GILENYA
NOVARTIS
fingolimode
Tratamento da esclerose múltipla.
Apresentações.
GilenyaTM 0,5 mg - embalagem contendo 28 cápsulas.
VIA ORAL
USO ADULTO
Composição.
Cada cápsula de GilenyaTM contém 0,56 mg de cloridrato de fingolimode, equivalente a 0,5 mg de fingolimode. Excipientes: manitol, estearato de magnésio, óxido de ferro amarelo, dióxido de titânio, gelatina, tinta de impressão preta e tinta de impressão amarela.
Indicações.
GilenyaTM é indicado como uma terapia modificadora da doença para o tratamento de pacientes com esclerose múltipla remitente recorrente para reduzir a frequência de reincidências e retardar a progressão da incapacidade.
Resultados de eficácia.
A eficácia de GilenyaTM(1) foi demonstrada em dois estudos que avaliaram doses diárias de 0,5 mg e 1,25 mg de GilenyaTM em pacientes com esclerose múltipla remitente recorrente. Os dois estudos incluíram pacientes que haviam apresentado pelo menos 2 recidivas clínicas durante os 2 anos antes da randomização ou pelo menos 1 recidiva clínica durante o 1° ano antes da randomização, e que haviam apresentado uma Escala de Estado de Incapacidade Expandida (EDSS) entre 0 e 5,5. Um terceiro estudo visando a mesma população de pacientes foi concluído após o registro de GilenyaTM(2).
Estudo D2301 (FREEDOMS)
O estudo D2301 (FREEDOMS) foi um estudo de fase III de 2 anos, randomizado, duplo-cego, placebo controlado em pacientes com esclerose múltipla recidiva-remitente que não haviam recebido betainterferona ou acetato de glatirâmer ao longo de pelo menos 3 meses anteriores e que não haviam recebido natalizumabe ao longo de pelo menos 6 meses anteriores. Avaliações neurológicas foram realizadas na Seleção, a cada 3 meses e no momento de suspeita da recidiva. Avaliações de MRI foram realizadas na seleção, mês 6, mês 12 e mês 24. O desfecho primário foi a taxa de recidiva anual. A idade média era de 37 anos, a duração mediana da doença era de 6,7 anos e a pontuação mediana basal na EDSS foi de 2,0. Os pacientes foram randomizados para receber GilenyaTM 0,5 mg (n = 425), GilenyaTM 1,25 mg (n = 429) ou placebo (n = 418) por até 24 meses. O tempo mediano recebendo o medicamento no estudo foi de 717 dias com 0,5 mg, 715 dias com 1,25 mg e 718,5 dias com placebo. A taxa de recidiva anual foi significativamente menor em pacientes tratados com GilenyaTM do que em pacientes que receberam placebo. O principal desfecho secundário foi o tempo até a progressão da incapacidade confirmada em 3 meses conforme medida por pelo menos um aumento de 1 ponto a partir do valor basal na EDSS (aumento de 0,5 ponto para pacientes com valor de basal de 5,5 na EDSS) mantido por 3 meses. O tempo até o início da progressão da incapacidade confirmada em 3 meses foi significativamente retardado com o tratamento com GilenyaTM em comparação com placebo. Não houve diferença significativa entre as doses de 0,5 mg e 1,25 mg em ambos os desfechos.
Os resultados para esse estudo estão demonstrados na Tabela 1 e Figuras 1 e 2.
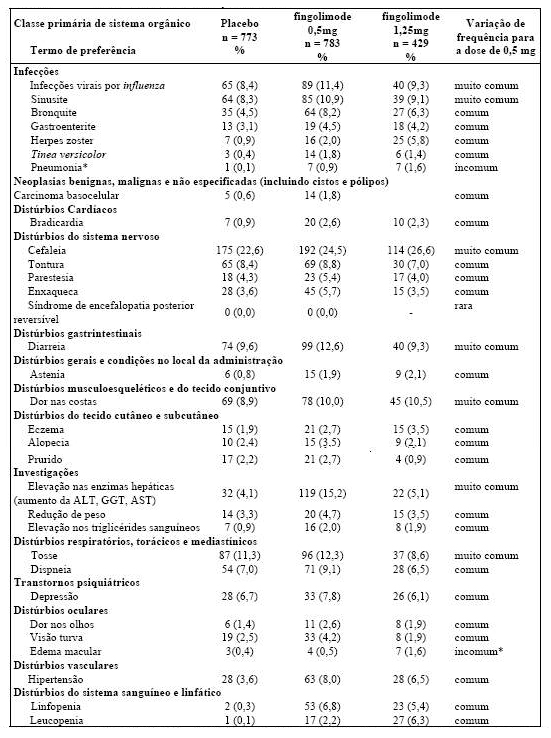
Tabela 1 Resultados clínicos e de MRI do Estudo FREEDOMS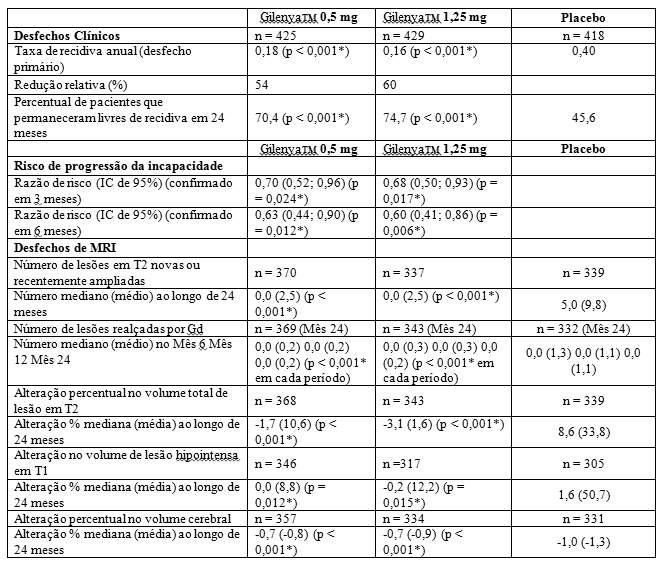
Todas as análises de desfechos clínicos foram intencionadas para tratar. As análises MRI usaram o conjunto de dados avaliável.
* Indica significância estatística versus placebo nível 0,05 bilateral.
Determinação de valores-p: ARR agregado por regressão binominal negativa ajustada por tratamento, país agrupado, número de recidivas nos últimos 2 anos e EDSS basal; percentual de pacientes mantendo regressão logística livre de recidiva ajustada por tratamento, país, número de recidiva nos últimos 2 anos, e EDSS basal; tempo até a progressão de deficiência confirmada em 3 meses/6 meses pelo modelo de riscos proporcionais de Cox ajustado por tratamento, país agrupado, EDSS basal, e idade; lesões em T2 novas/recentemente ampliadas por regressão binominal negativa ajustada por tratamento e país agrupado; lesões realçadas por Gd pelo rank ANCOVA ajustado por tratamento, país agrupado, e número basal das lesões realçadas por Gd; e alteração % na lesão e volume cerebral pelo rank ANCOVA ajustado por tratamento, país agrupado, e valor basal correspondente.
Figura 1 Gráfico de Kaplan-Meier do tempo até a primeira recidiva confirmada até o Mês 24 - Estudo FREEDOMS (população ITT)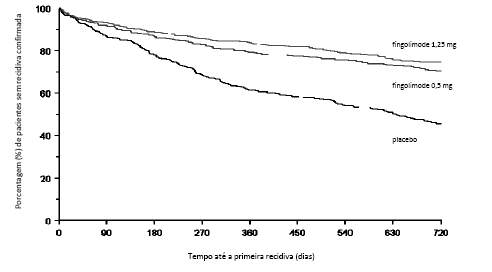
Figura 2
Gráfico cumulativo do tempo até a progressão da deficiência confirmada em 3 meses - Estudo FREEDOMS (população ITT)
Pacientes que completaram o estudo FREEDOMS (D2301) tinham a opção de entrar no estudo de extensão duplo-cego D2301E1(3). 920 pacientes do estudo principal entraram na extensão e foram todos tratados com fingolimode (n = 331 continuaram com 0,5 mg, 289 continuaram com 1,25 mg, 155 trocaram de placebo para 0,5 mg e 145 trocaram do placebo para 1,25 mg). 811 desses pacientes (88,2%) tiveram pelo menos 18 meses de acompanhamento na fase de extensão. A duração máxima da exposição cumulativa a fingolimode 0,5 mg (principal + extensão) foi de 1782 dias. No mês 24 do estudo de extensão, os pacientes que receberam placebo no estudo principal tiveram reduções em taxa de recidiva anual de 55% após a mudança para fingolimode 0,5 mg (razão taxa de recidiva anual 0,45, 95% CI 0,32-0,62, p < 0,001). A taxa de recidiva anual para os pacientes que foram tratados com fingolimode 0,5 mg no estudo principal manteve-se baixa durante o estudo de extensão (taxa de recidiva anual de 0,10 no estudo de extensão).
Estudo D2309 (FREEDOMS II)
O estudo D2309 (FREEDOMS II)(4) teve um desenho semelhante ao do estudo D2301 (FREEDOMS): o estudo foi de 2 anos, randomizado, duplo-cego, placebo-controlado, fase III, em pacientes com esclerose múltipla remitente recorrente da esclerose múltipla que não receberam qualquer betainterferona ou acetato de glatirâmer, pelo menos nos 3 meses anteriores e não receberam qualquer natalizumabe durante pelo menos os seis meses anteriores. Foram realizadas avaliações neurológicas durante a triagem, a cada 3 meses, e no momento da suspeita de recidiva. Avaliações de MRI foram realizadas na triagem, mês 6, mês 12 e mês 24. O objetivo primário foi a taxa de recidiva anual.
A idade média foi 40,5 anos, a duração média da doença foi de 8,9 anos e a média da pontuação da EDSS no início do estudo foi de 2,5. Os pacientes foram randomizados para receber tratamento com GilenyaTM 0,5 mg (n = 358) ou GilenyaTM 1,25 mg (n = 370) ou placebo (n = 355) por até 24 meses.
O tempo médio com a medicação de estudo foi de 719 dias com 0,5 mg e 719 dias com placebo.
Os pacientes randomizados para o braço com dose de fingolimode de 1,25 mg foram trocados de forma cega para receber fingolimode 0,5 mg quando os resultados do estudo de 2301 ficaram disponíveis e confirmaram um melhor risco benefício da dose mais baixa. A dose foi alterada para 113 pacientes (30,5%) nesse braço da dose, o tempo médio de fingolimode 1,25 mg neste braço foi de 496,1 dias e 209,8 dias para fingolimode 0,5 mg.
A taxa de recidiva anual foi significativamente inferior nos pacientes tratados com GilenyaTM em relação aos pacientes que receberam placebo. O primeiro objetivo secundário chave foi a mudança no volume cerebral do valor basal. A perda de volume cerebral foi significativamente inferior no tratamento com GilenyaTM em comparação com o placebo. O outro objetivo secundário chave foi a confirmação da progressão da incapacidade no tempo de três meses, medida por pelo menos aumento de 1 ponto do valor basal na EDSS (aumento de 0,5 ponto para pacientes com EDSS valor basal de 5,5) sustentado por 3 meses. O risco de progressão de incapacidade para GilenyaTM e placebo não foram estatisticamente diferentes.
Não houve diferenças significativas entre as doses de 0,5 mg e 1,25 mg em qualquer um dos objetivos. Os resultados deste estudo são apresentados na Tabela 2 e na Figura 3.
Tabela 2 Resultados clínicos e de MRI do Estudo FREEDOMS II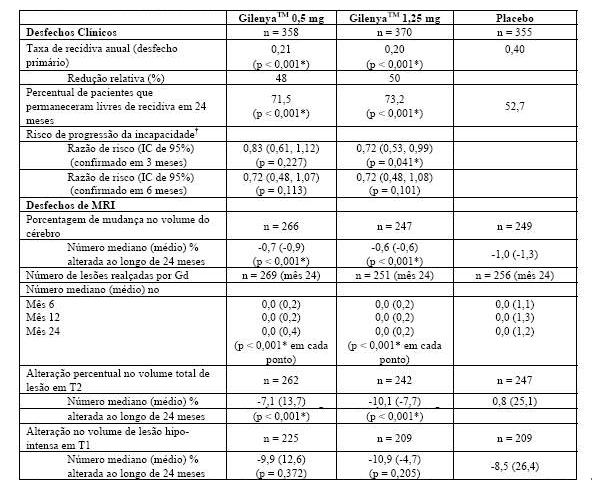
Todas as análises de desfechos clínicos foram intencionadas para tratar. As análises MRI usaram o conjunto de dados avaliável.
* Indica significância estatística versus placebo nível 0,05 bilateral.
Determinação de valores-p: ARR agregado por regressão binominal negativa ajustada por tratamento, país agrupado, número de recidivas nos últimos 2 anos e EDSS basal; percentual de pacientes mantendo regressão logística livre de recidiva ajustada por tratamento, país, número de recidiva nos últimos 2 anos, e EDSS basal; tempo até a progressão de deficiência confirmada em 3 meses/6 meses pelo modelo de riscos proporcionais de Cox ajustado por tratamento, país agrupado, EDSS basal, e idade; lesões em T2 novas/recentemente ampliadas por regressão binominal negativa ajustada por tratamento e país agrupado; lesões realçadas por Gd pelo rank ANCOVA ajustado por tratamento, país agrupado, e número basal das lesões realçadas por Gd; e alteração % na lesão e volume cerebral pelo rank ANCOVA ajustado por tratamento, país agrupado, e valor basal correspondente.
† Análises adicionais revelaram que resultados na população total não foram significativos devido às progressões falso positivas no subgrupo de pacientes com EDSS basal = 0 (n = 62, 8,7% da população do estudo). Em pacientes com EDSS > 0 (n = 651, 91,3% da população do estudo), fingolimode 0,5 mg demonstrou uma redução clinicamente relevante e estatisticamente significativa em relação ao placebo (HR = 0,70, IC (0,50, 0,98), p = 0,040), de acordo com estudo FREEDOMS.
Figura 3
Gráfico Kaplan-Meier para o tempo até a primeira recidiva confirmada até o Mês 24 - Estudo FREEDOMS II (população ITT)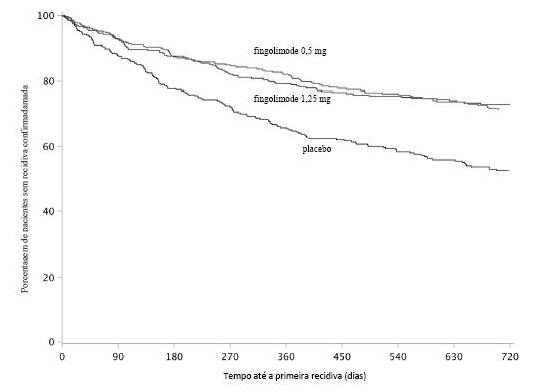
Estudo D2302 (TRANSFORMS)
O Estudo D2302 (TRANSFORMS) foi um estudo de Fase III de 1 ano, randomizado, duplo-cego, duplo-mascarado, ativo-controlado (betainterferona 1a, 30 mcg, intramuscular, uma vez por semana) em pacientes com esclerose múltipla remitente recorrente que não haviam recebido natalizumabe nos últimos 6 meses. A terapia anterior com betainterferona ou acetato de glatirâmer até o momento da randomização foi permitida.
Avaliações neurológicas foram realizadas na seleção, a cada 3 meses e no momento das suspeitas recidivas. Avaliações por MRI foram realizadas na seleção e no mês 12. O desfecho primário foi a taxa de recidiva anual.
A idade média era de 36 anos, a duração mediana da doença era de 5,9 anos e a pontuação mediana na EDSS basal foi de 2,0. Os pacientes foram randomizados para receber GilenyaTM 0,5 mg (n = 431) ou GilenyaTM 1,25 mg (n = 426) ou 30 microgramas de betainterferona 1a pela via intramuscular uma vez por semana (n = 435) por até 12 meses. O tempo médio do estudo recebendo o medicamento de 365 dias com GilenyaTM 0,5 mg, 354 dias com GilenyaTM 1,25 mg e 361 dias com betainterferona 1a.
A taxa de recidiva anual foi significativamente menor em pacientes tratados com GilenyaTM do que em pacientes que receberam betainterferona 1a IM. Não houve diferença significativa entre as doses de 0,5 mg e 1,25 mg de GilenyaTM . Os principais desfechos secundários foram o número de lesões em T2 novas ou recentemente ampliadas e o tempo até o início da progressão da deficiência confirmada em 3 meses conforme medida por pelo menos um aumento de 1 ponto a partir do valor de basal na EDSS (aumento de 0,5 ponto para aqueles com valor basal de 5,5 na EDSS) mantido por 3 meses. O número de lesões em T2 novas ou recentemente ampliadas foi significativamente menor em pacientes tratados com GilenyaTM do que em pacientes que receberam betainterferona 1a IM. Não houve diferença significativa no tempo até a progressão de deficiência confirmada em 3 meses entre pacientes tratados com GilenyaTM e betainterferona 1a IM em 1 ano. Não houve diferença significativa entre as doses de 0,5 mg e 1,25 mg em quaisquer desfechos.
Os resultados para esse estudo são demonstrados na Tabela 3 e Figura 4.
Tabela 3 Resultados clínicos e de MRI do Estudo TRANSFORMS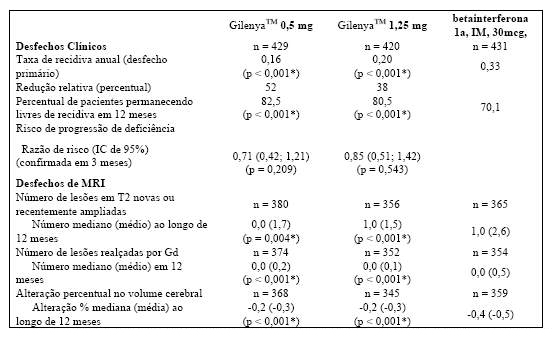
Todas as análises de desfechos clínicos foram intencionadas para tratar. As análises MRI usaram o conjunto de dados avaliável.
* Indica significância estatística versus betainterferona 1a IM no nível 0,05 bilateral.
Determinação de valores-p: ARR agregado por regressão binominal negativa ajustada por tratamento, país, número de recidivas nos últimos 2 anos e EDSS basal; porcentagem de pacientes mantendo regressão logística livre de recidiva ajustada por tratamento, país, número de recidiva nos últimos 2 anos, e EDSS basal; risco de progressão da deficiência pelo modelo de riscos proporcionais de Cox ajustado por tratamento, país, EDSS basal, e idade; lesões em T2 novas/recentemente ampliadas por regressão binominal negativa ajustada por tratamento, país, número de recidivas nos últimos 2 anos e EDSS basal; lesões realçadas por Gd pelo rank ANCOVA ajustado por tratamento, país, e número basal das lesões realçadas por Gd; e alteração % no volume cerebral pelo teste de soma de postos do rank Wilcoxon.
Figura 4 Gráfico Kaplan-Meier para o tempo até a primeira recidiva confirmada até o Mês 12 - Estudo TRANSFORMS (população ITT)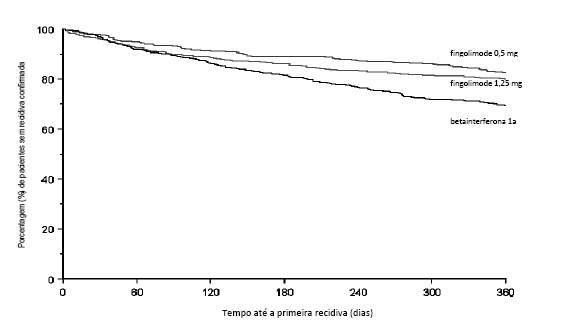
Os pacientes que completaram o estudo TRANSFORMS (D2302) tinham a opção de entrar na extensão de dose-cega. 1030 pacientes do estudo principal entraram na extensão (estudo D2302E1)(5), e foram tratados com fingolimode (n = 357 continuaram em 0,5 mg, 330 continuaram em 1,25 mg, 167 trocaram de betainterferona-1a para 0,5 mg e 176 trocaram de betainterferona-1a para 1,25 mg). 882 desses pacientes (85,9%) tiveram pelo menos 12 meses de acompanhamento na fase de extensão. A duração máxima da exposição cumulativa a fingolimode 0,5 mg (estudo principal + extensão) foi de 1594 dias.
No mês 12 de extensão do estudo, os pacientes que receberam betainterferona-1a i.m. no estudo principal tiveram reduções relativas na taxa de recidiva anual de 30% após mudar para o fingolimode 0,5 mg (taxa de recidiva anual = 0,70, p = 0,06). A taxa de recidiva anual para os pacientes que foram tratados com fingolimode 0,5 mg no estudo principal foi baixa durante a combinação do estudo principal e da extensão (taxa de recidiva anual de 0,18 até ao mês 24).
Os resultados agrupados dos estudos D2301 (FREEDOMS) e D2302 (TRANSFORMS) demonstraram uma redução consistente da taxa na recidiva anual de GilenyaTM em comparação com o comparador em subgrupos definidos por sexo, idade, terapia anterior para esclerose múltipla, atividade da doença ou níveis de deficiência basal(6).
Referências
(1) Summary of Clinical Efficacy (dados em arquivo).
(2) 2.5 Clinical Overview Rationale for changes to Core Data Sheet (CDS) - Updated efficacy data based on studies CFTY720D2309, CFTY720D2301E1 and CFTY720D2302E1. Novartis. 27-Aug-2013. (dados em arquivo).
(3) [D2301E1 report (2012)] An extension of the 24-month, double-blind, randomized, multicenter, placebo-controlled, parallel-group study comparing efficacy and safety of FTY720 1.25 mg and 0.5 mg administered orally once daily versus placebo in patients with relapsing-remitting multiple sclerosis. Novartis. 12-Jan-2012. (dados em arquivo).
(4) [TRANSFORM II Study D2309 report (2012)] A 24-month double-blind, randomized, multicenter, placebo controlled, parallel-group study comparing the efficacy and safety of 0.5 mg and 1.25 mg fingolimod (FTY720) administered orally once daily versus placebo in patients with relapsing-remitting multiple sclerosis. Novartis. 13-Jan2012. (dados em arquivo).
(5) [D2302E1 report (2012)] A 12-month double-blind, randomized, multicenter, activecontrolled, parallel-group study comparing the efficacy and safety of 0.5 mg and 1.25 mg fingolimod (FTY720) administered orally once daily versus interferon ß-1a (Avonex®) administered i.m. once weekly in patients with relapsing-remitting multiple sclerosis with optional Extension. Novartis. 10-Jan-2012 (dados em arquivo).
(6) Clinical Overview section 4.5 (dados em arquivo).
Caract. farmacológicas.
Código ATC: L04AA27
Mecanismo de ação
O fingolimode é um modulador do receptor esfingosina-1-fosfato. O fingolimode é metabolizado pela esfingosinaquinase ao metabólito ativo fingolimode-fosfato. O fingolimode-fosfato se liga em concentrações nanomolares baixas aos receptores esfingosina-1-fosfato (S1P) 1, 3, e 4 localizados nos linfócitos, e cruza prontamente a barreira hematoencefálica para se ligar aos receptores S1P 1, 3, e 5 localizados nas células neurais no sistema nervoso central (SNC). Agindo como um antagonista funcional de S1PR nos linfócitos, o fingolimode-fosfato bloqueia a capacidade dos linfócitos de egressar dos linfonodos, causando uma redistribuição, ao invés da depleção dos linfócitos. Essa redistribuição reduz a infiltração de células linfocíticas, incluindo células pró-inflamatórias Th17, patogênicas no SNC, no qual elas seriam envolvidas em inflamação nervosa e dano de tecido nervoso. Estudos em animais e experimentos in vitro indicam que o fingolimode pode também exercer efeitos benéficos na esclerose múltipla através da interação com receptores S1P em células neurais.
O fingolimode penetra no SNC, tanto em seres humanos como em animais, e demonstrou reduzir a astrogliose, desmielinização e perda neuronal. Além disso, o tratamento com fingolimode aumenta os níveis do fator neurotrópico derivado do cérebro (BDNF) no córtex, hipocampo e corpo estriado do cérebro para apoiar a sobrevivência neuronal e melhorar funções motoras.
Propriedades farmacodinâmicas
Sistema imune
Efeitos sobre os números de células imunes no sangue. Dentro de 4-6 horas após a primeira dose de 0,5 mg de fingolimode, a contagem de linfócitos reduz para aproximadamente 75% do valor basal. Com a continuação da dosagem diária, a contagem de linfócitos continua a reduzir ao longo de um período de duas semanas, alcançando uma contagem nadir de aproximadamente 500 células/mL ou aproximadamente 30% do valor basal. Dezoito por cento dos pacientes alcançaram um nadir ≤ 200 células/mL em pelo menos uma ocasião. Baixas contagens de linfócitos são mantidas com a dosagem crônica diária. A maioria dos linfócitos T e B regularmente circula através dos órgãos linfoides e essas são as células principalmente afetadas pelo fingolimode. Aproximadamente 15-20% dos linfócitos T possuem um fenótipo de memória efetora, células que são importantes para a vigilância periférica imune. Uma vez que esse subgrupo de linfócitos geralmente não circula para os órgãos linfoides, ele não é afetado pelo fingolimode. Aumentos na contagem de linfócitos periféricos são evidentes dentro de dias após a interrupção do tratamento com fingolimode e, geralmente, contagens normais são atingidas dentro de um a dois meses. A dosagem crônica de fingolimode leva a uma leve redução na contagem de neutrófilos para aproximadamente 80% do valor basal. Monócitos não são afetados pelo fingolimode.
Frequência e ritmo cardíaco
O fingolimode causa uma redução temporária na frequência cardíaca e na condução atrioventricular no início do tratamento (vide item Reações Adversas). O declínio máximo da frequência cardíaca é observado nas primeiras 4-5 horas pós-dose, com 70% do efeito cronotrópico negativo atingido no primeiro dia. A frequência cardíaca retorna progressivamente aos valores basais dentro de um mês do tratamento crônico.
Respostas autonômicas do coração, incluindo a variação diurna da frequência cardíaca e a resposta ao exercício, não são afetadas pelo tratamento com fingolimode.
Com o início do tratamento com fingolimode, ocorre um aumento nas contrações atriais prematuras, mas não há qualquer taxa aumentada de fibrilação/flutter atrial ou arritmias ventriculares ou ectopia. O tratamento com fingolimode não está associado com a redução no débito cardíaco.
A redução na frequência cardíaca induzida pelo fingolimode pode ser revertida pela atropina, isoprenalina ou salmeterol.
Potencial para prolongar o intervalo QT
Em um estudo minucioso sobre o intervalo QT das doses de 1,25 ou 2,5 mg de fingolimode em estado de equilíbrio, quando um efeito cronotrópico negativo de fingolimode ainda estava presente, o tratamento com fingolimode resultou em um prolongamento do QTcI, com o limite superior do IC de 90% ≤ 13,0 msec. Não há relação da dose-resposta ou da exposição-resposta de fingolimode com prolongamento de QTcI. Não há sinais consistentes da incidência aumentada de valores discrepantes no QTcI, sejam eles absolutos ou alterados em relação ao valor basal, associados com o tratamento com fingolimode. Nos estudos de esclerose múltipla, não houve qualquer prolongamento clinicamente relevante do intervalo QT.
Função pulmonar
O tratamento com fingolimode com dose única ou doses múltiplas de 0,5 e 1,25 mg por duas semanas não está associado com um aumento detectável na resistência das vias aéreas, medida pelo VEF1 e fluxo expiratório forçado durante a expiração de 25 a 75% da capacidade vital forçada (CVF25-75). No entanto, doses únicas de fingolimode ≥ 5 mg (10 vezes a dose recomendada) estão associadas a um aumento dose-dependente da resistência das vias aéreas. O tratamento com fingolimode com doses múltiplas de 0,5, 1,25, ou 5 mg não está associado a oxigenação prejudicada ou dessaturação de oxigênio com exercícios ou aumento da responsividade das vias aéreas à metacolina. Pacientes submetidos ao tratamento com fingolimode têm resposta broncodilatadora normal aos b -agonistas inalados.
Propriedades Farmacocinéticas
Absorção
A absorção do fingolimode é lenta (tmáx de 12-16 horas) e extensiva (³ 85%, baseados na quantidade de radioatividade excretada na urina e na quantidade de metabólitos extrapolados nas fezes para o infinito). A biodisponibilidade oral absoluta aparente é alta (93%). A ingestão de alimentos não altera a Cmáx ou a exposição (AUC) de fingolimode ou fingolimode-fosfato. Portanto, GilenyaTM pode ser tomado independentemente das refeições (vide item Posologia e Modo de Usar).
Concentrações sanguíneas em estado de equilíbrio são atingidas dentro de 1 a 2 meses da administração de uma vez ao dia e os níveis em estado de equilíbrio são aproximadamente 10 vezes maiores do que com a dose inicial.
Distribuição
O fingolimode se distribui altamente nos glóbulos vermelhos, com a fração de 86% nas células sanguíneas. O fingolimode-fosfato apresenta uma menor captação em células sanguíneas de < 17%. O fingolimode e o fingolimodefosfato são altamente ligados a proteínas ( > 99,7%). A ligação proteica de fingolimode e fingolimode-fosfato não é alterada por danos renais ou hepáticos.
O fingolimode é extensivamente distribuído aos tecidos do corpo com um volume de distribuição de cerca de 1200 ± 260 L. Um estudo realizado em quatro indivíduos saudáveis que receberam uma única dose intravenosa de fingolimode marcado com radio iodo demonstraram que o fingolimode penetra no cérebro. Num estudo com 13 pacientes do sexo masculino com esclerose múltipla que receberam GilenyaTM 0,5 mg/dia, no estado de equilíbrio, a quantidade de fingolimode (e fingolimode-fosfato) na ejaculação seminal era mais do que 10000 vezes menor do que a dose administrada (0,5 mg).
Metabolismo
A biotransformação de fingolimode em humanos ocorre por três vias principais; por fosforilação estereo seletiva reversível para o (S)-enantiômero farmacologicamente ativo de fingolimode-fosfato, por biotransformação oxidativa catalisada principalmente pela CYP4F2 e possivelmente outras isoenzimas CYP4F e subsequente degradação semelhante à do ácido graxo para metabólitos inativos, e pela formação de análogos de ceramida não-polares farmacologicamente inativos de fingolimode.
Após a administração oral única de [14C]-fingolimode, os principais componentes no sangue relacionados ao fingolimode, conforme julgados pela sua contribuição à AUC de até 816 horas pós-dose do total de componentes radiomarcados, são o próprio fingolimode (23,3%), fingolimode-fosfato (10,3%), e metabólitos inativos (metabólito do ácido carboxílico M3 (8,3%), metabólito da ceramida M29 (8,9%) e metabólito da ceramida M30 (7,3%)).
Eliminação
O clearance sanguíneo de fingolimode é de 6,3 ± 2,3 L/h, e a meia-vida (t1/2) terminal aparente média é de 6-9 dias. Os níveis sanguíneos de fingolimode-fosfato reduzem em paralelo com fingolimode na fase terminal, produzindo meiasvidas semelhantes para ambos.
Após administração oral, cerca de 81% da dose é lentamente excretada na urina na forma de metabólitos inativos. O fingolimode e o fingolimode-fosfato não são excretados intactos na urina, mas são os principais componentes nas fezes, com quantidades representando menos que 2,5% da dose, cada. Após 34 dias, a recuperação da dose administrada é de 89%.
Linearidade
As concentrações de fingolimode e de fingolimode-fosfato aumentam de uma maneira aparentemente proporcional à dose após múltiplas doses de uma vez ao dia de 0,5 mg ou 1,25 mg de fingolimode.
Populações Especiais
Disfunção Renal
O comprometimento renal grave aumenta a Cmáx e a AUC de fingolimode em 32% e 43%, respectivamente, e a Cmáx e a AUC de fingolimode-fosfato em 25% e 14%, respectivamente. A meia-vida de eliminação aparente não é alterada para ambos os analitos. Nenhum ajuste de dose de GilenyaTM é necessário em pacientes com comprometimento renal.
Disfunção Hepática
A farmacocinética do fingolimode em dose única (1 ou 5 mg), quando avaliada em indivíduos com comprometimentos hepáticos leve, moderado e grave (Child-Pugh classe A, B e C), não demonstrou qualquer alteração na Cmáx de fingolimode, mas um aumento na AUC em 12%, 44% e 103%, respectivamente. A meia-vida de eliminação aparente não é alterada pelo comprometimento hepático leve, mas é prolongada em 49-50% pelo comprometimento hepático moderado e grave. Em pacientes com insuficiência hepática grave (Child-Pugh classe C), o Cmáx do fingolimode-fosfato foi reduzido em 22% e a AUC aumentada em 38%. A farmacocinética do fingolimode-fosfato não foi avaliada em pacientes com insuficiência hepática leve a moderada. Embora o comprometimento hepático tenha provocado alterações na disposição de fingolimode e fingolimode-fosfato, a magnitude dessas alterações sugere que a dose de fingolimode não precisa ser ajustada em pacientes com comprometimento hepático leve ou moderado (Child-Pugh classe A e B). O fingolimode deve ser usado com cautela em pacientes com comprometimento hepático grave (Child-Pugh classe C).
Pediatria
A segurança e eficácia de GilenyaTM em pacientes pediátricos abaixo de 18 anos não foram estudadas. GilenyaTM não é indicado para o uso em pacientes pediátricos.
Geriatria
O mecanismo de eliminação e os resultados da população farmacocinética sugerem que o ajuste de dose não seria necessário em pacientes idosos. Entretanto, a experiência clínica em pacientes com mais de 65 anos de idade é limitada.
Etnia
Os efeitos de origem étnica na farmacocinética de fingolimode e fingolimode-fosfato não são de relevância clínica.
Sexo
O sexo não exerce influência sobre a farmacocinética de fingolimode e fingolimode-fosfato.
Dados de segurança pré-clínicos
O perfil de segurança pré-clínico de fingolimode foi avaliado em camundongos, ratos, cães e macacos. Os principais órgãos-alvo foram o sistema linfoide (linfopenia e atrofia linfoide), pulmões (aumento de peso, hipertrofia do músculo liso na junção bronquioalveolar), e coração (efeito cronotrópico negativo, aumento na pressão arterial, alterações perivasculares e degeneração do miocárdio) em diversas espécies; vasos sanguíneos (vasculopatia) apenas em ratos; e pituitária, pré-estômago, fígado, adrenais, trato gastrintestinal e sistema nervoso apenas em altas doses (frequentemente associados com sinais de toxicidade geral) em diversas espécies.
Nenhuma evidência de carcinogenicidade foi observada em um bioensaio de 2 anos em ratos em doses orais de fingolimode até a dose máxima tolerada de 2,5 mg/kg, representado uma margem de aproximadamente 50 vezes com base na exposição sistêmica humana (AUC) na dose de 0,5 mg. Entretanto, em um estudo de 2 anos em camundongos, uma incidência elevada de linfoma maligno foi observada em doses de 0,25 mg/kg e superiores, representando uma margem de aproximadamente 6 vezes com base na exposição sistêmica humana (AUC) em uma dose diária de 0,5 mg. O fingolimode não foi mutagênico em um teste Ames e em uma linhagem celular de linfoma L5178Y de camundongo in vitro. Nenhum efeito clatogênico foi observado in vitro em células pulmonares V79 de hamster chinês. O fingolimode induziu aberrações cromossômicas numéricas (poliploide) em células V79 em concentrações de 3,7 mcg/mL e superiores. O fingolimode não foi clastogênico nos teste de micronúcleo in vivo em camundongos e ratos.
O fingolimode não teve qualquer efeito na contagem ou motilidade de esperma, nem na fertilidade em ratos machos e fêmeas até a dose mais alta testada (10 mg/kg), representando uma margem de aproximadamente 150 vezes com base na exposição sistêmica humana (AUC) em uma dose diária de 0,5 mg.
O fingolimode foi teratogênico no rato quando administrado a doses de 0,1 mg/kg ou superiores. As malformações fetais mais comuns incluíram tronco arterial persistente e defeito do septo ventricular. Um aumento na perda pós-implante foi observado em ratos a 1 mg/kg e doses superiores e uma redução nos fetos viáveis foi observada a 3 mg/kg. O fingolimode não foi teratogênico nos coelhos, nos quais uma mortalidade embriofetal elevada foi observada a doses de 1,5 mg/kg e superiores, e uma redução nos fetos viáveis, bem como retardamento no crescimento fetal foram observados a 5 mg/kg.
Em ratos, a sobrevida das crias de geração F1 foi reduzida no início do período pós-parto a doses que não causaram toxicidade materna. Entretanto, os pesos corporais, desenvolvimento, comportamento e fertilidade de F1 não foram afetados pelo tratamento com fingolimode. Em um estudo de toxicidade em ratos jovens, nenhum órgão-alvo adicional de toxicidade foi observado em comparação com os ratos adultos. Estímulos repetidos com hemocianina do molusco lapa californiana (KLH) demonstraram uma resposta moderadamente reduzida durante o período de tratamento, mas reações imunes totalmente em funcionamento no final de um período de recuperação de 8 semanas.
O fingolimode foi excretado no leite de animais tratados durante a lactação. O fingolimode e seus metabólitos cruzaram a barreira placentária em coelhas prenhas.
Contraindicações.
GilenyaTM é contraindicado em pacientes com:
- conhecida hipersensibilidade ao fingolimode ou a qualquer um dos excipientes.
- ocorrência recente (últimos 06 meses) de infarto do miocárdio, derrame, angina instável, ataque isquêmico transitório, insuficiência cardíaca descompensada necessitando hospitalização, insuficiência cardíaca classe III/IV.
-histórico ou presença de bloqueio atrioventricular de 2° ou 3° grau com Mobitz tipo II, doença do nó sinusal (exceto o paciente que faz uso de marca-passo), hipertensão arterial não controlada, apneia do sono grave não tratada.
-uso de drogas antiarrítmicas classe Ia ou classe III.
- intervalo de QT maior ou igual a 500 ms.
-insuficiência hepática grave (Child-Pugh classe C).
Advertências e precauções.
Infecções
Um efeito farmacodinâmico fundamental de GilenyaTM é a redução dose-dependente da contagem de linfócitos periféricos para 20 - 30% dos valores basais. Isso se deve ao sequestro reversível de linfócitos em tecidos linfoides. (vide item Características Farmacológicas).
Os efeitos de GilenyaTM no sistema imune (vide item Características Farmacológicas) podem aumentar o risco de infecções, incluindo infecções oportunistas (vide item Reações Adversas).
Antes de iniciar o tratamento com GilenyaTM , uma contagem recente de células brancas do sangue deve estar disponível (por exemplo, dentro de 6 meses ou após a descontinuação da terapia prévia).
O início do tratamento com GilenyaTM deve ser postergado em pacientes com infecção severa ativa até sua resolução. Estratégias diagnósticas e terapêuticas eficazes devem ser empregadas em pacientes com sintomas de infecção durante a terapia. Considerando que a eliminação de fingolimode após a descontinuação pode levar até dois meses, a vigilância quanto à infecção deve ser continuada ao longo desse período (vide subitem Interrompendo a terapia).
Terapias antineoplásicas, imunomoduladoras ou imunossupressoras (incluindo corticosteroides) devem ser administradas concomitantemente com cautela devido ao risco de efeitos adicionais no sistema imune. Decisões específicas quanto à dosagem e duração do tratamento com corticosteroides devem ser baseadas na avaliação clínica. A coadministração de um tratamento de curta duração com corticosteroides (até 5 dias, conforme protocolos de estudo) não aumentou a taxa global de infecção em pacientes tratados com fingolimode em estudos clínicos de fase III, em comparação com placebo. Com base nestes dados, os tratamentos curtos de corticosteroides (até 5 dias) podem ser utilizados em combinação com GilenyaTM (vide itens Reações Adversas e Interações Medicamentosas).
Pacientes recebendo GilenyaTM devem ser instruídos a relatar sintomas de infecções aos seus médicos. A suspensão do tratamento com GilenyaTM deve ser considerada caso um paciente desenvolva uma infecção séria e o risco-benefício deve ser levado em consideração antes de reiniciar a terapia.
Casos de leucoencefalopatia multifocal progressiva (LMP) foram relatados na experiência pós-comercialização (vide item Reações Adversas). A LMP é uma infecção oportunista causada pelo vírus JC, que pode ser fatal ou resultar em incapacidade grave. Os médicos devem estar atentos aos sintomas clínicos ou resultados de imagem de ressonância magnética que podem ser sugestivos de LMP. Se houver suspeita de LMP, o tratamento GilenyaTM deve ser suspenso até que LMP tenha sido excluída.
Foram reportados na experiência pós-comercialização casos isolados de meningite criptococócica. Os pacientes com sinais e sintomas evidentes de meningite criptococócica devem submeter-se a uma rápida avaliação do diagnóstico. Se diagnosticado, o tratamento apropriado deve ser iniciado. Pacientes recebendo GilenyaTM devem ser instruídos a relatar sintomas de infecções aos seus médicos. A suspensão do tratamento com GilenyaTM deve ser considerada caso um paciente desenvolva uma infecção séria e o risco-benefício deve ser levado em consideração antes de reiniciar a terapia. Os pacientes devem ser avaliados quanto a sua imunidade à varicela (catapora) antes do tratamento com GilenyaTM . Recomenda-se que pacientes sem a confirmação por profissional de saúde de histórico de catapora ou comprovação de um curso completo de vacinação com a vacina contra varicela realizem testes de anticorpos para o vírus varicela zoster (VVZ), antes de iniciar o tratamento com GilenyaTM . Um curso completo de vacinação para pacientes anticorpos negativos com a vacina contra varicela é recomendado antes de iniciar o tratamento com GilenyaTM (vide Reações Adversas). O início do tratamento com GilenyaTM deverá ser adiado por um mês após a vacinação, para permitir que a plena eficácia da mesma possa ocorrer.
Vacinação
A vacinação pode ser menos eficaz durante e até dois meses após interromper o tratamento com GilenyaTM (vide Interrompendo a terapia). O uso de vacinas com vírus vivos atenuados deve ser evitado.
Edema Macular
O edema macular (vide item Reações Adversas) com ou sem sintomas visuais foi relatado em 0,5% dos pacientes tratados com GilenyaTM 0,5 mg, ocorrendo predominantemente nos primeiros 3-4 meses de terapia. Uma avaliação oftálmica é, portanto, recomendada 3-4 meses após o início do tratamento. Caso pacientes relatem distúrbios visuais a qualquer momento durante a terapia com GilenyaTM, uma avaliação de fundo do olho, incluindo a mácula, deve ser realizada.
Pacientes com histórico de uveíte e pacientes com diabetes mellitus estão em risco elevado de edema macular (vide item Reações Adversas). GilenyaTM não foi estudado em pacientes com esclerose múltipla concomitante com diabetes mellitus. Recomenda-se que pacientes com esclerose múltipla e diabetes mellitus ou com histórico de uveíte sejam submetidos a uma avaliação oftálmica antes do início da terapia com GilenyaTM e tenham avaliações de acompanhamento enquanto recebem a terapia com GilenyaTM .
A continuação de GilenyaTM em pacientes com edema macular não foi avaliada. Na decisão sobre se a terapia com GilenyaTM deve ou não ser descontinuada, se faz necessário levar em consideração os possíveis riscos e benefícios para cada paciente.
Bradiarritmia
O início do tratamento com GilenyaTM resulta em uma redução temporária na frequência cardíaca. Após a primeira dose, a redução da frequência cardíaca começa dentro de uma hora e a redução máxima é atingida em até 6 horas.
Com a continuação da dosagem, a frequência cardíaca retorna ao valor basal dentro de um mês de tratamento crônico (vide item Características Farmacológicas, Frequência e ritmo cardíaco). Em pacientes recebendo GilenyaTM 0,5 mg, a redução na frequência cardíaca medida pelo pulso, é em média 8 batimentos por minuto (bpm) aproximadamente. Frequências cardíacas abaixo de 40 bpm foram raramente observadas (vide item Reações Adversas). Pacientes que apresentaram bradicardia mostraram-se geralmente assintomáticos, mas alguns pacientes apresentaram sintomas leves a moderados, incluindo hipotensão, tontura, fadiga e/ou palpitações, que se resolveram dentro das primeiras 24 horas de tratamento.
O início do tratamento com GilenyaTM foi associado com atrasos na condução atrioventricular, geralmente bloqueios atrioventriculares de primeiro grau (intervalo PR prolongado no eletrocardiograma). Bloqueios atrioventriculares de segundo grau, geralmente Mobitz tipo I (Wenckebach), foram observados em menos de 0,2% de paci