FUZEON®
ROCHE
enfuvirtida
Agente anti-retroviral.
USO ADULTO E PEDIÁTRICO.
Apresentações.
Caixa com: 60 frascos-ampola contendo pó branco a quase branco, estéril, liofilizado. 60 frascos-ampola contendo 2,0 mL de água estéril para injeção. 60 seringas de 3 mL. 60 seringas de 1 mL. 180 saches de algodão. Após reconstituição aplicar injeção subcutânea.
Composição.
Princípio ativo: frasco-ampola contendo 108 mg de enfuvirtida que após reconstituição com 1,1 mL de água estéril para injeção, forma solução contendo 90 mg/mL de enfuvirtida. Excipientes: carbonato de sódio, manitol, hidróxido de sódio e ácido clorídrico.
Informações ao paciente.
Solicitamos a gentileza de ler cuidadosamente as informações a seguir. Caso não esteja seguro a respeito de determinado item, favor informar ao seu médico. 1. AÇÃO DO MEDICAMENTO: Enfuvirtida é o primeiro membro de classe terapêutica chamada inibidor de fusão. É um inibidor do rearranjo estrutural da gp41 do HIV que se liga especificamente à proteína gp 41 do vírus HIV bloqueando a entrada do vírus na célula. Enfuvirtida não requer ativação intracelular. A atividade antiviral da enfuvirtida resulta de sua associação com a região HR1 da gp 41 do HIV na superfície viral. 2. INDICAÇÕES DO MEDICAMENTO: FUZEON® é indicado para o tratamento da infecção por HIV-1 em combinação com outros agentes anti-retrovirais em pacientes com tratamento prévio e com evidência de replicação do HIV-1 a despeito da terapia anti-retroviral. Não há estudos sobre o uso de FUZEON® em pacientes virgens de tratamento anti-retroviral. 3. RISCOS DO MEDICAMENTO: Contra-indicações: FUZEON® está contra-indicado em pacientes com hipersensibilidade conhecida à enfuvirtida ou a qualquer um de seus componentes. Os pacientes que desenvolvam sinais ou sintomas sugestivos de reação de hipersensibilidade devem descontinuar o tratamento com FUZEON® e procurar avaliação médica imediatamente. Cada frasco e cada seringa devem ser usados uma única vez, sendo que as porções não utilizadas devem ser eliminadas. Não é permitido misturar qualquer outra substância à solução com FUZEON® para ser injetado conjuntamente. Informe seu médico sobre qualquer medicamento que esteja usando antes do início ou durante o tratamento. Foram relacionados com um risco maior de pneumonia bacteriana os usuários de drogas intravenosas, os fumantes, os pacientes com história de doença pulmonar prévia e aqueles com um baixo número inicial de linfócitos CD4. Estes grupos de pacientes devem ser acompanhados de perto pelo médico, caso apresentem sinais e sintomas sugestivos de pneumonia. NÃO TOME REMÉDIO SEM O CONHECIMENTO DO SEU MÉDICO. PODE SER PERIGOSO PARA A SAÚDE. Advertências: Um pequeno número de casos de reações de hipersensibilidade sistêmica foi associado à terapia com FUZEON®: rubor, febre, náuseas, vômitos, calafrios, tremores, hipotensão, elevação de enzimas hepáticas, reação primária de imunocomplexos, distúrbio respiratório e glomerulonefrite. Houve o relato de um caso de Síndrome de Guillain-Barré observado nos estudos clínicos. Os pacientes devem ser orientados a descontinuar o tratamento com FUZEON® e procurar avaliação médica imediatamente, caso desenvolvam sinais ou sintomas sugestivos de reação de hipersensibilidade. A terapia com FUZEON® não deve ser reiniciada após sinais e sintomas consistentes com uma reação de hipersensibilidade relacionada ao uso de FUZEON®. Observou-se uma incidência aumentada de pneumonia bacteriana, algumas vezes fatal, nos estudos clínicos em pacientes tratados com FUZEON®. Os pacientes devem ser monitorados quanto a sinais e sintomas de infecção, especialmente se apresentarem os seguintes fatores de risco: baixa contagem inicial de linfócitos CD4, elevada carga viral inicial, uso de drogas intravenosas, tabagismo e história pulmonar prévia. A administração de FUZEON® a indivíduos não infectados (em profilaxia pós-exposição, por exemplo) pode induzir à formação de anticorpos antienfuvirtida que reagem de forma cruzada com a gp41 do HIV. Isto pode resultar em falso positivo em teste anti-HIV ELISA. Síndrome da reconstituição imune (também conhecida como Síndrome da reativação imune, Doença da restauração imune ou Síndrome da inflamação reconstituição imune): Síndrome da reconstituição imune tem sido relatada em pacientes tratados com terapia anti-retroviral combinada, incluindo FUZEON®. Durante a fase inicial do tratamento anti-retroviral em combinação, pacientes com sistema imunológico responsivo podem desenvolver uma resposta inflamatória a infecções oportunistas residuais ou indolentes (tais como infecção por Mycobaterium avium, citomegalovirus, pneumonia por Pneumocystis jirovecii [PCP], tuberculose ou outras) e podem necessitar de avaliação e tratamento imediatos. O paciente deve ser sempre bem orientado quanto aos: cuidados e manuseio correto de seringas e agulhas; sobre a importância do descarte destes materiais no recipiente adequado e sobre o local de retorno deste recipiente para que o mesmo seja submetido à destruição adequada. Os materiais do kit e o recipiente de descarte sempre devem ser mantidos fora do alcance das crianças. Caso alguma outra pessoa auxilie o paciente a aplicar a medicação, esta deve ser orientada a utilizar luvas e a procurar serviço médico imediatamente caso sofra algum acidente com material pérfuro-cortante. Principais interações medicamentosas: Com base nos resultados de um estudo microssomal humano in vitro, FUZEON® não é um inibidor das enzimas CYP450 e, portanto, não alterará o metabolismo das drogas metabolizadas pelas enzimas CYP450. Em um estudo do metabolismo humano in vivo, FUZEON®, na dose recomendada de 90 mg duas vezes ao dia, não inibe o metabolismo de substratos pelo CYP3A4 (dapsona), CYP2D6 (debrisoquina), CYP1A2 (cafeína), CYP2C19 (mefentoína) e CYP2E1 (clorzoxazona). Em estudos isolados de interação farmacocinética, as co-administrações de ritonavir, saquinavir e rifampicina, não resultaram em interações farmacocinéticas clinicamente significantes com FUZEON®. Gravidez e amamentação: Informe ao seu médico a ocorrência de gravidez na vigência do tratamento ou após seu término. Informe ao médico se está amamentando. Você não deverá amamentar durante o tratamento com FUZEON®. Não há, até o momento, estudos adequados e controlados em mulheres grávidas, portanto FUZEON® só deverá ser usado durante a gravidez se o benefício potencial justificar o risco ao feto. Este medicamento não deve ser utilizado por mulheres grávidas ou que estejam amamentando sem orientação médica ou do cirurgião-dentista. Efeitos sobre a capacidade de dirigir e operar máquinas, quando for o caso. Não foram realizados estudos sobre a capacidade de dirigir veículos ou de operar máquinas durante a administração de FUZEON®. Não há evidências de que FUZEON® possa alterar a capacidade dos pacientes para dirigir veículos e de usar máquinas, porém, o perfil de eventos adversos de FUZEON® deve ser levado em conta (Tabela 6). Não existem dados que estabeleçam dose de FUZEON® em crianças menores de 6 anos de idade. Informe ao médico ou cirurgião-dentista o aparecimento de reações indesejáveis. Informe ao seu médico ou cirurgião-dentista se você está fazendo uso de algum outro medicamento. Não use medicamento sem o conhecimento do seu médico. Pode ser perigoso para a sua saúde. 4. MODO DE USO. Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Recomenda-se que a primeira injeção subcutânea ocorra sob a supervisão de um profissional de saúde devidamente qualificado. Um profissional de saúde deverá reavaliar periodicamente se os procedimentos estão sendo feitos corretamente. FUZEON® é fornecido em pó liofilizado, que precisa ser reconstituído em solução com água estéril para injeção. Todos os componentes necessários à injeção de FUZEON® são fornecidos em um kit: 60 frascos de FUZEON® (frasco com o pó branco); 60 frascos de água estéril para injeção (frasco com líquido transparente); Chumaços embebidos em álcool; 60 seringas de 3 mL (para preparar a solução); 60 seringas de 1 mL (para a injeção subcutânea); Recipiente para descarte de seringas. Preparação: Aspiração da água para injeção: As mãos devem ser lavadas com água e sabão antes que os frascos sejam preparados. Devem ser usadas luvas descartáveis se outra pessoa estiver aplicando a droga ao paciente. Se FUZEON® estiver sendo armazenado em geladeira, retire-o junto com a ampola de água para injeção e aguarde até que atinjam a temperatura ambiente. Após retirar as tampas plásticas dos frascos de FUZEON® e de água, faça antissepsia nas tampas de borracha de cada frasco com o chumaço embebido em álcool fornecido e deixe secar naturalmente.
Retire a água estéril com a seringa de 3 mL: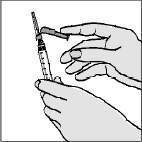
Pegue a seringa maior de 3 mL. Usando o dedo indicador, empurre para trás o protetor colorido da agulha em direção à seringa (figura 1).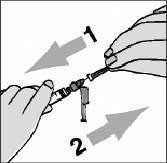
Para remover a capa plástica incolor da agulha, empurre-a e puxe-a sem fazer muita força (figura 2)
É preciso tomar cuidado durante os passos seguintes para garantir que não se formem bolhas na solução e que não fique ar retido na seringa antes da injeção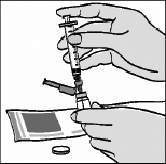
Inicialmente aspire 1,1 mL de ar com a seringa de 3 mL. Introduza a agulha da seringa na tampa de borracha do frasco de água para injeção e aperte o êmbolo para injetar o ar da seringa (figura 3)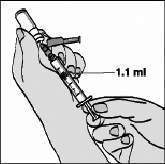
Vire o frasco de água para baixo, mantendo a ponta da agulha abaixo do nível da água. Puxe o êmbolo de volta lentamente até que a água chegue um pouco além da marca de 1,1 mL. ATENÇÃO: O frasco contém 2 mL, ou seja, mais água do que a quantidade necessária para a diluição. Você só deve aspirar 1,1 mL do frasco, para que a medicação seja preparada adequadamente. Bata levemente na seringa para fazer com que bolhas de ar, eventualmente presentes, subam para a superfície. Aperte suavemente o êmbolo para expelir o ar e certifique-se de que exatamente 1,1 mL de água estéril fique na seringa. Retire a agulha do frasco, tomando cuidado para não encostar a agulha nos dedos ou em qualquer outro objeto. Descarte o frasco de água com o restante que não foi aspirado. A ÁGUA QUE SOBROU NO FRASCO DILUENTE NÃO DEVE SER REUTILIZADA. Injetando água no frasco de FUZEON® em pó: A água deve ser acrescentada ao frasco de FUZEON®. Antes de injetar a água, bata levemente no frasco de FUZEON® para que o pó se desprenda das paredes do frasco.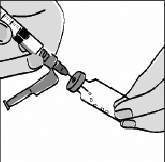
Introduza a agulha da seringa com uma leve inclinação através da tampa de borracha do frasco de FUZEON®. Aperte lentamente o êmbolo fazendo com que a água escorra lentamente pela lateral interna do frasco, para evitar a formação de espuma. Depois de acrescentar toda a água, retire a seringa do frasco (figura 5).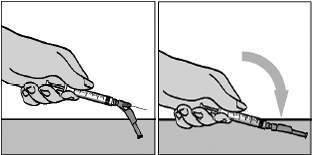
Para proteger a agulha, segure o corpo da seringa e pressione o protetor colorido contra a mesa, conforme figura 6. Você ouvirá um clique. Não use a outra mão para auxiliá-lo, tão pouco cubra a agulha com a capa incolor, pois você pode se machucar durante essa tentativa. Despreze a seringa usada no recipiente fornecido para o descarte das seringas. Misturando a água com o pó de FUZEON®: Para misturar, bata suavemente no frasco de FUZEON® com um dedo para ajudar a dissolver o pó. A solução não deve ser agitada nem virada para misturar, porque isto provoca formação de bolhas. Esfregar o frasco suavemente pode ajudar a misturar. Aguarde até que o pó se dissolva completamente e que eventuais bolhas restantes desapareçam. Isto habitualmente demora 10 a 20 minutos, mas pode demorar até 45 minutos. É muito importante que todo o pó da droga se dissolva para que seja aplicada a dose. Avalie visualmente a solução e devolva o produto ao profissional de saúde antes de usar, caso a solução tenha material particulado. Não deverá haver aderências sólidas nas paredes ou na base do frasco. Caso persistam bolhas na solução, bata ligeiramente no frasco. Depois de reconstituído, FUZEON® deve ser aplicado imediatamente. Você pode preparar um frasco com antecedência ou preparar dois frascos ao mesmo tempo. Neste caso, não se esqueça de utilizar um Kit diferente para cada frasco (água, seringas, agulhas e algodão). O frasco que será usado mais tarde deve ser colocado sob refrigeração de 2° a 8°C (nunca guarde FUZEON® na seringa) logo após a reconstituição, podendo ser usada em até 24 horas. Alguns minutos antes da aplicação, retire o frasco da geladeira já que é necessário que ele fique em temperatura ambiente antes da injeção. Uso de FUZEON® diluído: Use a seringa de segurança de 1 mL. Proceda com a seringa de 1 mL da mesma forma que o orientado nas figuras 1 e 2. Aspire 1 mL de ar. Cuidado para não ultrapassar a marca limite de 1 mL da seringa. Introduza a agulha na tampa de borracha do frasco de FUZEON®, injete o ar da seringa e vire suavemente o frasco. Mantenha a ponta da agulha abaixo da superfície da solução para evitar bolhas de ar. Puxe o êmbolo lentamente até retirar o maior volume possível da solução de FUZEON®. Cuidado para não ultrapassar a marca limite de 1 mL da seringa (figura 7).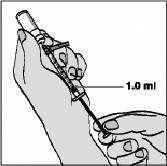
Bata suavemente na seringa para fazer com que as bolhas de ar subam para a superfície. Aperte o êmbolo para expelir o ar de volta para o frasco, certificando-se de deixar 1 mL de FUZEON® no corpo da seringa. Aplicação: O uso de FUZEON® é por injeção subcutânea (logo abaixo da pele). O local de injeção deve ser rodiziado para evitar injeções repetidas na mesma área. Os locais de injeção mais comuns são abdômen, parte anterior da coxa e lateral-externa do braço (conforme figura 8). Essas áreas tendem a apresentar mais gordura no subcutâneo e por isso reduzem o risco de injetar inadvertidamente em local profundo demais (no músculo em vez de sob a pele), o que pode provocar reações mais severas. Evite a área do abdômen em torno do umbigo e áreas que já apresentem reações. Não é recomendável que FUZEON® seja injetado na face (figura 8).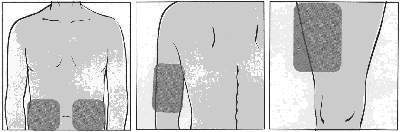
As aplicações da manhã e da noite não devem ser nunca na mesma área. Limpe a área de injeção com o chumaço embebido em álcool e pegue uma prega de pele.
Introduza a agulha na pele fazendo um ângulo de 45° com a seringa.
Ao introduzir metade da agulha na pele, solte a prega da pele e passe a segurar o corpo da seringa para evitar que se desloque. Pressione lentamente o êmbolo para baixo para injetar FUZEON® (figura 9).
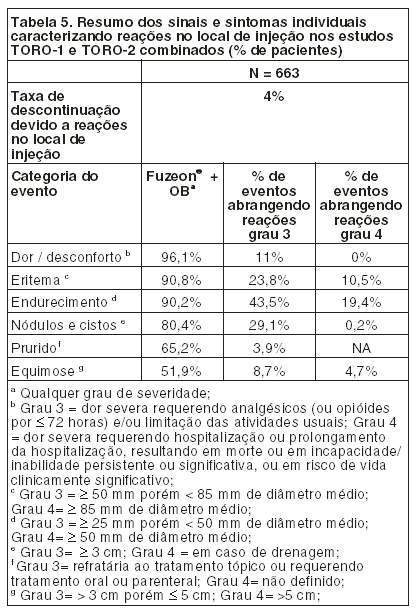
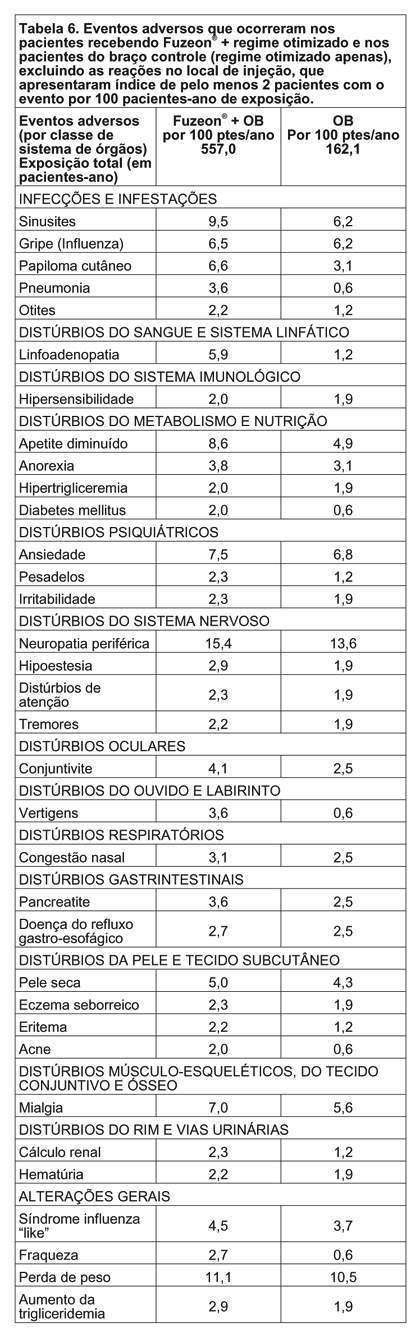
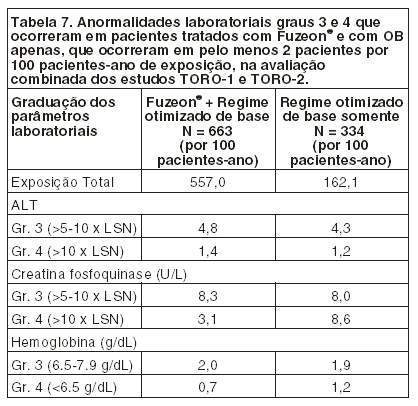
Após o término da injeção de FUZEON®, para proteger a agulha, segure o corpo da seringa e pressione o protetor colorido contra a mesa, conforme figura 6. Você ouvirá um clique. Não use a outra mão para auxiliá-lo, tão pouco cubra a agulha com a capa incolor, pois você pode se machucar durante essa tentativa. Despreze a seringa usada no recipiente fornecido para o descarte das seringas. ATENÇÃO: CUIDADO<33> DESCARTE AS SERINGAS NO RECIPIENTE FORNECIDO. É expressamente proibido o descarte das seringas utilizadas e materiais sujos de sangue fora dos recipientes plásticos fornecidos em função dos riscos de transmissão do HIV e de outras doenças transmissíveis a outras pessoas. O descarte das seringas deve ser feito no recipiente fornecido e, quando atingida a sua capacidade volumétrica, o recipiente deve ser recolhido à unidade de saúde mais próxima para a sua completa destruição, conforme orientação de seu médico. Mantenha o recipiente de descarte e o material do kit fora do alcance das crianças. Caso alguém que o esteja auxiliando a aplicar a medicação, se machuque com a agulha da seringa de aplicação (1 mL), ele deve contatar um profissional de saúde imediatamente. Os frascos, tanto de FUZEON® quanto de água, devem ser utilizados uma única vez. O que sobrar em qualquer um dos frascos deve ser descartado. Interrupção do tratamento: Não interrompa o tratamento sem o conhecimento de seu médico. FUZEON® deve ser administrado em conjunto com outros anti-retrovirais para obter melhores resultados. ADVERTÊNCIA IMPORTANTE: Uma elevada incidência de pneumonia bacteriana, algumas vezes fatal, ocorreu em pacientes tratados com FUZEON® em estudos clínicos. Foram relacionados com um risco maior de pneumonia bacteriana os usuários de drogas intravenosas, os fumantes, os pacientes com história de doença pulmonar prévia e aqueles com um baixo número inicial de linfócitos CD4. Estes grupos de pacientes devem ser acompanhados de perto pelo médico, caso apresentem sinais e sintomas sugestivos de pneumonia. Informe imediatamente ao seu médico caso surjam sinais ou sintomas sugestivos de pneumonia (tosse, dor no peito, febre ou falta de ar) mesmo que você não se identifique com este grupo de pacientes. Siga a orientação de seu médico, respeitando sempre os horários, as doses e a duração do tratamento. Não interrompa o tratamento sem o conhecimento do seu médico. Não use o medicamento com o prazo de validade vencido. Antes de usar observe o aspecto do medicamento. 5. REAÇÕES ADVERSAS. Informe o seu médico sobre o aparecimento de reações desagradáveis. Pacientes adultos: As reações adversas mais comuns relacionadas a FUZEON® foram as reações no local de injeção que, em sua maioria, ocorreram na primeira semana de tratamento e foram associadas a dor leve a moderada ou desconforto no local da injeção, vermelhidão, endurecimento, surgimento de nódulos e cistos, prurido e equimose. Um pequeno número de reações de hipersensibilidade sistêmica foi associado à terapia com FUZEON®: rubor, febre, náuseas, vômitos, calafrios, tremores, hipotensão, elevação de enzimas hepáticas, reação primária de imunocomplexos, distúrbio respiratório e glomerulonefrite. Foi relatado um caso de Síndrome de Guillain-Barré. Observou-se uma incidência aumentada de pneumonia bacteriana nos estudos clínicos em pacientes tratados com FUZEON®. Superdosagem: A maior dose administrada para 12 pacientes em estudos clínicos (estudo T20-501) foi de 180 mg em uma única dose por via subcutânea. Estes pacientes não apresentaram nenhum evento adverso diferente daqueles encontrados com a dose recomendada. Em estudos de acesso expandido um paciente fez uso de uma dose de 180 mg. Ele também não apresentou nenhum evento adverso novo. Pacientes pediátricos: FUZEON® foi estudado em 69 pacientes pediátricos entre 4 e 16 anos de idade com duração de exposição ao FUZEON® variando de 1 dose para > 48 semanas. As reações adversas observadas durante os estudos clínicos foram similares às observadas em adultos sujeitos ao tratamento. TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS. Atenção: este é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis para comercialização, efeitos indesejáveis e não conhecidos podem ocorrer. Neste caso, informe ao seu médico. 6. CONDUTA EM CASO DE SUPERDOSE: Não há antídoto específico para superdosagem com FUZEON® e o tratamento deve consistir de medidas gerais de suporte. Não há relatos de superdosagem com FUZEON® em seres humanos. A maior dose administrada foi de 180 mg em um estudo clínico para 12 pacientes como dose única subcutânea. Estes pacientes não apresentaram evento adverso que não fosse observado com a dose recomendada. Em estudos de acesso expandido um paciente fez uso de uma dose de 180 mg. Ele também não apresentou nenhum evento adverso novo. 7. CUIDADOS DE CONSERVAÇÃO. Os frascos não abertos de FUZEON® podem ser armazenados em temperatura ambiente controlada de 15° a 30°C. Se não for possível garantir o controle da temperatura, recomenda-se armazenar sob refrigeração de 2° a 8°C. Caso FUZEON® não seja injetado imediatamente após ser dissolvido, o mesmo poderá ser usado até, no máximo, 24 horas se mantido sob refrigeração de 2° a 8°C. Prazo de validade: Este medicamento possui prazo de validade a partir da data de fabricação (vide embalagem externa do produto). Não tome o medicamento após a data de validade indicada na embalagem, pode ser prejudicial à saúde. Os frascos, tanto de FUZEON® quanto de água, devem ser utilizados uma única vez. O que sobrar em qualquer um dos frascos deve ser descartado. Todo medicamento deve ser mantido fora do alcance das crianças. Este medicamento, depois de reconstituído, somente poderá ser utilizado em até 24 horas.
Informações técnicas.
1. CARACTERÍSTICAS FARMACOLÓGICAS. FUZEON® é o primeiro membro da classe terapêutica denominada inibidores da fusão. Trata-se de um peptídeo de 36 aminoácidos que se liga, fora da célula, especificamente à cadeia de repetição heptavalente (HR1) da glicoproteína gp41 do HIV, inibindo o seu rearranjo estrutural e, desta forma, bloqueando a entrada do vírus na célula. FUZEON® não requer ativação intracelular. Cada frasco contém um pó branco a quase branco, estéril, liofilizado, contendo 108 mg de enfuvirtida, 2,87 mg de carbonato de sódio como agente amortecedor, 27,05 mg de manitol como excipiente a granel, hidróxido de sódio como agente alcalinizante, e ácido clorídrico como agente acidificante. Atividade antiviral in vitro: A atividade antiviral in vitro de FUZEON® foi demonstrada em infecção aguda de linhagens celulares T linfoblastóides, monócitos, macrófagos e células mononucleares sanguíneas periféricas primárias (PBMC) através de isolados HIV-1 laboratoriais e clínicos. FUZEON® demonstrou atividade seletiva anti-HIV-1 contra ambos os isolados virais, o prototípico e o primário. A susceptibilidade dos isolados do vírus 130 PBMC à enfuvirtida no baseline de pacientes tratados com FUZEON® foi determinada em um ensaio cMAGI (estudos clínicos fase II). FUZEON® apresentou uma média geométrica da EC50 de 0,016 mcg/mL (DP = 0,057) e faixa de < 0,001 a 0,480 mcg/mL contra estes isolados do vírus. FUZEON® também inibiu a fusão célula-célula mediada pelo envelope HIV-1. Os estudos de combinação de FUZEON® com membros representativos das diversas classes anti-retrovirais (inibidores de transcriptase reversa nucleosídeos, inibidores de transcriptase reversa não nucleosídeos e inibidores da protease) incluindo zidovudina, lamivudina, nelfinavir, indinavir e efavirenz, apresentaram efeitos sinérgicos aditivos e ausência de antagonismo. A relação entre a susceptibilidade in vitro do HIV-1 para FUZEON® e a inibição da replicação do HIV-1 em humanos não foram estabelecidas. Devido aos diferentes alvos enzimáticos e conforme implícito pela atividade de FUZEON® contra as cepas HIV que apresentam resistência a outras classes de agentes anti-retrovirais, os isolados HIV resistentes à enfuvirtida permanecerão sensíveis aos inibidores de transcriptase reversa (nucleosídeos e não nucleosídeos) e aos inibidores da protease. Resistência in vitro: Os isolados HIV-1 com susceptibilidade reduzida a FUZEON® foram selecionados in vitro. Estes isolados virais apresentavam substituições nos aminoácidos 36-38 do ectodomínio da gp41. Estas substituições foram correlacionadas com níveis variáveis de susceptibilidade reduzida a FUZEON® em HIV mutantes. Resistência in vivo: O aparecimento de resistência a FUZEON® é influenciado pela eficácia da totalidade do regime de tratamento. As substituições nos aminoácidos 36-45 da gp41 foram observadas em vírus de pacientes recebendo FUZEON® em estudos clínicos Fase II e Fase III. Em ordem decrescente, as substituições observadas encontravam-se nas posições 38, 43, 36, 40, 42 e 45. Não foi estabelecida a relação destas substituições com a eficácia in vivo. Geralmente, as substituições em aminoácidos 36-45 da gp41, durante o tratamento, implicaram em susceptibilidade fenotípica in vitro diminuída a FUZEON® em isolados de vírus do paciente. Resistência cruzada: FUZEON® é igualmente ativo in vitro contra os isolados laboratoriais e clínicos do tipo selvagem, e contra aqueles com resistência genotípica a 0, 1, 2 ou 3 classes de anti-retrovirais (inibidores de transcriptase reversa nucleosídeos, inibidores de transcriptase reversa não nucleosídeos e inibidores da protease). Estes isolados tiveram a resistência genotípica identificada especificamente para: zidovudina, lamivudina, estavudina, didanosina, zalcitabina, abacavir, nevirapina, delavirdina, efavirenz, indinavir, saquinavir, nelfinavir, ritonavir e amprenavir. Todos estes foram sensíveis a FUZEON®. Não é esperado, entretanto, que mutações que induzam resistência a FUZEON® apresentem resistência cruzada a outras classes de anti-retrovirais. Farmacocinética: As propriedades farmacocinéticas da enfuvirtida foram avaliadas em adultos e em pacientes pediátricos infectados por HIV-1. Após uma dose subcutânea única de 90 mg de FUZEON® no abdômen de 12 pacientes infectados por HIV-1, a mediana da Cmáx foi de 4,59 mcg/mL ± 1,5 mcg/mL, a AUC (área sob a curva) foi de 55,8 mcg/mL ± 12,1 mcg.h/mL e a biodisponibilidade absoluta foi de 84,3% ± 15,5%, usando a dose intravenosa de 90 mg como referência. A absorção subcutânea de FUZEON® é proporcional à dose administrada, na faixa de dosagem de 45 a 180 mg. A absorção subcutânea com a dose de 90 mg é comparável quando injetada no abdômen, coxas ou braço. As concentrações plasmáticas medianas em estado de equilíbrio dinâmico (steady state) de enfuvirtida 90 mg encontram-se na Figura 10.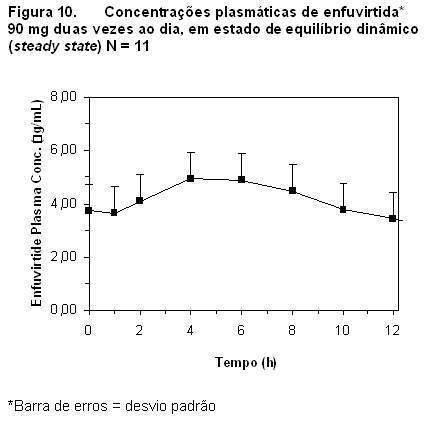
Em quatro diferentes estudos (N = 9 a 12), as concentrações plasmáticas medianas de vale, em estado de equilíbrio dinâmico (steady state), variaram de 2,6 a 3,4 mcg/mL. Distribuição: A mediana do volume de distribuição em estado de equilíbrio dinâmico (steady-state), após a administração de uma dose de 90 mg de FUZEON® (N = 12) foi de 5,5 ± 1,1 litro. FUZEON® está ligado em 92% às proteínas em plasma infectado por HIV, para uma faixa de concentração de 2 a 10 mcg/mL. Sua ligação efetua-se predominantemente à albumina e, em menor extensão, à glicoproteína a-1 ácida. FUZEON® não foi deslocado de seus sítios de ligação pelo saquinavir, nelfinavir, lopinavir, efavirenz, nevirapina, amprenavir, itraconazol, midazolam ou varfarina. FUZEON® não deslocou a varfarina, midazolam, amprenavir ou efavirenz de seus sítios de ligação. Os níveis de enfuvirtida no fluído cerebroespinal, medidos em um pequeno número de pacientes infectados por HIV, mostraram-se insignificantes. A molécula pode ser grande demais para atravessar a barreira sanguínea cerebral. Metabolismo: Por tratar-se de um peptídeo, espera-se que FUZEON® seja submetido a catabolismo de seus aminoácidos constituintes, com a subsequente reciclagem dos aminoácidos em seu conjunto corpóreo. Estudos microssomais humanos realizados in vitro indicam que FUZEON® não é um inibidor das enzimas CYP450. Observou-se em estudos microssomais e em hepatócitos humanos realizados in vitro, que a hidrólise do grupo amido do aminoácido C-terminal fenilalanina, resultou em um metabólito desamidado e a formação deste metabólito não é NADPH-dependente. Este metabólito (M3) é detectado no plasma humano após a administração de FUZEON®, com uma AUC variando de 2,4 a 15% da AUC do FUZEON®. Eliminação: Os estudos de balanço de massa para determinar a(s) via(s) de eliminação de enfuvirtida não foram realizados em humanos. Porém, estudos realizados em roedores usando 3H-enfuvirtida indicaram a recuperação incompleta na excreção da radioatividade administrada 7 dias após a dose. Houve retenção da radioatividade nos músculos esqueléticos. Após uma dose subcutânea de 90 mg (n = 12) a mediana da meia-vida de eliminação de FUZEON® foi de 3,8 ± 0,6 horas e a mediana do clearance de 1,7 ± 0,4 L/h. Farmacocinética em situações clínicas especiais: Insuficiência hepática: A farmacocinética de FUZEON® não foi estudada em pacientes com insuficiência hepática. Insuficiência renal: Análises dos dados de concentração plasmática dos pacientes dos estudos clínicos indicaram que o clearance de FUZEON® não é afetado de forma clinicalmente relevante em pacientes com clearance de creatinina superior a 35 mL/min. Os resultados de um estudo de insuficiência renal indicaram que o clearance da enfuvirtida foi reduzido em aproximadamente 38% em pacientes com insuficiência renal grave e em aproximadamente 14 - 28% em pacientes com doença renal em estágio final mantidos sob diálise, comparados a pacientes com função renal normal. Os resultados estavam dentro da faixa observada em pacientes com função renal normal em estudos pilotos. A hemodiálise não alterou significantemente o clearance da enfuvirtida. Portanto, não são requeridas ajustes de doses para pacientes com insuficiência renal. Sexo e peso: As análises dos dados de concentração plasmática de pacientes dos estudos clínicos indicaram que o clearance de enfuvirtida é 20% mais lento nas mulheres do que nos homens. O aumento de peso, independentemente do sexo, está relacionado com maior clearance de enfuvirtida (20% maior em um paciente com peso corpóreo de 100 Kg e 20% mais lento em um paciente com peso corpóreo de 40 Kg, em relação a um paciente de referência pesando 70 Kg). Porém, estas mudanças não são clinicamente significativas e não requerem ajuste de dose. Raça: As análises dos dados de concentração plasmática de pacientes dos estudos clínicos indicaram que o clearance de FUZEON® não foi diferente em negros comparado com caucasianos. Outros estudos de farmacocinética sugerem que não há diferença entre asiáticos e caucasianos, após o ajuste da exposição de acordo com o peso corpóreo. Idosos: Estudos clínicos de FUZEON® não incluíram um número suficiente de pacientes com 65 anos de idade ou mais para determinar se eles respondem de maneira diferente dos pacientes mais jovens. Pacientes pediátricos: A farmacocinética da enfuvirtida foi estudada em 32 pacientes com idade de 3 à 16 anos com doses variando de 0,5 à 2,5 mg/Kg. A dose de 2 mg/Kg duas vezes ao dia (no máximo 90 mg duas vezes ao dia) levaram a concentrações plasmáticas de enfuvirtida similar ao obtido com pacientes adultos recebendo dosagem de 90 mg duas vezes ao dia. Em 25 pacientes variando em idade de 5 a 16 anos e recebendo a dose de 2 mg/Kg duas vezes ao dia, a média no nível da AUC foi 54,3 + 23,5 mg.h/mL, Cmáx foi 6,14 + 2,48 mg/mL e Cbasal foi 2,93 + 1,55 mg/mL. Segurança pré-clínica: Carcinogênese: Não foram realizados estudos de longo prazo de carcinogenicidade em animais. Mutagênese: FUZEON® não foi mutagênico nem clastogênico em uma série de ensaios in vivo e in vitro, incluindo o ensaio de mutação reversa bacteriana de Ames, um ensaio de mutação genética sobre células de mamífero em células do ovário do Hamster Chinês AS52 ou em um ensaio de micronúcleos murinos in vivo. Fertilidade prejudicada: FUZEON® não causou efeitos adversos sobre a fertilidade de ratos machos e fêmeas, com doses de enfuvirtida de 0,7; 2,5; e 8,3 vezes a dose diária máxima recomendada para humanos adultos, com base em mg/Kg, administrada através de injeção subcutânea. Reações no local de injeção: Em macacos cynomolgus tratados com doses de enfuvirtida de 5 e 10 mg/Kg duas vezes ao dia durante nove meses, foram observadas as mudanças no local de injeção que incluíram inchaço, edema e formação de abscesso, aparecendo geralmente no mês 5. Por ocasião da necropsia, foram observados com destaque a descoloração, espessamento e cistos nos locais de injeção de FUZEON®. Observados ao microscópio, estes achados estão correlacionados com hemorragia subcutânea, edema, infiltrado inflamatório e se encontravam presentes com incidência e severidade significativamente maiores nos locais de injeção de FUZEON®. O infiltrado inflamatório misto foi predominantemente linfocítico e formado frequentemente por folículos linfóides. Foi observado um componente celular plasmático substancial, consistente com a observação de produção crônica de anticorpos antienfuvirtida e um componente eosinofílico polimorfonuclear do infiltrado, sugestivo de reação de hipersensibilidade. Em cobaias tratadas durante até duas semanas com FUZEON® (com 4 injeções/dia de 50 mg/mL ou 2 injeções/dia de 100 mg/mL), foram observadas massas subcutâneas dentro de 8 dias de injeções diárias repetidas. As observações ao microscópio de biópsias por punção coletadas nos locais de injeção nos dias 8 e 15 incluíram edema, inflamação aguda e/ou crônica, necrose, fibrose e degeneração do colágeno na derme. Foram observadas mudanças significativas da derme e do subcutâneo, incluindo a presença de células gigantes estranhas ao corpo e granulomas. 2. RESULTADOS DE EFICÁCIA. Estudos com pacientes submetidos a tratamento anti-retroviral anterior: Os estudos T20-301 (TORO-1) e T20-302 (TORO-2) encontram-se em andamento, sendo estudos randomizados, controlados, abertos, multicêntricos em pacientes infectados com HIV-1, com pelo menos 3 a 6 meses de tratamento anterior com inibidores de transcriptase reversa nucleosídeos e não nucleosídeos e inibidores da protease. Todos os pacientes receberam um regime otimizado de base (OB), consistindo de 3 a 5 agentes anti-retrovirais selecionados com base na história anterior do paciente, como também nas avaliações de resistência viral genotípica e fenotípica no baseline. Os pacientes foram randomizados na proporção de 2:1 para FUZEON® (90 mg SC duas vezes ao dia) + OB ou somente OB. Os estudos TORO-1 e TORO-2 são bastante similares na população de pacientes estudada, desenho, critérios de seleção de pacientes (com exceção da duração prévia de exposição aos antiretrovirais e número de inibidores de protease), condução, monitoramento e planejamento das análises. Desta forma a análise conjunta dos dados dos dois estudos foi prospectivamente planejada. Os resultados da análise de 48 semanas são apresentados, portanto, de forma combinada possibilitando a análise de uma amostra maior e uma estimativa mais precisa nas diferenças de tratamento do grupo com e sem FUZEON®. As características demográficas combinadas da população de intenção de tratamento (ITT) dos pacientes incluídos nos estudos TORO-1 e TORO-2 estão demonstrados na Tabela 1. Os pacientes foram previamente expostos a uma mediana de 12 anti-retrovirais, por um tempo mediano de 7 anos.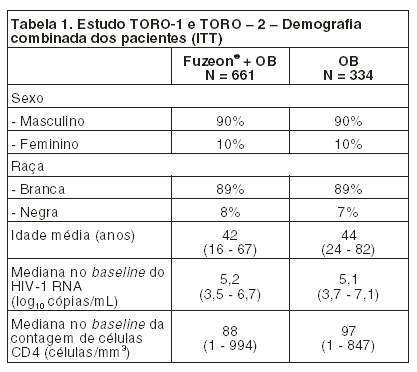
As mudanças na semana 48 em relação ao baseline dos estudos TORO-1 e TORO-2 combinados estão resumidas na Tabela 2 e todas foram significantemente maiores no grupo de tratamento com FUZEON®.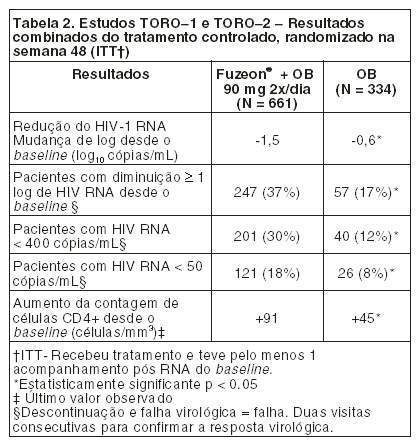
A proporção de pacientes que alcançaram uma carga viral menor que 400 cópias/mL até a semana 48 foi de 30% entre os pacientes no regime com FUZEON® + OB, comparado com 12% entre os pacientes que receberam apenas regime otimizado (Tabela 2). A análise do tempo de falha virológica está demonstrada na Figura 11. O tempo médio para falha virológica no grupo com FUZEON® + OB foi de 32 semanas, significativamente maior do que no grupo com OB apenas (11 semanas).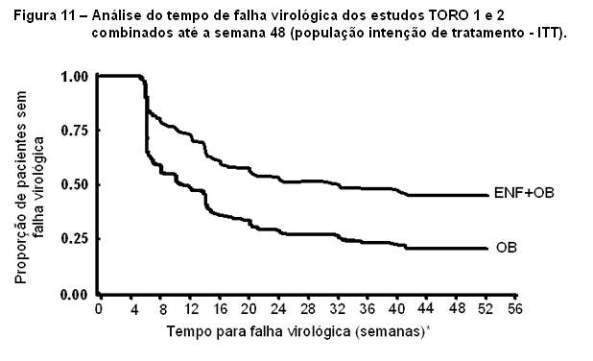
*Definição de falha virológica pelo protocolo (baseado em 2 medidas consecutivas): redução menor que 0,5log10 HIV-1 RNA do baseline até a semana 8; redução menor 1.0 log10 HIV-1 RNA até a semana 16; aumento maior ou igual a 1.0 log10 HIV-1 RNA após resposta de, pelo menos, 2.0 log10 HIV-1 RNA. Os resultados dos estudos também foram analisados em subgrupos, e a terapia com FUZEON® foi associada a uma maior proporção de pacientes que alcançaram HIV-1 RNA menor que 400 cópias/mL em todos os subgrupos; baseado na contagem de CD4+ no baseline, baseado na medida de HIV-1 RNA no baseline; pelo número prévio de anti-retrovirais e pelo número de anti-retrovirais ativos no regime otimizado. No entanto, pacientes com CD4+ > 100 células/mL, HIV-1 RNA inicial < 5.0 log10 cópias/mL, experiência com menos de 10 anti-retrovirais prévios e uma ou mais drogas ativas no regime anti-retroviral foram mais propensos a alcançar uma carga viral menor que 400 cópias/mL em ambos os tratamentos (Tabela 3).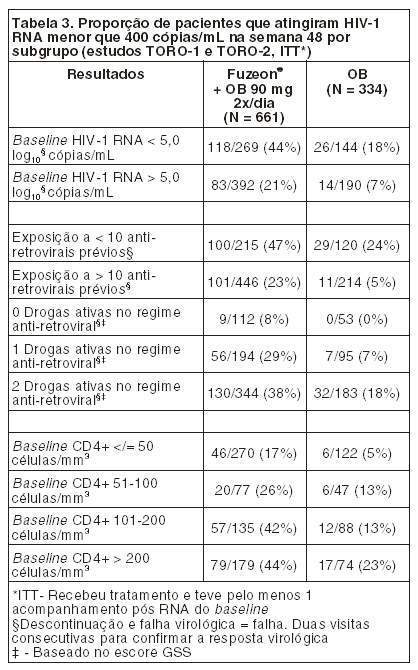
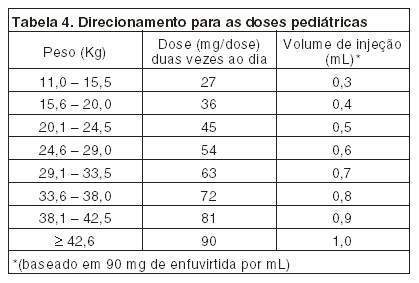
Uso pediátrico: Dados limitados de eficácia do FUZEON® estão disponíveis para crianças acima de 3 anos de idade. O estudo T20-204 é um estudo aberto e multicêntrico que avalia a atividade antiviral, segurança e farmacocinética do FUZEON® em 14 pacientes pediátricos com idade de 3 à 12 anos e experiência com no máximo 2 classes de antiretrovirais registrados(1). No estudo T20-2004 pacientes receberam ou 30 ou 60 mg/m2/dose de FUZEON®duas vezes ao dia associados ao seu regime anti-retroviral de base. Após 7 dias, seus regimes de tratamento de base foram alterados para 3 novos ou anti-retrovirais sensíveis e a dosagem de FUZEON® foi continuada. Os pacientes tinham uma idade média de 8 anos de idade (variando de 3,7 a 11,9 anos). A linha de base média da contagem de células CD4 foi 523 células/mL e a linha de base média de RNA HIV-1 foi 4,4 log10 c/mL. Seguindo análise da atividade antiviral, farmacocinética e segurança no dia 7, todos exceto 1 paciente mudaram para uma dose de FUZEON® de 60 mg/m2/dose. A mudança média da linha de base de RNA HIV-1 em 7 dias foi 1,15 log10 cópias/mL para 10 crianças recebendo a dose de 60 mg/m2 (1, 2). Todos exceto 3 pacientes completaram 48 semanas de terapia crônica. Na semana 48, 6/14 (43%) dos pacientes tinha declínio na RNA HIV-1 > 1 log10 e 4/14 (29%) dos pacientes estavam abaixo de 400 cópias/mL de RNA HIV-1. As mudanças médias da linha de base (para a população considerada como tratada) na contagem de células CD4 e RNA HIV-1 foram 237 células/mL e 1,24 log10 c/mL respectivamente (3). Referências Bibliográficas: 1) Carlyon T Paediatric U