma dose inicial de 400 mg. Os pacientes foram distribuídos em três categorias principais: falha hematológica (29%), falha citogenética (35%) ou intolerância à interferona (36%). Os pacientes haviam recebido previamente uma média de 14 meses de tratamento com interferona em doses ≥ 25 x 106 UI/semana e encontravam-se todos em fase crônica tardia, com um tempo médio de diagnóstico de 32 meses. A variável primária de eficácia foi a taxa de resposta citogenética Maior (resposta citogenética completa + resposta citogenética parcial, ou seja, 0 a 35% de metáfases Ph+ na medula óssea).
Neste estudo, 65% dos pacientes atingiram resposta citogenética Maior que foi completa em 53% dos pacientes (Tabela 2). Foi atingida resposta hematológica completa em 95% dos pacientes 3,20,21.
Fase acelerada: foram admitidos 235 pacientes com doença em fase acelerada. Os primeiros 77 pacientes iniciaram tratamento com 400 mg, o protocolo foi emendado subsequentemente para permitir a administração de doses mais elevadas e os 158 pacientes remanescentes iniciaram com 600 mg.
A variável primária de eficácia foi a taxa de resposta hematológica, relatada como resposta hematológica completa, sem evidência de leucemia (isto é, clareamento de blastos da medula e do sangue, mas sem recuperação total do sangue periférico como nas respostas completas), ou retorno à fase crônica da LMC. Foi atingida uma resposta hematológica confirmada em 71,5% dos pacientes (Tabela 3). Importante referir que, 27,7% dos pacientes também atingiram resposta citogenética Maior, a qual foi completa em 20,4% dos pacientes. Para os pacientes tratados com 600 mg, a estimativa atual para as medianas de sobrevida livre de progressão de doença e de sobrevida global foi de 22,9 e 42,5 meses, respectivamente. Em uma análise multivariada, a dose de 600 mg foi associada com a melhora do tempo para a progressão de doença, independente da contagem de plaquetas, blastos sanguíneos e hemoglobina ≥ 10 g/L2,20,22.
Crise blástica mieloide: foram admitidos 260 pacientes com crise blástica mieloide. Noventa e cinco pacientes (37%) haviam recebido quimioterapia prévia para tratamento de fase acelerada ou de crise blástica ("pacientes pré-tratados") enquanto que 165 (63%) não haviam recebido quimioterapia prévia ("pacientes não tratados"). Os primeiros 37 pacientes iniciaram o tratamento com a dose de 400 mg, o protocolo foi emendado subsequentemente para permitir a administração de doses mais elevadas e os 223 pacientes remanescentes iniciaram o tratamento com 600 mg.
A variável primária de eficácia foi a taxa de resposta hematológica relatada como uma resposta hematológica completa, sem evidência de leucemia, ou retorno à fase crônica da LMC utilizando os mesmos critérios usados para o estudo em fase acelerada. Neste estudo, 31% dos pacientes atingiram resposta hematológica (36% em pacientes não tratados previamente e 22% tratados previamente). A taxa de resposta também foi mais elevada nos pacientes tratados com 600 mg (33%), quando comparados aos pacientes tratados com 400 mg (16%, p = 0,0220). A estimativa atual de sobrevida mediana dos pacientes não tratados e dos tratados previamente foi de 7,7 e 4,7 meses, respectivamente 1,20,11.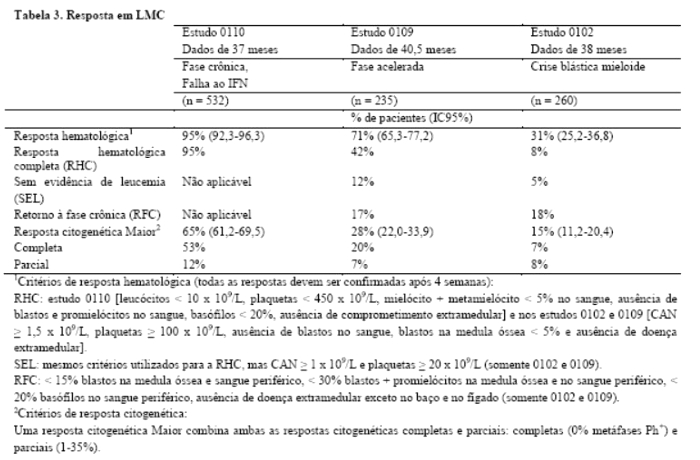
Pacientes pediátricos29,30: um total de 51 pacientes pediátricos com LMC recém diagnosticada e não previamente tratada em fase crônica participaram do estudo clínico de fase II, aberto, multicêntrico de braço único e foram tratados com mesilato de imatinibe 340 mg/m2/dia.. O tratamento com GLIMATIN levou a uma resposta rápida em pacientes pediátricos com LMC recém diagnosticada com RHC de 78% após 8 semanas de terapia e uma resposta citogenética completa (RCC) de 65%, (comparável aos resultados em adultos) após 3 a 10 meses de tratamento.
Um total de 31 pacientes pediátricos, intensamente tratados previamente, (45% com TMO prévio e 68% recebendo múltiplos agentes quimioterápicos previamente) com LMC em fase crônica (n= 15) ou em crise blástica ou leucêmia linfoblástica aguda Ph+ LLA (n=16) participaram de um estudo clínico fase I de escalonamento de dose. Os pacientes foram tratados com GLIMATIN com doses variando de 260 mg/m2/dia, e 570 mg/m2/dia. Dentre os 13 pacientes com LMC e dados de citogenética disponíveis, 7 (54%) e 4 (31%) alcançaram resposta citogenética completa e parcial, respectivamente, com taxa de RCM de 85%32.
Estudos Clínicos em LLA Ph+2, 33-37.
Um total de 851 pacientes com LLA Ph+ recentemente diagnosticada ou com doença recidivada/refratária foram incluídos em 11 estudos clínicos, dos quais dez foram estudos não-controlados e um estudo foi randomizado. Dos 851 pacientes, 93 eram pacientes pediátricos (incluindo 4 pacientes com mais de 18 e menos de 22 anos de idade) tratados em um estudo de fase III, aberto, multicêntrico e não randomizado33.
LLA Ph+ pacientes adultos recentemente diagnosticada
Em um estudo controlado (ADE10) de GLIMATIN versus quimioterapia de indução em 55 pacientes recentemente diagnosticados com idade de 55 anos ou mais, GLIMATIN foi usado como agente único e induziu uma taxa significantemente maior de resposta hematológica completa do que de quimioterapia (96,3% vs. 50%; p = 0,0001). Quando a terapia de resgate com GLIMATIN foi administrada em pacientes que não responderam ou que tiveram resposta pobre à quimioterapia, 9 (81,8%) de 11 pacientes atingiram uma resposta hematológica completa. Este efeito clínico foi associado com uma redução em transcritos BCR-ABL maior nos pacientes tratados com GLIMATIN do que no braço de quimioterapia após 2 semanas de terapia (p = 0,02). Todos os pacientes receberam GLIMATIN e quimioterapia de consolidação após indução e os níveis de transcritos BCR-ABL foram idênticos nos dois braços nas 8 semanas. Como esperado com base no desenho do estudo, nenhuma diferença foi observada na duração da remissão, sobrevida livre de doença ou sobrevida global, embora pacientes com resposta molecular completa e que permaneceram com doença residual mínima tiveram um resultado melhor em termos de duração da remissão (p = 0,01) e sobrevida livre de doença (p = 0,02).
Os resultados observados em uma população de 211 pacientes com LLA Ph+ recentemente diagnosticada em quatro estudos clínicos não-controlados (AAU02, ADE04, AJP01 e AUS01) são consistentes com os resultados descritos acima, conforme demonstrado na Tabela 4. O GLIMATIN, em combinação com quimioterapia de indução, resultou em uma taxa de resposta hematológica completa de 93% (147 de 158 pacientes avaliáveis) e em uma taxa de resposta citogenética Maior de 90% (19 de 21 pacientes avaliáveis). A taxa de resposta molecular completa foi 48% (49 de 102 pacientes avaliáveis).
Similarmente, em dois estudos clínicos não-controlados (AFR09 e AIT04) nos quais 49 pacientes com LLA Ph+ recentemente diagnosticada com idade de 55 anos ou mais receberam GLIMATIN combinado com esteroides com ou sem quimioterapia, houve uma taxa de resposta hematológica completa de 89% nessa população e uma taxa de resposta molecular completa de 26% em 39 pacientes avaliáveis. A sobrevida livre de doença (SLD) e a sobrevida global (SG) excederam um ano e foram superiores ao controle histórico (SLD p < 0,001; SG p < 0,01) em três estudos (AJP01, AUS01 e AFR09).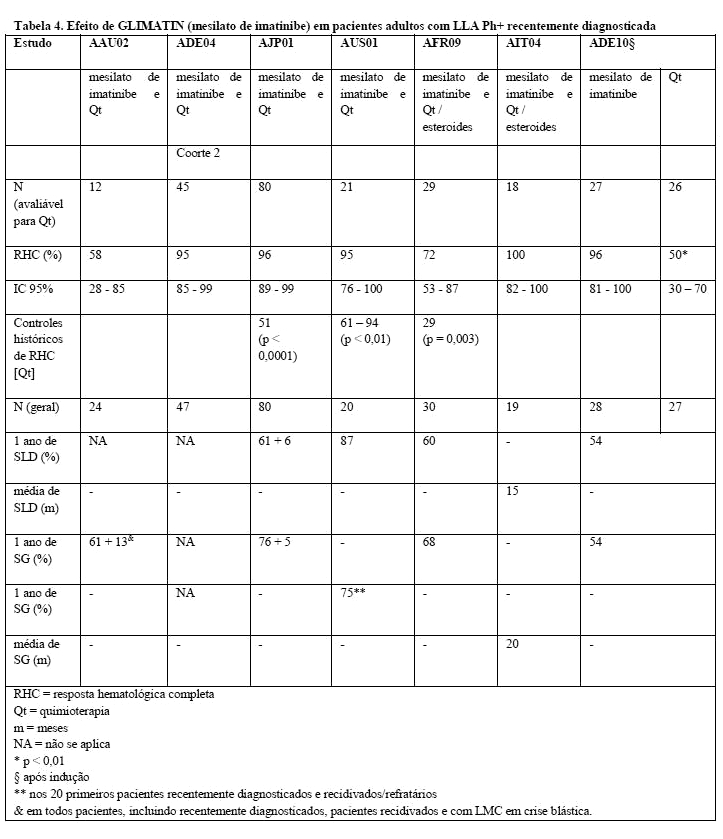
LLA Ph+ pacientes pediátricos33-37: No estudo I2301, um total de 93 pacientes pediátricos, adolescentes e adultos jovens (incluindo 4 pacientes com mais de 18 e menos de 22 anos de idade) com LLA Ph+ foram incluídos no estudo de fase III, aberto, multicêntrico, em coortes sequenciais (coorte 1 a 5) e não randomizado, e foram tratados com mesilato de imatinibe (340 mg/m2/dia) em combinação com quimioterapia intensiva após terapia de indução. GLIMATIN foi administrado intermitentemente nos grupos de 1 a 5, com aumento da duração e diferença no tempo de início do tratamento com Glivec de grupo para grupo; grupo 1 recebeu a intensidade mais baixa e grupo 5 recebeu a intensidade mais alto de Glivec (duração mais longa em dias com uso continuo diário de GLIMATIN durante o primeiro curso do tratamento quimioterápico). Exposição diária contínua a GLIMATIN, iniciada precocemente no curso do tratamento, em combinação com quimioterapia nos pacientes do grupo 5 (n=50) melhorou a sobrevida livre de eventos (SLE) de 4 anos comparado aos controles históricos (n=120), que receberam quimioterapia padrão sem Glivec® (69,9% vs. 31,6%, respectivamente). A sobrevida global estimada de 4 anos nos pacientes do grupo 5 foi 83,6% comparado a 44,8% nos controles históricos.
Estudos Clínicos em GIST metastático ou inoperável24,25
Foram conduzidos dois estudos de Fase III internacionais, randomizados e abertos (SWOG, EORTC) em pacientes com tumores estromais gastrintestinais (GIST) metastáticos ou inoperáveis. O desenho destes dois estudos foi similar permitindo uma análise combinada pré-definida de segurança e eficácia. Um total de 1640 pacientes foram recrutados nos dois estudos e randomizados 1:1 para receber 400 mg ou 800 mg oralmente uma vez ao dia continuadamente até a progressão de doença ou toxicidade inaceitável. Pacientes no grupo de tratamento de 400 mg uma vez ao dia que tiveram progressão de doença foram permitidos migrar para receber o tratamento do grupo de 800 mg uma vez ao dia. Os estudos foram desenhados para comparar as taxas de resposta, sobrevida livre de progressão e sobrevida global entre os grupos de dose. A idade mediana dos pacientes admitidos foi 60 anos (faixa entre 17 a 94 anos, idade 25 - 75 anos percentil 50 a 69). Homens compreenderam 58% dos pacientes admitidos. Todos os pacientes tiveram um diagnóstico patológico de GIST metastático ou inoperável CD117 positivo.
O objetivo primário dos dois estudos foi avaliar sobrevida livre de progressão (SLP), com um objetivo secundário de sobrevida global, (SG) em um estudo (EORTC) e sobrevida global com um objetivo secundário de SLP no outro estudo (SWOG). Foi conduzida uma análise planejada de ambas SG e SLP do conjunto de dados combinado destes dois estudos. Os resultados desta análise combinada são mostrados na Tabela 5.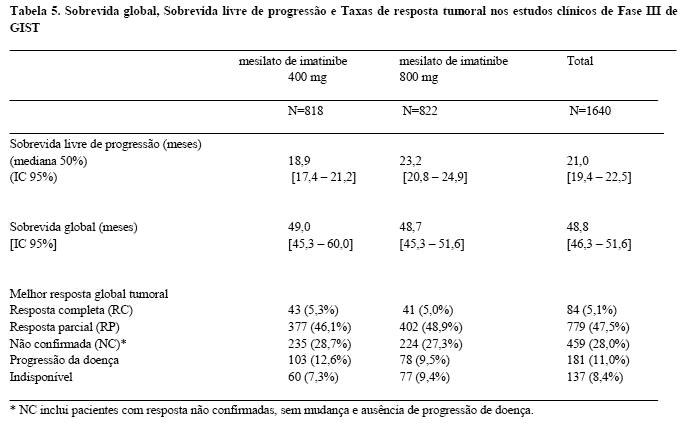
A mediana de acompanhamento para os estudos combinados foi de 37,5 meses (25 a 75 percentil 19 a 46 meses). Houve uma melhora estatisticamente significante na SLP no grupo de tratamento de 800 mg (23,2 meses [IC de 95%; 20,8 a 24,9]) comparada com o grupo de tratamento de 400 mg (18,9 meses [IC de 95%; 17,4 a 21,2]) (p=0,03). Entretanto, não foram observadas diferenças na sobrevida global entre os grupos de tratamento (p=0,98). A SLP global estimada para todos os 1640 pacientes nestes estudos de Fase III foi de 21 meses [IC de 95%; 19,4 a 22,5] e a SG estimada de 48,8 meses [IC de 95%; 46,3 a 51,6]. 5,1% dos pacientes atingiram uma resposta completa confirmada e 47,5% atingiram uma resposta parcial. O tratamento em qualquer uma das doses foi geralmente bem tolerado e no geral 5,4% dos pacientes interromperam o tratamento devido a toxicidade.
Pacientes que, após progressão da doença, migraram do grupo de tratamento 400 mg/dia para o grupo de tratamento de 800 mg/dia (n=347) tiveram uma mediana de 3,4 meses e exposição média de 7,7 meses ao GLIMATIN após migração. A sobrevida global de pacientes após migração foi de 14,3 meses [IC de 95%; 12,2 a 16,7] e 19,3% destes pacientes ainda estão vivos aos 48 meses.
Foi conduzido um estudo multinacional, randomizado, aberto, de fase II em pacientes com tumor estromal gastrintestinal (GIST) metastático ou inoperável. Neste estudo foram incluídos 147 pacientes, os quais foram randomizados para tratamento com 400 mg ou 600 mg por via oral uma vez ao dia, por até 36 meses. Estes pacientes tinham entre 18 e 83 anos e apresentavam diagnóstico patológico de GIST c-Kit-positivo, metastático e/ou inoperável.
A evidência primária de eficácia foi estabelecida com base nas taxas de resposta objetiva. Os tumores deviam ser mensuráveis em pelo menos um sítio da doença e a caracterização da resposta baseou-se nos critérios do Southwestern Oncology Group (SWOG). Em um estudo, 83% dos pacientes atingiram tanto a resposta completa, resposta parcial ou doença estável. Os resultados estão apresentados na Tabela 6.
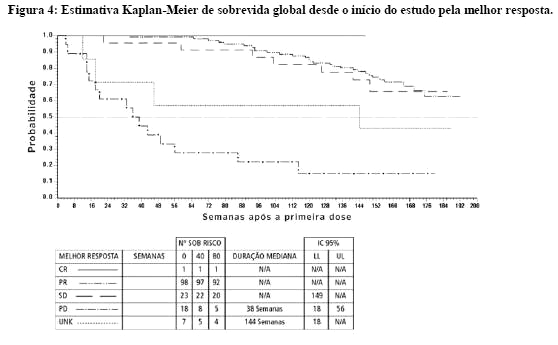
Não foram encontradas diferenças nas taxas de resposta entre os dois grupos de dosagem. Um número significante de pacientes que obteve estabilização da doença no momento da análise interina atingiu uma resposta parcial com o prolongamento do tratamento (mediana de acompanhamento de 31 meses). O tempo mediano para resposta foi de 13 semanas (IC de 95% 12 a 23). O tempo mediano para a falha de tratamento em respondedores foi de 122 semanas (IC de 95% 106 a 147), enquanto que na população geral do estudo foi de 84 semanas (IC de 95% 71 a 109). A mediana de sobrevida global não foi alcançada. A estimativa Kaplan-Meier para sobrevida após 36 meses de acompanhamento é de 68% (Figura 3). Adicionalmente, não há diferença da sobrevida entre pacientes que atingiram a doença estável e pacientes que atingiram a resposta parcial (Figura 4)25.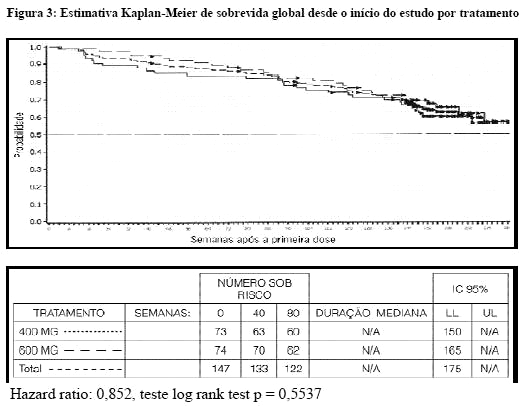
Estudos Clínicos de Adjuvância em GIST26, 31
Em adjuvância, GLIMATIN foi investigado em um estudo clínico de Fase III controlado por placebo, de longa duração, duplo-cego, multicêntrico (Z9001) envolvendo 713 pacientes. A idade destes pacientes variou de 18 a 91 anos. Pacientes incluídos tiveram um diagnóstico histológico de GIST primário expressando proteína KIT por imuno-histoquímica e um tamanho de tumor ≥ 3 cm de dimensão no máximo, com ressecção macroscópica completa de GIST primário dentro de 14 a 70 dias antes do registro no estudo. Após ressecção de GIST primário, pacientes foram randomizados para um dos dois braços: mesilato de imatinibe com 400 mg/dia ou placebo por um ano.
O objetivo primário do estudo foi a sobrevida livre de recorrência (SLR) definida como o tempo da data de randomização até a data de recorrência ou morte por qualquer causa.
O GLIMATIN prolongou significativamente a SLR estando 75% dos pacientes livres de recorrência aos 38 meses no grupo de GLIMATIN vs. 20 meses no grupo placebo (ICs 95%; [30 não-estimáveis]; [14 não-estimáveis], respectivamente); (hazard ratio = 0,398 [0,259 a 0,610], p < 0,0001). Em um ano a SLR global foi significativamente melhor para GLIMATIN (97,7%) vs. placebo (82,3%), (p < 0,0001) reduzindo, portanto, o risco de recorrência em aproximadamente 89% quando comparado com placebo (hazard ratio = 0,113 [0,049 a 0,264]).
Um segundo estudo clínico aberto de fase III (SSG XVIII/AIO) comparou tratamento por 12 meses com GLIMATIN versus tratamento por 36 meses em pacientes após ressecção cirúrgica de GIST e um dos seguintes: tumor com diâmetro > 5 cm e contagem mitótica > 5/50 campos de grande aumento (CGA); ou diâmetro do tumor > 10 cm e qualquer contagem mitótica ou tumor de qualquer tamanho com contagem mitótica > 10/50 CGA ou tumores rompidos dentro da cavidade peritoneal. Houve um total de 397 pacientes consentidos e randomizados no estudo (199 pacientes no braço de 12 meses e 198 pacientes no braço de 36 meses), a idade mediana foi de 61 anos (intervalo de 22 a 84 anos). O tempo mediano de acompanhamento foi 54 meses (da data de randomização até a data de corte), com um total de 83 meses entre o primeiro paciente randomizado e a data de corte.
O objetivo primário do estudo foi a SLR definida como o tempo da data de randomização até a data de recorrência ou morte por qualquer causa.
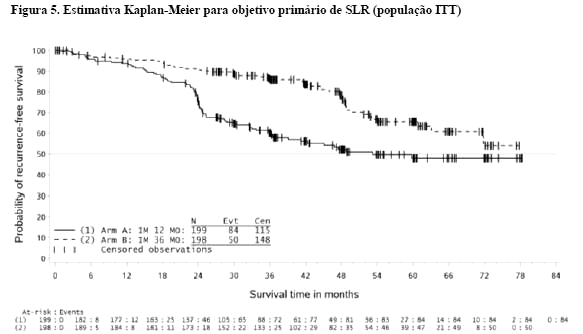
O tratamento com GLIMATIN por trinta e seis (36) meses prolongou significativamente a SLR comparado ao tratamento com GLIMATIN por 12 meses (com hazard ratio global= 0,46 [0,32, 0,65], p < 0,0001 e um hazard ratio de 0,42 [0,28, 0,61] além do mês 12) (Tabela 7, Figura 5). Houve 84 (42%) e 50 (25%) de eventos totais de SLR para os braços de tratamento por 12 meses e por 36 meses respectivamente.
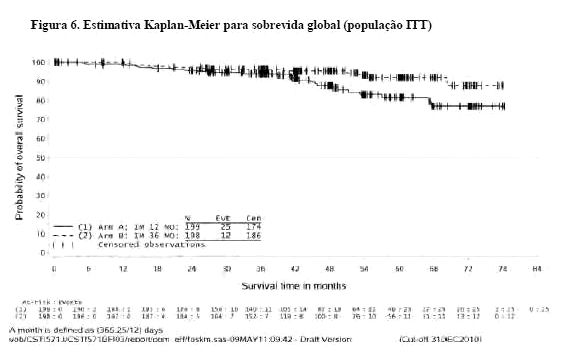
Adicionalmente, o tratamento com GLIMATIN por trinta e seis (36) meses prolongou significativamente a sobrevida global (SG) comparado ao tratamento com GLIMATIN por 12 meses (hazard ratio = 0,45 [0,22, 0,89], p = 0,0187) (Tabela 7, Figura 6). O número total de mortes foi 25 para o braço do tratamento por 12 meses e 12 para o braço de tratamento por 36 meses.
Estudos clínicos em insuficiência hepática
Em um estudo de pacientes com graus variados de insuficiência hepática (leve, moderada e grave - vide Tabela 8 abaixo, para classificação da função hepática), a exposição média ao imatinibe (AUC normalizada por dose) não aumentou comparada aos pacientes com função hepática normal. Neste estudo, 500 mg ao dia foi usado com segurança em pacientes com insuficiência hepática leve e 300 mg ao dia foi usado em outros pacientes. Embora tenha-se utilizado somente a dose de 300 mg ao dia em pacientes com insuficiência hepática moderada e grave, a análise farmacocinética projeta que 400 mg pode ser usado com segurança27 (vide "Posologia e modo de usar", "Advertências e precauções", "Reações adversas", "Características farmacológicas").
Estudos clínicos em insuficiência renal
Em um estudo de pacientes com graus variáveis de disfunção renal (leve, moderada e grave - vide Tabela 9 abaixo para classificação da função renal), a exposição significativa a imatinibe (dose normalizada pela área sob a curva - AUC) aumentou 1,5 a 2,0 vezes comparada a pacientes com função renal normal, os quais apresentaram um nível elevado de glicoproteína-alfa ácida plasmática, uma proteína na qual o imatinibe se liga fortemente. Nenhuma relação entre a exposição de imatinibe e a gravidade da deficiência renal foi observada. Neste estudo, 800 mg por dia foram usados com segurança em pacientes com disfunção renal leve e 600 mg por dia foram usados em disfunção renal moderada. As doses de 800 mg não foram testadas em pacientes com disfunção renal moderada devido ao número limitado de pacientes admitidos. Da mesma maneira, apenas dois pacientes com disfunção renal grave foram admitidos nas doses baixas (100 mg) e as doses maiores não foram testadas. Nenhum dos pacientes em hemodiálise foi admitido neste estudo. Dados de literatura mostraram que uma dose diária de 400 mg foi bem tolerada em um paciente em hemodiálise com doença renal em estágio terminal. A farmacocinética de exposição plasmática neste paciente caiu dentro da faixa de valores de imatinibe e seus metabólitos CGP74588 observados em pacientes com função renal normal. A diálise não mostrou intervir na cinética plasmática do imatinibe. Como a excreção renal representa a menor via de eliminação do imatinibe, pacientes com insuficiência renal grave e em diálise poderiam receber tratamento de dose inicial de 400 mg. Entretanto, recomenda-se cautela com estes pacientes. A dose pode ser reduzida se houver intolerância, ou aumentada em caso de falta de eficácia28 (vide "Posologia e modo de usar", "Advertências e precauções" e "Características farmacológicas").
Referências Bibliográficas
1. Protocol 0102, A Phase II open-label study to determine the safety and anti-leukemic effects of STI571 in patients with Philadelphia chromosome-positive chronic myeloid leukemia in myeloid blast crisis. Novartis Pharma AG. Basel, Switzerland. 06 Feb 01, Part IV, Volume 5, Page No.064. [1] (dados em arquivo)
2. Protocol 0109, A Phase II study to determine the safety and anti-leukemic effects of STI 571 in adult patients with Philadelphia chromosome positive leukemia including acute lymphoblastic leukemia, acute myeloid leukemia, lymphoid blast crisis chronic myeloid leukemia and accelerated phase chronic myeloic leukemia. Novartis Pharma AG. Basel, Switzerland. 07 Feb 01, Part IV, Volume 13, Page No.124. [2] (dados em arquivo)
3. Protocol 0110, A Phase II study to determine the efficacy and safety of STI571 in patients with chronic myeloid leukemia who are refractory to or intolerant of interferon-alpha. Novatis Pharma AG. Basel, Switzerland. 02 Feb 01, Part IV, Volume 23, Page No.276. [3] (dados em arquivo)
4. A phase III study of STI571 versus Interferon-a (IFN-a) combined with Cytarabine (Ara-C) in patients with newly diagnosed previously untreated Philadelphia chromosome positive (Ph+) chronic myelogenous leukemia in chronic phase (CML-CP). Study No. CSTI571 0106. Novartis Pharma AG. Basel, Switzerland. 28 May 02, Part IV1, Volume 1, Page No.111. [48] (dados em arquivo)
5. Study CSTI571 0106 Amendment 3, A phase III of STI571 versus Interferon-alpha (IFN-alpha) combined with Cytarabine (Ara-C) in patients with Newly diagnosed previously untreated Ph+ chronic myelogeneous leukemia in chronic phase. Novartis Pharma AG. Basel, Switzerland. 23 Jan 02. [66] (dados em arquivo)
6. Clinical Overview to update MESILATO DE IMATINIBE /Gleevec labeling for the treatment of chronic myeloid leukaemia. Novartis Pharma AG. Basel, Switzerland. 18 Jul 06. [102] (dados em arquivo)
7. Study No. CSTI571 0106 A phase III of STI571 versus Interferon-alpha (IFN-alpha) combined with Cytarabine (Ara-C) in patients with newly diagnosed previously untreated Ph+ chronic myelogeneous leukemia in chronic phase - Data update (cut-off 31-Jan-08): 84-month data. Novartis Pharma AG. Basel, Switzerland. 20 Jun 08. [117] (dados em arquivo)
8. Study No. CSTI571 0106, A phase III of STI571 versus Interferon-alpha (IFN-alpha) combined with Cytarabine (Ara-C) in patients with Newly diagnosed previously untreated Ph+ chronic myelogeneous leukemia in chronic phase - Data update (cut-off 31-Jul-03): 30-months data. Novartis Pharma AG. Basel, Switzerland. 18 Dec 03, Module 5, Volume 6, Section 5.3.5.1, Page No.1. [63] (dados em arquivo)
9. [Study No. CSTI571 0106] A phase III of STI571 versus Interferon-alpha (IFN-alpha) combined with Cytarabine (Ara-C) in patients with newly diagnosed previously untreated Ph+ chronic myelogeneous leukemia in chronic phase - Data update (cut-off 31-Jan-06): 60-month data. Novartis Pharma AG. Basel, Switzerland. 03 Jul 06. [103] (dados em arquivo)
10. Study No. CSTI571 0106, A phase III of STI571 versus Interferon-alpha (IFN-alpha) combined with Cytarabine (Ara-C) in patients with Newly diagnosed previously untreated Ph+ chronic myelogeneous leukemia in chronic phase- Quantitative PCR analysis of Minimal Residual Disease (cut-off 31-Jul-03). Novartis Pharma AG. Basel, Switzerland. 27 Jul 04, Module 5, Volume 8, Section 5.3.5.1, Page No.7236. [67] (dados em arquivo)
11. Clinical Study Report Study No. CSTI571 0102: A Phase II open-label study to determine the safety and anti-leukemic effects of STI571 in patients with Philadelphia chromosome-positive chronic myeloid leukemia in myeloid blast crisis. Novartis Pharma AG. Basel, Switzerland. 16 Dec 02. [61] (dados em arquivo)
12. [Carroll M, Ohno-Jones S, Tamura S, et al. (1997)] CGP 57148, a tyrosine kinase inhibitor, inhibits the growth of cells expressing BCR-ABL, TEL-ABL, and TEL-PDGFR fusion proteins. Blood 90:4947-52. [90]
13. Clinical Overview - MESILATO DE IMATINIBE in the treatment of life threatening diseases known to be associated with one or more imatinib-sensitive tyrosine kinases - Systemic Mastocytosis (SM) without the D816V c-Kit mutation. Novartis Pharma AG. Basel, Switzerland. 07 Feb 06. [95] (dados em arquivo)
14. Clinical Overview - MESILATO DE IMATINIBE in the treatment of life threatening diseases known to be associated with one or more imatinib-sensitive tyrosine kinases - Myelodisplastic / Myeloproliferative Diseases associated with PDGFR gene re-arrangement. Novartis Pharma AG. Basel, Switzerland. 23 Nov 05. [82] (dados em arquivo)
15. [Pardanani A, Reeder T, Porrata LF, et al. (2003)] Imatinib therapy for hypereosinophilic syndrome and other eosinophilic disorders. Blood; 101(9):3391-7. [89]
16. [Roy L, Guilhot J, Krahnke T, et al (2006)] Survival advantage from imatinib compared to the combination interferon-alpha plus cytarabine in chronic phase CML: historical comparison between two phase III trials. Blood; 108(5):1478-84. [104]
17. Addendum - Study No. CSTI571 0106 A phase III of STI571 versus Interferon-alpha (IFN-alpha) combined with Cytarabine (Ara-C) in patients with newly diagnosed previously untreated Ph+ chronic myelogeneous leukemia in chronic phase - Data update (cut-off 31-Jan-08): 84-month data, Additional analysis for BPI update. Novartis Pharma AG. Basel, Switzerland. 24