FLYKATO
DR. REDDY'S
midostaurina
Antineoplásico. Inibidor da proteína quinase.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
FLYKATO® 25 mg - embalagens contendo 112 cápsulas moles.
VIA ORAL
USO ADULTO
Composição.
Cada cápsula contém 25 mg de midostaurina.
Excipientes: óleo de rícino hidrogenado etoxilado, macrogol, monoestearato de glicerina, racealfatocoferol, álcool etílico anidro, gelatina, glicerol, dióxido de titânio, óxido de ferro amarelo, água purificada.
Informações técnicas.
1. INDICAÇÕES
FLYKATO® (midostaurina) é indicado em combinação com a quimioterapia padrão de indução com daunorrubicina e citarabina e de consolidação com citarabina em altas doses, e para pacientes em resposta completa seguida por monoterapia de manutenção com FLYKATO®, para pacientes adultos com leucemia mieloide aguda (LMA) recém diagnosticada com mutação de FLT3 (vide seção Posologia e Modo de Usar).
FLYKATO® (midostaurina) é indicado como monoterapia para o tratamento de pacientes adultos com mastocitose sistêmica agressiva (MSA), mastocitose sistêmica associada com doença clonal hematopoética de linhagem não mastocitária (MS-ADHNM) ou leucemia das células mastocíticas (LCM).
2. RESULTADOS DE EFICÁCIA
Eficácia clínica e segurança
LMA
Estudo A2301 (RATIFY)
A eficácia e a segurança de midostaurina em combinação com quimioterapia padrão versus placebo em combinação com quimioterapia padrão e como monoterapia de manutenção foi investigada em 717 pacientes (18 a 60 anos de idade) em um estudo de fase III, randomizado, duplo-cego. Os pacientes com LMA recém diagnosticada com mutação de FLT3 foram determinados aleatoriamente (1:1) para receber midostaurina 50 mg duas vezes ao dia (n=360) ou placebo (n=357) sequencialmente em combinação com daunorrubicina padrão (60 mg/m2 diários nos dias 1- 3)/indução de citarabina (200 mg/m2 por dia nos dias 1-7) e consolidação de citarabina em altas doses (3g/m2 a cada 12 horas nos dias 1, 3, 5), seguida de tratamento contínuo com midostaurina ou placebo de acordo com a atribuição inicial de até 12 ciclos adicionais (28 dias/ciclo). Embora o estudo inclua pacientes com várias alterações citogenéticas relacionadas à LMA, pacientes com leucemia promielocítica aguda (M3) ou LMA relacionada à terapia foram excluídos. Os pacientes foram estratificados pelo perfil da mutação FLT3: TKD, ITD com razão alélica < 0,7 e ITD com razão alélica ≥ 0,7.
Os dois grupos de tratamento estavam, de maneira geral, equilibrados em relação aos dados demográficos das características da doença na linha basal. A mediana de idade dos pacientes foi 47 anos (faixa: 18 a 60 anos), a maioria dos pacientes apresentou escala de desempenho do ECOG de 0 ou 1 (88,3%), e muitos pacientes apresentaram LMA de novo (95%). Dos pacientes com informação de raça relatada, 88,1% eram caucasianos. A maioria dos pacientes (77,4%) apresentava mutações FLT3-ITD, muitos deles (47,6%) com uma taxa alélica baixa ( < 0,7), e 22,6% dos pacientes apresentavam mutações FLT3-TKD. Quarenta e oito por cento eram homens no braço da midostaurina e 41% no braço placebo.
Os pacientes submetidos a transplante de células-tronco hematopoiéticas (TCT) deixaram de receber o tratamento do estudo antes do início do regime de condicionamento para TCT. A taxa global de TCT foi de 59,4% (214/360) dos pacientes no braço de midostaurina associada à quimioterapia padrão versus 55,2% (197/357) no braço de placebo associado à quimioterapia padrão. Todos os pacientes foram acompanhados quanto à sobrevida.
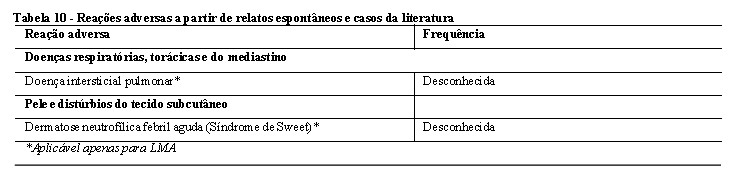
O desfecho primário do estudo foi a sobrevida global (SG), medida a partir da data da randomização até a morte por qualquer causa. A análise primária foi realizada após um acompanhamento mínimo de aproximadamente 3,5 anos após a randomização do último paciente. O estudo demonstrou um aumento estatisticamente significativo na SG com uma redução de risco de morte de 23% para midostaurina associada à quimioterapia padrão em comparação com placebo associado à quimioterapia padrão (vide Tabela 1 e Figura 1).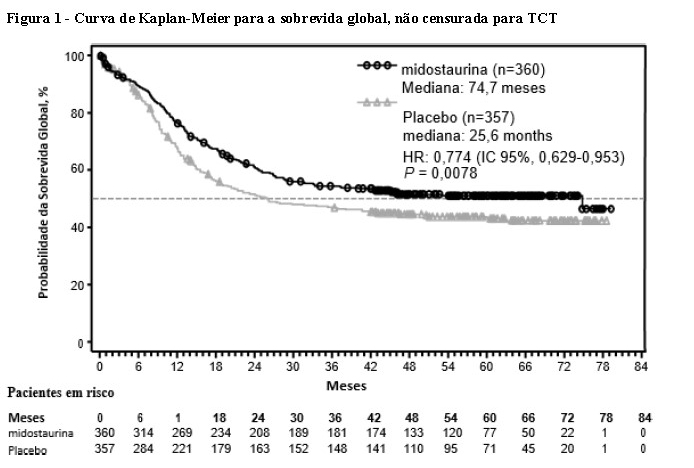
O desfecho secundário chave do estudo foi a sobrevida livre de eventos (SLE; um evento SLE é definido como falha em obter remissão completa (RC) dentro de 60 dias do início da terapia de protocolo, recidiva ou morte por qualquer causa). Houve um aumento estatisticamente significativo na SLE para midostaurina associada à quimioterapia padrão em comparação com placebo associado à quimioterapia padrão (HR: 0,78 [IC de 95%, 0,66 a 0,93] p = 0,0024), e uma SLE mediana de 8,2 meses e 3,0 meses, respectivamente; vide Tabela 1.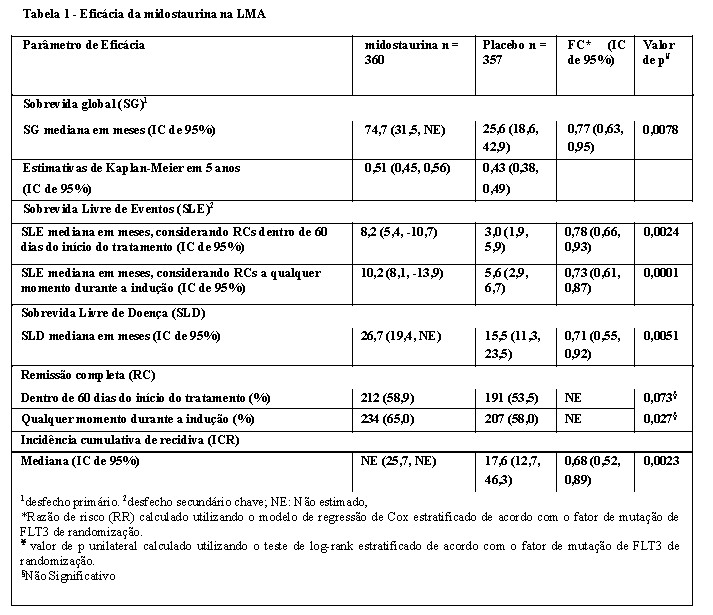
Houve uma tendência favorável à midostaurina em relação à taxa de RC no dia 60 para o braço de midostaurina (58,9% vs. 53,5%; p = 0,073), que permaneceu quando consideradas todos as RCs durante a indução (65,0% vs. 58,0%; p = 0,027). Além disso, em pacientes que obtiveram remissão completa durante a indução, a incidência cumulativa de recidiva aos 12 meses foi de 26% no braço midostaurina versus 41% no grupo placebo.
As análises de sensibilidade para SG e SLE quando censuradas no momento da TCT também corroboraram o benefício clínico da midostaurina associada à quimioterapia padrão em relação ao placebo.
Os resultados para SG pelo status TCT são mostrados na Figura 2. Para a SLE, considerando remissões completas dentro de 60 dias do início do tratamento de estudo, a HR foi 0,602 (IC de 95%: 0,372, 0,974) para pacientes com TCT e 0,827 (IC de 95%: 0,689, 0,993) para pacientes sem TCT, favorecendo a midostaurina.
Em uma análise de subgrupo, nenhum benefício de SG aparente foi observado em mulheres, no entanto, um benefício do tratamento foi observado em mulheres em todos os desfechos secundários de eficácia (vide Tabela 2).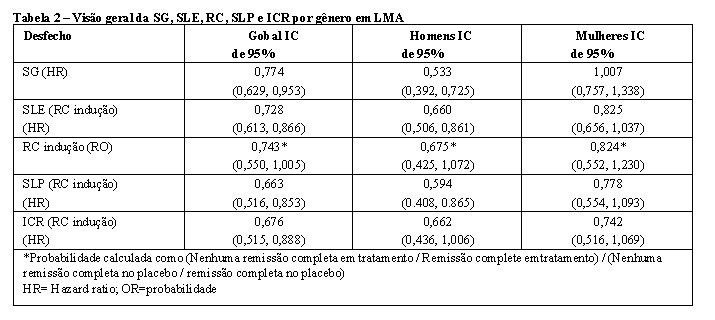
A eficácia e segurança em pacientes com idade entre 60 e 70 anos foram avaliadas em um estudo fase II, braço único, iniciado por investigador de midostaurina em combinação com indução intensiva, consolidação incluindo TCT alogênico e manutenção como monoterapia em pacientes com LMA com mutação FLT3-ITD. Com base em uma análise interina, a taxa de SLE aos 2 anos (desfecho primário) foi 27,1% (IC de 95%: 16,6%, 44,1%) e a SG mediana foi 15,5 meses em pacientes com mais de 60 anos de idade (46 de 145 pacientes).
MSA, MS-ADHNM e LCM*
* A nomenclatura leucemia das células mastocíticas (LCM) está alinhada às diretrizes mais recentes da Organização Mundial da Sáude.
Estudo D2201
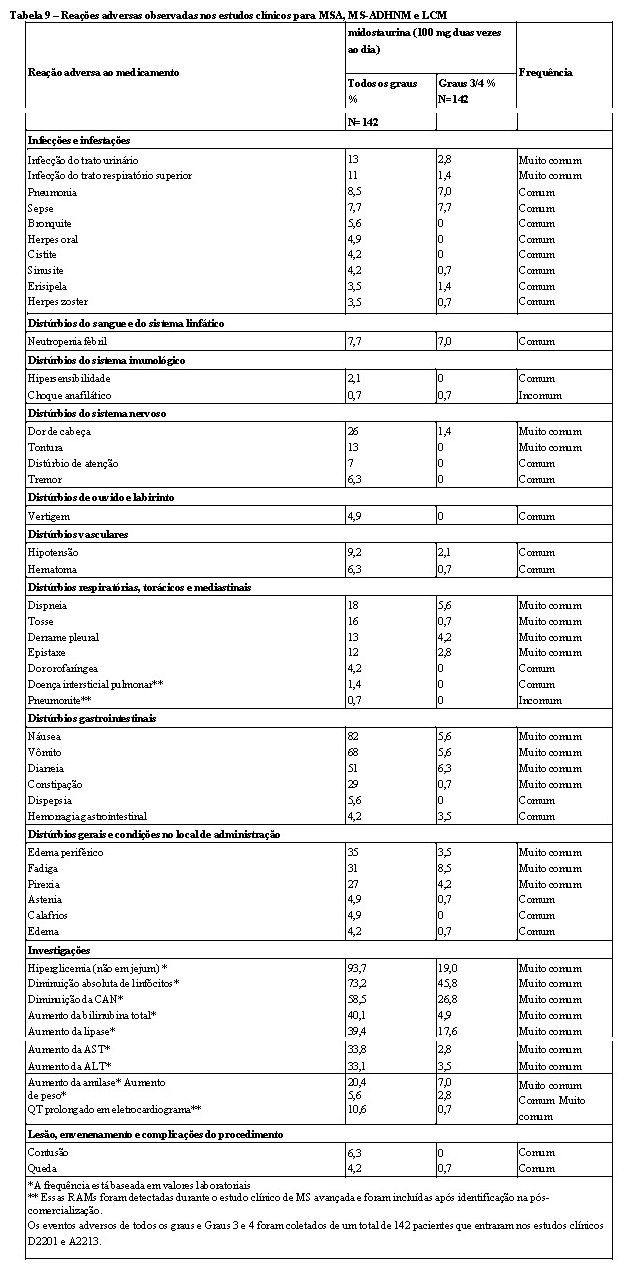
A eficácia de midostaurina em pacientes com MSA, MS-ADHNM e LCM, coletivamente referida como mastocitose sistêmica avançada (MS), foi avaliada em dois estudos abertos, braço único e multicêntrico (142 pacientes ao todo).
O estudo pivotal foi um estudo fase II multicêntrico, braço único com 116 pacientes com MS avançada (Estudo CPKC412D2201).
Adultos (≥ 18 anos) com mastocitose sistêmica agressiva, associada a neoplasia hematológica não mastocitária ou leucemia das células mastocíticas foram elegíveis para entrada no estudo. Todos os pacientes que entraram no estudo tinham performance status entre 0 e 3 pelo ECOG, além de funções hepática e renal normais. Pacientes que tinham recebido três ou mais tratamentos para mastocitose ou que tinham fração de ejeção < 50% foram excluídos. A midostaurina foi administrada oralmente a 100 mg duas vezes ao dia até a progressão da doença ou toxicidade intolerável. Dos 116 pacientes incluídos, 89 foram considerados elegíveis para avaliação da resposta e constituíram a população de eficácia primária (que tiveram pelo menos um achado C mensurável relacionado à MS no período basal de acordo com os critérios modificados de Valent / Cheson, além de atender aos critérios diagnósticos para MSA ou LCM). Destes, 73 pacientes tiveram MSA (57 com um ADHNM) e 16 pacientes tiveram LCM (6 com um ADHNM). A idade mediana na população de eficácia primária foi 64 anos com aproximadamente metade dos pacientes com ≥65 anos. Aproximadamente um terço (36%) receberam terapia antineoplásica prévia para MSA, MS-ADHNM ou LCM. Na linha basal na população de eficácia primária, 65% dos pacientes tiveram > 1 achado C mensurável (trombocitopenia, hipoalbuminemia, anemia, bilirrubina total elevada, anemia dependente de transfusão, perda de peso, neutropenia, ALT elevada ou AST elevada). A mutação KIT D816V foi detectada em 82% dos pacientes.
O desfecho primário foi taxa de resposta global (TRG) (melhor resposta global obtida nos primeiros 6 ciclos de tratamento, de 28 dias cada, e mantida por mais 8 semanas). As taxas de resposta foram avaliadas com base no critério de Valent e Cheson modificados e as respostas foram adjudicadas por um comitê de direção do estudo. As respostas obtidas foram confirmadas na população de eficácia primária, quanto a obtenção de resposta maior (ou seja, resolução completa de um ou mais achados C e sem progressao confirmada de outro achado C) e resposta parcial (ou seja, melhora mensurável em um ou mais achados C e sem progressão confirmada em outros achados C).
Os desfechos secundários incluíram duração da resposta, tempo para a resposta, sobrevida global, sobrevida livre de progressão, resposta histopatológica, alteração percentual dos níveis séricos de triptase, melhora na organomegalia (fígado e baço). As respostas à midostaurina estão apresentadas na Tabela 4. A atividade foi observada independentemente do número de terapias anteriores e presença ou ausência de uma ADHNM. O status mutacional na população de eficácia (n=89) era 77 (87%) KIT D816V positivos, 10 (11%) negativos e 2 (2%) com status desconhecido. Respostas confirmadas foram observadas em pacientes com ambas as mutações KIT D816V positiva (TRG=63%) e KIT D816V do tipo selvagem ou com status desconhecido (TRG=43,8%). No entanto, a sobrevida mediana para pacientes com KIT D816V positiva foi mais longa, por exemplo 33,9 meses (IC de 95%: 20,7, 42), do que para pacientes com KIT D816V do tipo selvagem ou com status desconhecido, por exemplo 10 meses (IC de 95%: 6,9, 17,4). Quarenta e seis por cento dos pacientes tiveram uma diminuição na infiltração da medula óssea que excederam 50% e 58% dos pacientes tiveram uma diminuição nos níveis séricos de triptase que excederam 50%. O volume do baço diminuiu em ≥10% em 68,9% dos pacientes com pelo menos uma avaliação após a linha basal (26,7% dos pacientes tiveram uma redução de ≥35%, que se correlaciona com uma diminuição de 50% pela palpação).
O tempo mediano de tratamento foi 11,4 meses (intervalo de variação: 0,3 a 68,3 meses) e tempo mediano para resposta foi 0,3 meses (intervalo de variação: 0,1 a 3,7 meses). A duração mediana de acompanhamento foi 43 meses (intervalo de variação: 29 a 70 meses).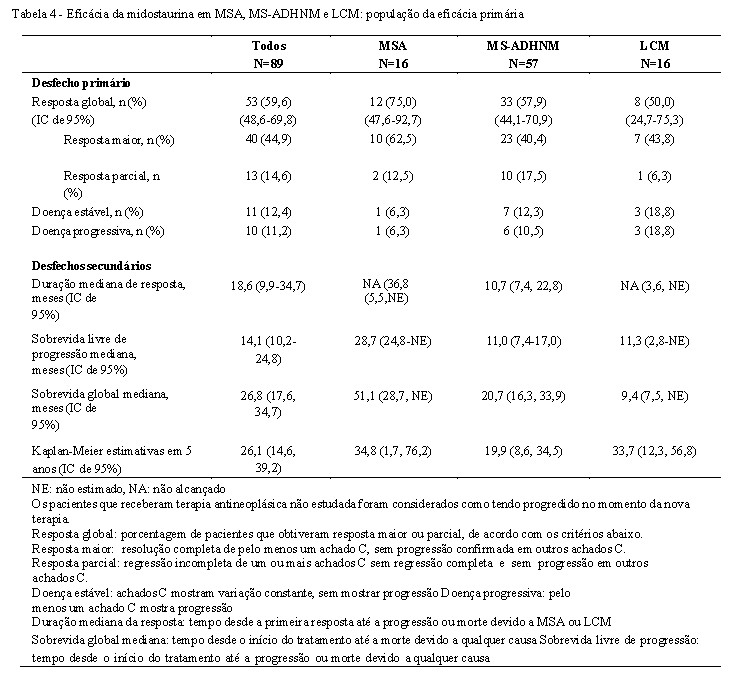
Outros desfechos secundários foram possíveis de ser avaliados em 72 pacientes. Entre estes, 41 (57%) tiveram redução ≥ 50% na carga de mastócitos na medula óssea; 24 (33%) mantiveram esta redução em pelo menos duas biópsias consecutivas. A porcentagem mediana de alteração na carga de mastócitos foi de -59% (-96% a 160%). A mediana da melhor alteração do nível de triptase sérica foi -58% (-99% a 185%). Redução em ambos, carga de mastócitos e triptase sérica, ocorreu em 78% dos pacientes. Entre os 39 pacientes que tinham esplenomegalia no início do estudo e pelo menos uma avalição posterior, 30 (77%) tiveram redução do volume do baço sendo 10 (26%) redução de pelo menos 35% em relação ao volume inicial.
Embora o estudo tenha sido desenhado para ser avaliada com o critério de Valent e Cheson modificados, com uma análise explanatória post-hoc, a eficácia também foi avaliada segundo os critérios do consenso do Grupo de Trabalho Internacional de Pesquisa e Tratamento de Neoplasias Mieloproliferativas da Rede de Competência Europeia sobre Mastocitose 2013 (2013 International Working Group Myeloproliferative Neoplasms Research and Treatment European Competence Network on Mastocytosis - IWG MRT ECNM). A resposta a midostaurina foi determinada usando um algoritmo computacional aplicado sem qualquer adjudicação. Dos 116 pacientes, 113 tiveram achados C conforme definido pelo critério de resposta do IWG (excluindo ascites como um achado C). Todas as respostas foram consideradas e exigiram uma confirmação de 12 semanas (veja a Tabela 5).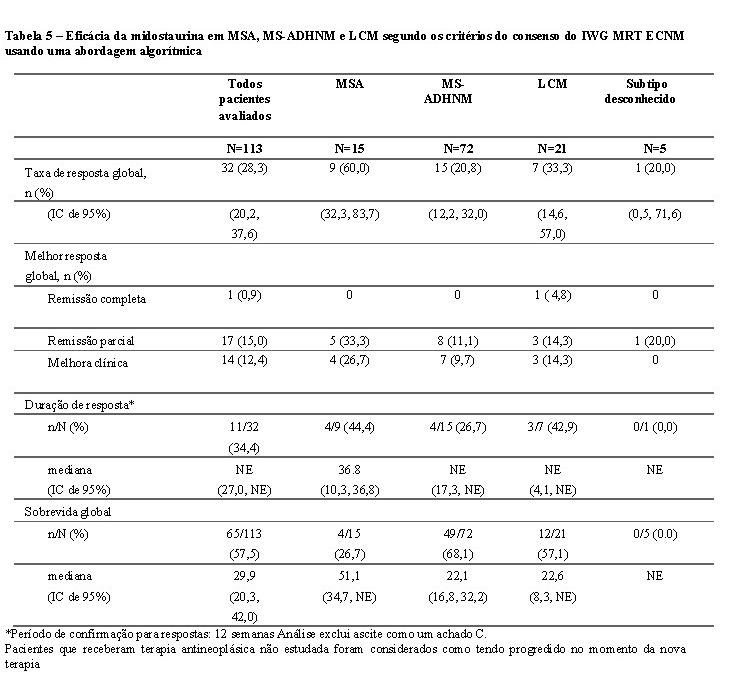
Estudo A2213
O estudo de suporte foi um estudo de fase II braço único, multicêntrico, aberto de 26 pacientes com MSA, MS-ADHNM e LCM (CPKC412A2213). A midostaurina foi administrada oralmente 100 mg duas vezes ao dia em ciclos de 28 dias. A falta de resposta maior (RM) ou resposta parcial (RP) ao final do segundo ciclo resultou em descontinuação do tratamento em estudo. Vinte pacientes (76,9%) tiveram MSA (17 [85%] ADHNM) e 6 pacientes (23,1%) tiveram LCM (2[33,3%] ADHNM). A idade mediana foi de 64,5 anos com metade dos pacientes com ≥65 anos. Na linha basal, 88,5% tiveram > 1 achado C e 69,2% tinham recebido pelo menos um regime antineoplásico prévio.
O desfecho primário foi a TRG avaliada pelo critério de Valent durante os primeiros dois ciclos de tratamento. Dezenove pacientes (73,1%; IC de 95% = [52,2, 88,4]) alcançaram a resposta durante os primeiros dois ciclos de tratamento (13 RM; 6 RP). A duração mediana de acompanhamento foi 73 meses, e a duração mediana de resposta não foi alcançada. A mediana de sobrevida global foi 40,0 meses (pacientes foram acompanhados somente por um ano após a descontinuação do tratamento para sobrevida).
Referências bibliográficas
Summary of Clinical Efficacy] PKC412 - 2.7.3 Summary of Clinical Efficacy in FLT3-mutated Acute Myeloid Leukemia (LMA). Novartis. 2016.
[Summary of Clinical Efficacy] PKC412 - 2.7.3 Summary of Clinical Efficacy - in Aggressive Systemic Mastocytosis (ASM) and Mast Cell Leukemia (MCL). Novartis. 2016.
Gotlib et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. New England Journal of Medicine. 2016; 374:2530-2541
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Agentes antineoplásicos, inibidores da proteína quinase. Código ATC: L01XE39
Mecanismo de ação
A midostaurina inibe múltiplos receptores de tirosinaquinase, incluindo as quinases FLT3. A midostaurina inibe a sinalização do receptor FLT3 e induz a parada do ciclo celular e a apoptose em células leucêmicas que expressam os receptores com mutação FLT3 ITD ou TKD ou os receptores com superexpressão de FLT3 do tipo selvagem. Dados in vitro indicam que a midostaurina inibe receptores KIT com mutação D816V a níveis de exposição alcançados em pacientes (média atingida em exposição mais alta que o IC50). Dados in vitro indicam que os receptores KIT do tipo selvagem são inibidos a um grau muito menor a essas concentrações (média atingida em exposição menor que o IC50). A midostaurina interfere com a sinalização mediada por D816V KIT aberrante e inibe a proliferação de mastocitócitos, sobrevivência e liberação de histamina.
Além disso, a midostaurina inibe vários outros receptores de tirosina-quinase tais como PDGFR (receptor do fator de crescimento derivado de plaquetas) ou VEGFR2 (receptor do fator de crescimento endotelial vascular 2), bem como, membros da família PKC de serina/treonina quinase (proteína quinase C). A midostaurina se liga ao domínio catalítico destas quinases e inibe a sinalização mitogênica dos respectivos fatores de crescimento nas células, resultando na parada do crescimento.
A midostaurina em combinação com agentes quimioterápicos (citarabina, doxorrubicina, idarrubicina e daunorrubicina) resultou em inibição sinérgica do crescimento em linhagens celulares de LMA com expressão de FLT3-ITD.
Efeitos farmacodinâmicos
Dois metabólitos principais foram identificados em modelos murinos e humanos, isto é, CGP62221 e CGP52421. Em ensaios de proliferação com células que expressam FLT3-ITD, CGP62221 mostrou potência semelhante em comparação com o composto de original, no entanto CGP52421 foi aproximadamente 10 vezes menos potente.
Eletrofisiologia cardíaca
Um estudo dedicado ao intervalo QT em 192 indivíduos saudáveis com uma dose de 75 mg duas vezes ao dia não revelou prolongamento clinicamente significativo do QT por midostaurina e CGP62221, mas a duração do estudo não foi suficientemente longa para estimar os efeitos do prolongamento QTc do metabólito de ação prolongada CGP52421. Portanto, a mudança na linha basal em QTcF com a concentração de midostaurina e ambos os metabólitos foi mais explorada em um estudo fase II em 116 pacientes com MSA, MS-ADHNM ou LCM. No pico mediano das concentrações Cmín atingidas na dosagem de 100 mg duas vezes ao dia, nem a midostaurina, nem CGP62221 ou CGP52421 mostraram um potencial para causar prolongamento QTcF clinicamente significativo, uma vez que os limites superiores da mudança prevista nestes níveis de concentração eram inferiores a 10 ms (5,8, 2,4 e 4,0 ms, respectivamente). Na população com MSA, MS-ADHNM ou LCM, 25,4% dos pacientes tiveram pelo menos uma medição de ECG com um QTcF superior a 450 ms e em 4,7% dos pacientes foi superior a 480 ms.
Propriedades farmacocinéticas
A midostaurina é um composto com boa absorção e fraca solubilidade. Dois dos seus metabólitos demonstraram atividades farmacológicas (CGP52421 e CGP62221). Após doses múltiplas, a farmacocinética de midostaurina e CGP62221 foi dependente do tempo, com um aumento inicial observado na primeira semana seguido por um declínio das concentrações até atingir o estado de equilíbrio no dia 28. As concentrações de CGP52421 não parecem diminuir tão significativamente quanto para midostaurina e CGP62221.
Absorção
A biodisponibilidade absoluta de midostaurina após administração oral não é conhecida.
Em humanos, a absorção de midostaurina foi rápida após administração oral, com Tmáx de radioatividade total observado em 1-3 horas após a dose. A análise da população farmacocinética indicou que a absorção em pacientes foi menor do que a dose proporcional a doses > 50 mg duas vezes ao dia.
Em indivíduos saudáveis, após administração de uma dose única de 50 mg de midostaurina com alimento, a AUC de midostaurina aumentou para 20800 ng*h/ml e Cmáx diminuiu para 963 ng/ml (vide seção "Interações Medicamentosas"). Similarmente, para os metabólitos CGP52421 e CGP62221 a ASC aumentou para 19000 e 29200 ng*h/ml e Cmáx diminuiu para 172 e 455 ng/ml, respectivamente. O tempo até o pico de concentração também foi retardado na presença de uma refeição rica em gordura. O Tmáx foi retardado para todos os indivíduos, oTmáx mediano da midostaurina foi 3 h., e, para CGP52421 e CGP62221, o Tmáx foi retardado para 6 e 7 horas respectivamente.
Em estudos clínicos, a eficácia e a segurança de midostarina foram investigadas após administração com uma refeição leve. Após a administração oral de uma única dose de 100 mg de midostaurina em condições alimentadas em pacientes com MSA, MS- ADHNM e LCM, a AUCinf, Cmáx e Tmáx foram 49600 ng*h/ml, 2940 ng/ml e 3 h, respectivamente, para midostaurina. Para CGP52421, a AUC0-12h e Cmáx foram 2770 ng*h/ml e 299 ng/ml, respectivamente. A AUC0-12h e o Cmáx para CGP62221 foram 8700 ng*h/ml e 931 ng/ml, respectivamente. Após várias doses orais múltiplas de 100 mg de midostaurina, a Cmin,ss plasmática em pacientes com LMA e mastocitose sistêmica avançada (MSA, MS- ADHNM e LCM) foi 919 ng/ml e 1060 ng/ml, respectivamente. O Cmin,ss do CGP62221 nas populações com LMA e mastocitose sistêmica avançada (MSA, MS-ADHNM e LCM) foi 1610 ng/ml e 2020 ng/ml, respectivamente. O Cmin,ss do CGP52421 nas populações com LMA e mastocitose sistêmica avançada (MSA, MS- ADHNM e LCM) foi 8630 ng/ml e 2860 ng/ml, respectivamente.
Distribuição
A midostaurina tem uma distribuição tecidual de média geométrica de 95,2 L (Vz/F). A midostaurina e seus metabólitos são distribuídos principalmente no plasma ao invés de nos glóbulos vermelhos. Os dados in vitro mostraram que a midostaurina fica mais de 98% ligada às proteínas do plasma, tais como, albumina, a1 glicoproteína ácida (AGP) e lipoproteína.
Biotransformação
A midostaurina é metabolizada pelo CYP3A4 principalmente através de vias oxidativas. Os principais componentes plasmáticos incluíram midostaurina e dois principais metabólitos ativos, CGP62221 (via O- desmetilação) e CGP52421 (via hidroxilação), representando 27,7 ± 2,7% e 38,0± 6,6%, respectivamente, da exposição plasmática total às 96 horas após uma dose única de 50 mg de midostaurina.
Eliminação
As meia-vidas terminais medianas de midostaurina, CGP62221 e CGP52421 no plasma são aproximadamente 20,5, 32,3 e 482 horas. O clearance plasmático médio aparente (CL/F) foi de 2,4-3,1 L/h em indivíduos saudáveis. Em pacientes com LMA LMA e mastocitose sistêmica avançada (MSA, MS- ADHNM e LCM), as estimativas farmacocinéticas da população para clearance de midostaurina no estado de equilíbrio foram de 5,9L/h e 4,4L/h, respectivamente. Os resultados do estudo do Equilíbrio de Massa Humana indicaram que a excreção fecal é a principal via de excreção (78% da dose), e principalmente na forma de metabólitos (73% da dose), enquanto a midostaurina inalterada responde por 3% da dose. Apenas 4% da dose é recuperada na urina.
Linearidade/não-linearidade
Em geral, a midostaurina e os seus metabólitos não apresentaram desvio importante da proporcionalidade de dose após uma única dose variando de 25 mg a 100 mg. Contudo, houve um aumento na exposição menos do que proporcional à dose após doses múltiplas dentro da faixa de doses de 50 mg a 225 mg por dia.
Após doses orais múltiplas, a midostaurina apresentou uma farmacocinética dependente do tempo, com um aumento inicial das concentrações plasmáticas durante a primeira semana (pico Cmín), seguido de um declínio do tempo até o estado de equilíbrio após aproximadamente 28 dias (diminuição de 2,5 vezes). Apesar do mecanismo exato para a diminuição da concentração de midostaurina não ser claro, é provavelmente dado as propriedades de autoindução da midostaurina e seus dois metabólitos CGP52421 e CGP62221 sobre a CYP3A4. A farmacocinética do metabólito CGP62221 mostrou uma tendência semelhante. No entanto, as concentrações de CGP52421 aumentaram até 2,5 vezes para MSA, MS- ADHNM e LCM e até 9 vezes para LMA em comparação com a midostaurina após um mês de tratamento.
Avaliação in vitro do potencial de interação medicamentosa
Com base em dados in vitro, a midostaurina e seus metabólitos ativos, CGP52421 e CGP62221, são considerados inibidores do CYP1A2 e CYP2E1 e indutores do CYP1A2. Com base em dados in vitro, a midostaurina pode inibir a BSEP. Simulações usando modelos farmacocinéticos de base fisiológica (PBPK) previram que a midostaurina administrada em uma dose de 50 mg duas vezes ao dia no estado estacionário é improvável de causar inibição clinicamente relevante de OATP1B.
Populações especiais
Pacientes idosos
Com base nas análises farmacocinéticas populacionais, nenhum impacto significativo da idade na farmacocinética da midostaurina e dos seus dois metabólitos ativos foi identificado em pacientes com idade entre 65 e 85 anos. Em pacientes adultos com MSA, MS- ADHNM e LCM e LMA, não é necessário o ajuste de dose da midostaurina com base na idade.
Pacientes pediátricos
A midostaurina não é recomendado para crianças e adolescentes (vide seção "Posologia e Modo de administração"). A farmacocinética da midostaurina em pacientes pediátricos foi explorada em um estudo de fase I de monoterapia com escalonamento de dose com 22 pacientes (12 com idade entre 0-2 anos e 10 com idade entre 10-17 anos) com LMA ou LLA com rearranjo de MLL utilizando uma abordagem farmacocinética populacional. A farmacocinética da midostaurina foi menos do que proporcional à dose com as doses de 30 mg/m2 e 60 mg/m2 depois de doses únicas e múltiplas. Devido aos dados farmacocinéticos limitados em pacientes pediátricos, nenhuma comparação pôde ser feita com a farmacocinética de midostaurina em adultos.
Gênero
Com base nas análises do modelo farmacocinético populacional do efeito do gênero no clearance da midostaurina e dos seus metabólitos ativos, não houve nenhum achado estatisticamente significativo e as alterações esperadas na exposição ( < 20%) não foram consideradas clinicamente relevantes. Não é necessário o ajuste de dose da midostaurina com base no gênero.
Raça/etnia
Não existem diferenças no perfil farmacocinético entre indivíduos caucasianos e afrodescendentes. Com base em um estudo de fase I com voluntários japoneses saudáveis, os perfis farmacocinéticos da midostaurina e dos seus metabólitos (CGP62221 e CGP52421) são semelhantes em comparação com os observados em outros estudos farmacocinéticos realizados em caucasianos e afrodescendentes. Não é necessário o ajuste de dose da midostaurina com base na etnia.
Insuficiência hepática
Um estudo específico para comprometimento hepático avaliou a exposição sistêmica da midostaurina após administração oral de 50 mg duas vezes ao dia durante 6 dias em indivíduos com insuficiência hepática leve, moderada ou grave na linha basal (Classe A, B ou C de Child-Pugh) e indivíduos controle com função hepática normal.
A concentração máxima foi atingida entre 2 e 3 horas após a administração de doses únicas ou repetidas para todos os grupos. No dia 1, as AUC0-12 e Cmáx foram 8130 ng.h/mL e 1206 ng/mL, respectivamente, para indivíduos saudáveis. A AUC0-12 diminui em 39% e 36% em indivíduos com insuficiência hepática leve e moderada, respectivamente. No dia 7, a AUCCinf (exposição sob a curva de Cinf do dia 1 ao dia 7) era de 5410* ng.h/mL em indivíduos saudáveis e foi diminuída em 35% e 20% em indivíduos com insuficiência hepática leve e moderada, respectivamente. AUCtau diminui em 28% e 20%, no dia 7, respectivamente. Por fim, os dados de longo prazo dos pacientes foram analisados utilizando uma abordagem farmacocinética populacional. Nenhum impacto da insuficiência hepática pôde ser identificado em pacientes com insuficiência hepática leve ou moderada nas populações com MSA, MS-ADHNM e LCM e LMA.
Em geral, não houve aumento da exposição (AUC) à midostaurina plasmática e seus metabólitos (CGP62221 e CGP52421) em indivíduos com insuficiência hepática leve, moderada e grave em comparação com indivíduos com função hepática normal. Não é necessário qualquer ajuste posológico em pacientes com insuficiência hepática leve, moderada ou grave na linha basal.
Insuficiência renal
A excreção renal é uma via secundária de eliminação da midostaurina. Não foi realizado nenhum estudo específico para insuficiência renal para midostaurina. As análises farmacocinéticas populacionais foram realizadas com dados de estudos clínicos em pacientes com LMA (n = 180) e MSA, MS-ADHNM e LCM (n = 141). Dos 321 pacientes incluídos, 177 pacientes apresentaram insuficiência renal prévia leve (n = 113), moderada (n = 60) ou grave (n = 4) (15 mL/min ≤ clearance da creatinina < CrCl < 90 mL/min). Uma quantidade de 144 pacientes apresentou função renal normal (CrCL > 90 mL/min) na linha basal. Com base nas análises farmacocinéticas da população, o clearance da midostaurina não foi significativamente afetado pela insuficiência renal e, portanto, não é necessário qualquer ajuste posológico em pacientes com insuficiência renal leve ou moderada.
Dados de segurança pré-clínica
Devido à toxicidade limitada à dose, os níveis de exposição terapêutica clínica não puderam ser atingidos em animais. Todos os achados em animais descritos abaixo foram observados com em exposição à midostaurina significativamente inferior aos níveis terapêuticos.
Farmacologia de segurança e toxicidade de dose única/repetida
Estudos farmacológicos de segurança indicam que é improvável que a midostaurina interfira nas funções vitais do sistema nervoso central. In vitro, a midostaurina não inibiu a atividade do canal hERG até o limite de solubilidade de 12 mM. Os dois principais metabólitos humanos CGP52421 e CGP62221 (também testados no limite de solubilidade) inibiram a hERG corrente com margem de segurança moderada. Nos estudos de doses repetidas em cães, observou-se diminuição da frequência cardíaca, prolongamento do intervalo P-Q e bloqueios atrioventriculares esporádicos em animais específicos.
Nos estudos de dose repetida, os órgãos-alvo de toxicidade foram os do trato gastrointestinal (êmese em cães e macacos, diarreia e alteração da mucosa), testículos (diminuição da espermatogênese), medula óssea (hipocelularidade) e órgãos linfoides (depleção/atrofia). O efeito sobre a medula óssea e os órgãos linfoides foi acompanhado por alterações hematológicas de redução de glóbulos brancos, linfócitos e parâmetros eritrocitários. Observou-se um aumento nas enzimas hepáticas (ALT e AST) de forma consistente em ratos, cães e macacos em estudos de longo prazo com ≥ 3 meses de duração, sem correlatos histopatológicos.
Toxidade reprodutiva
Em um estudo de fertilidade em ratos, a midostaurina foi associada à redução da fertilidade, degeneração testicular e atrofia, motilidade espermática reduzida, oligospermia e aspermia, aumento da reabsorção, diminuição da taxa de fecundação, do número de implantes e de embriões vivos.
Em estudos de desenvolvimento embriofetal em ratos e coelhos, observou-se aumento do número de reabsorções tardias, redução do peso fetal e redução da ossificação esquelética.
Em um estudo de desenvolvimento pré e pós-natal, observaram-se distocia materna e tamanho reduzido da ninhada, pesos corporais inferiores, abertura dos olhos completa acelerada e ontogenia auricular atrasada.
Estudos com animais jovens
Em um estudo de toxicidade em ratos jovens, a midostaurina foi administrada dos dias 7 a 70 após o parto. Observou-se uma redução do peso corporal, hemorragia e infiltração de células misturadas nos pulmões, e eritrocitose/eritrofagocitose nos gânglios linfáticos mesentéricos. Não houve efeitos no desenvolvimento físico, na função sensorial ou comportamental. O índice de acasalamento, índice de fertilidade e taxas de concepção foram reduzidas a 0, 5 e 15 mg/kg/dia, mas não a 2 mg/kg/dia.
Genotoxicidade
Estudos de genotoxicidade in vitro e in vivo abrangendo os desfechos de genotoxicidade relevantes demonstraram que não há evidência de atividade mutagênica ou clastogênica. Não foram realizados estudos de carcinogenicidade.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes.
Administração concomitante de potentes indutores de CYP3A4, por exemplo, rifampicina, Erva de São João (Hypericum perforatum), carbamazepina, enzalutamida, fenitoína (vide seção "Interações Medicamentosas").
5. ADVERTÊNCIAS E PRECAUÇÕES
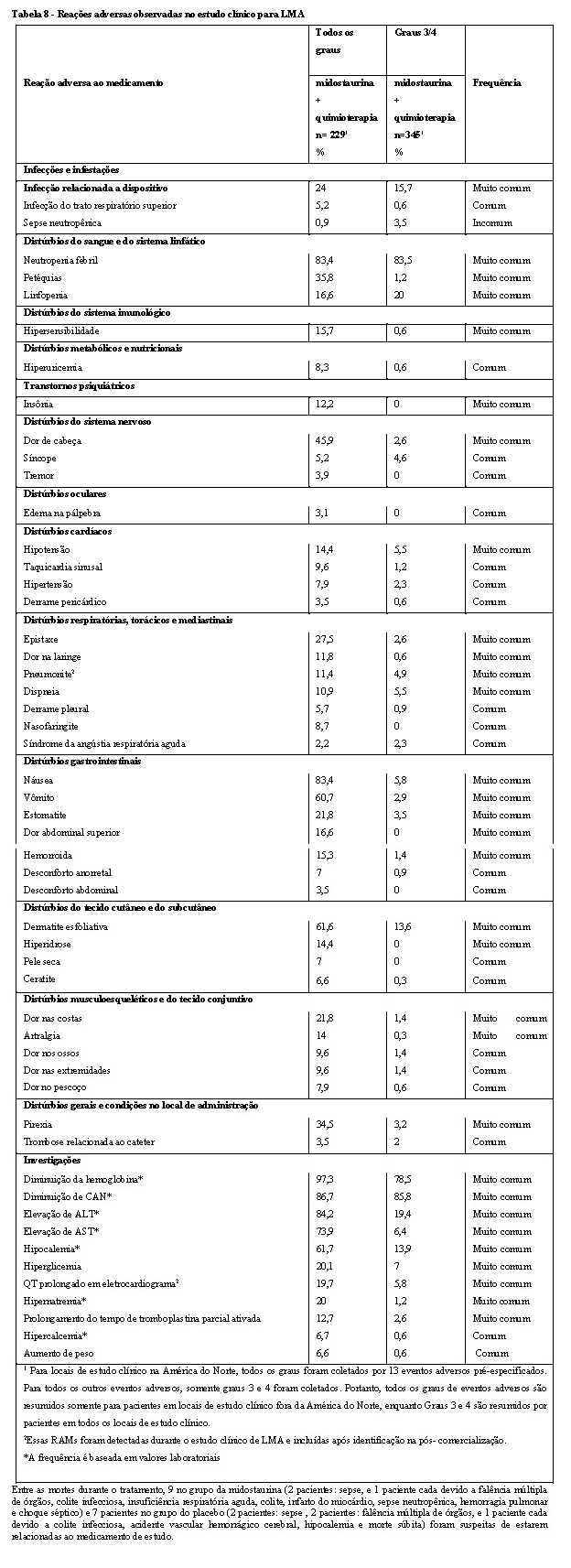
Neutropenia e infecções
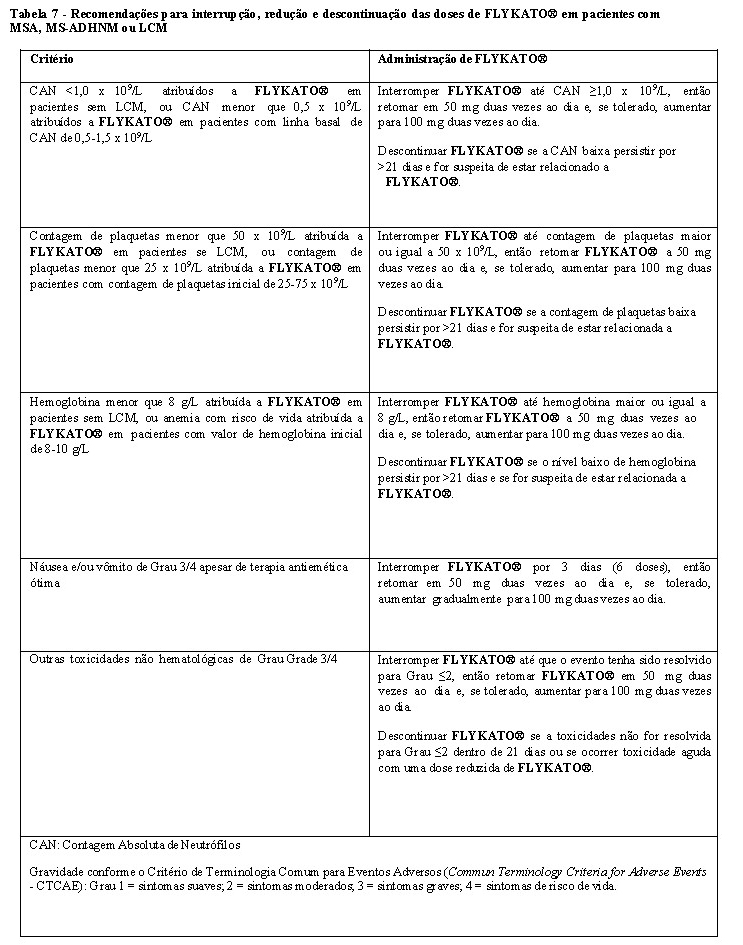
Ocorreu neutropenia em pacientes que receberam midostaurina em monoterapia e em associação com quimioterapia (vide seção "Reações Adversas"). A neutropenia grave (CAN < 0,5 × 109/L) foi geralmente reversível por suspensão de midostaurina até recuperação e descontinuação nos estudos de MSA, MS-ADHNM e LCM. As contagens de glóbulos brancos (WBCs) devem ser monitoradas regularmente, especialmente no início do tratamento.
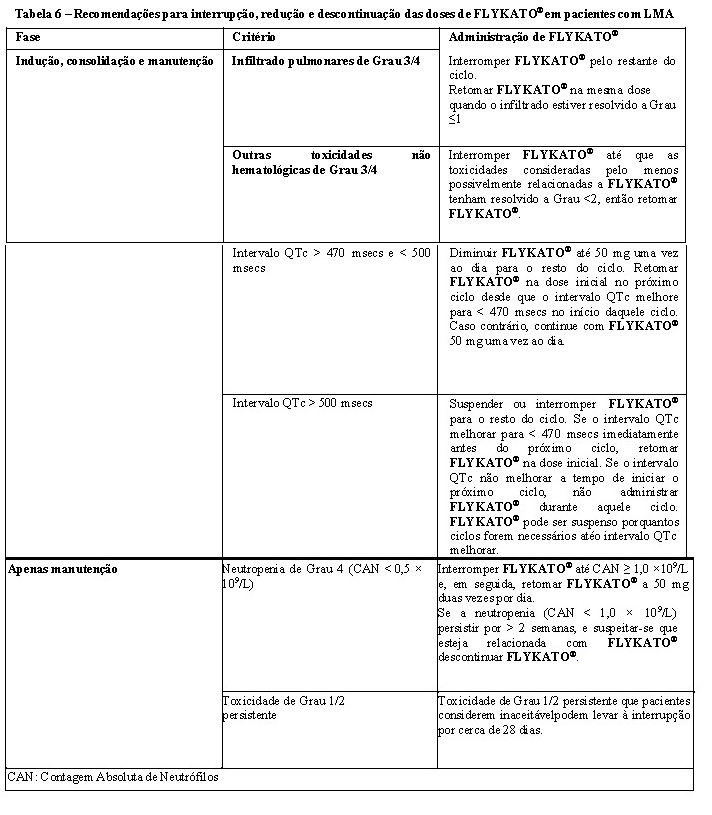
Nos pacientes que desenvolvem neutropenia grave inexplicada, o tratamento com midostaurina t deve ser interrompido até que a CAN seja ≥ 1,0 × 109/L, tal como recomendado nas Tabelas 6 e 7. A midostaurina deve ser interrompido em pacientes que desenvolvem neutropenia grave recorrente ou prolongada suspeita de estar relacionada com midostaurina (vide seção "Posologia e Modo de administração").
Qualquer infecção grave ativa deve estar sob controle antes de iniciar o tratamento com FLYKATO® em monoterapia. Os pacientes devem ser monitorados quanto a sinais e sintomas de infecção, incluindo qualquer infecção relacionada a dispositivo, e se um diagnóstico de infecção for feito, o tratamento adequado deve ser prontamente estabelecido, incluindo, se necessário, a descontinuação de FLYKATO®.
Disfunção cardíaca
Pacientes com insuficiência cardíaca congestiva sintomática foram excluídos dos estudos clínicos. Nos estudos de MSA, MS- ADHNM e LCM, ocorreram disfunções cardíacas tais como insuficiência cardíaca congestiva (ICC) (incluindo algumas fatalidades) e diminuições transientes na fração de ejeção do ventrículo esquerdo (FEVE). No estudo randomizado de LMA, não foi observada diferença na ICC entre os braços de quimioterapia + midostaurina e placebo + quimioterapia. Em pacientes em risco, FLYKATO® deve ser utilizado com cautela, e o paciente deve ser cuidadosamente monitorado através da avaliação FEVE quando clinicamente indicada (no início e durante o tratamento).
Um aumento na frequência do prolongamento QTc foi notado em pacientes tratados com midostaurina (vide seção "Reações Adversas"), no entanto, não foi encontrada explicação mecanicista para essa observação. Deve-se ter cautela com pacientes em risco de prolongamento QTc (por exemplo, devido a medicamentos concomitantes e/ou distúrbios eletrolíticos). Avaliações do intervalo QT por ECG devem ser consideradas se midostaurina for tomado concomitantemente com medicamentos que podem prolongar o intervalo QT.
Toxicidade pulmonar
A doença pulmonar intersticial (DPI) e a pneumonite, em alguns casos fatais, ocorreram em pacientes tratados com midostaurina em monoterapia ou em combinação com quimioterapia. Os pacientes devem ser monitorados quanto a sintomas pulmonares indicativos de DPI ou pneumonite, e FLYKATO® deve ser descontinuado em pacientes que apresentaram sintomas pulmonares indicativos de DPI ou pneumonite sem uma etiologia infecciosa que sejam ≥ Grau 3.
Toxicidade embriofetal e amamentação
Mulheres grávidas devem ser informadas quanto ao potencial risco ao feto; mulheres com potencial reprodutivo devem ser aconselhadas a utilizar teste de gravidez dentro de 7 dias antes de iniciar o tratamento com FLYKATO® e usar método contraceptivo durante o tratamento com FLYKATO® e por pelo menos 4 meses após parar o tratamento.
Devido ao potencial de reações adversas graves em lactentes de FLYKATO®, mulheres devem descontinuar a amamentação durante o tratamento com FLYKATO® e por pelo menos 4 meses após parar o tratamento.
Insuficiência hepática grave
Deve-se ter cautela quando considerada a administração de midostaurina em pacientes com insuficiência hepática grave. Os pacientes devem ser cuidadosamente monitorados para toxicidade (vide seção "Características Farmacológicas").
Insuficiência renal grave
Deve-se ter cautela quando considerada a administração de midostaurina em paciente com insuficiência renal grave ou doença em estado terminal. Os pacientes devem ser cuidadosamente monitorados para toxicidade (vide seção "Características Farmacológicas").
Interações
Deve-se ter cautela ao prescrever concomitantemente com midostaurina outros medicamentos que são inibidores potentes de CYP3A4, tais como, mas não limitado a,antifúngicos (por exemplo, cetoconazol), certos antivirais (por exemplo, ritonavir), antibióticos macrólidos (por exemplo, claritromicina) e nefazodona, porque podem causar aumento das concentrações do plasma de midostaurina especialmente quando (re-)iniciado com tratamento de midostaurina (vide seção "Interações Medicamentosas"). Medicamentos alternativos que não inibam fortemente a atividade de CYP3A4 devem ser considerados. Em situações nas quais não existam alternativas terapêuticas satisfatórias, os pacientes devem ser cuidadosamente monitorados para toxicidade relacionada a midostaurina.
Excipientes
FLYKATO® contém óleo de rícino hidrogenado etoxilado, que pode causar desconforto no estômago e diarreia. Uma dose de 100 mg de FLYKATO® contém aproximadamente 14 % de álcool, o que corresponde a 333 mg de álcool. Isso equivale a 8,4 mL de cerveja ou 3,5 mL de vinho. O álcool pode ser prejudicial em pacientes com problemas relacionados ao álcool, epilepsia ou problemas hepáticos ou durante a gravidez ou amamentação.
Este medicamento contém 14% de álcool (etanol) e pode causar intoxicação, especialmente em crianças.
Atenção: Contém os corantes dióxido de titânio, óxido de ferro amarelo que podem, eventualmente, causar reações alérgicas.
Fertilidade, gravidez e lactação
Mulheres em idade fértil
As mulheres em idade fértil devem ser informadas de que estudos com animais demonstraram que a midostaurina é prejudicial ao feto em desenvolvimento. As mulheres sexualmente ativas em idade fértil são aconselhadas a fazerem um teste de gravidez antes de iniciar o tratamento com FLYKATO® e a utilizarem contraceptivo eficaz (métodos que resultem em taxas de gravidez inferiores a 1%) quando estiver tomando FLYKATO® e durante pelo menos 4 meses após a interrupção do tratamento.
Gravidez
A midostaurina pode causar dano fetal quando administrado a grávidas. Não existem estudos adequados e bem controlados em grávidas. Estudos reprodutivos em ratos e coelhos demonstraram que a midostaurina induziu a fetotoxicidade (vide seção "Dados de segurança pré-clínica"). FLYKATO® não é recomendado durante a gravidez ou em mulheres em idade fértil que não utilizam contracepção. As grávidas devem ser avisadas do risco potencial para o feto.
Amamentação
Desconhece-se se a midostaurina ou os seus metabólitos ativos são transferidos para o leite humano. Dados animais disponíveis mostraram que a midostaurina e os seus metabólitos ativos passam para o leite de ratos lactantes.
A amamentação deve ser descontinuada durante o tratamento com FLYKATO® e durante, pelo menos, 4 meses após a interrupção do tratamento.
Uso contraindicado no aleitamento ou na doação de leite humano.
Fertilidade
Não há dados sobre o efeito de midostaurina na fertilidade humana. Estudos com midostaurina em animais demonstraram diminuição da fertilidade (vide seção "Dados de segurança pré-clínica").
Este medicamento pertence à categoria D de risco à gravidez e não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Efeitos sobre a capacidade de conduzir veículos e utilizar máquinas
Foram relatados casos de tontura e vertigem em pacientes que tomaram midostaurina, o que deve ser considerado ao avaliar a capacidade do paciente de conduzir veículos ou utilizar máquinas.
Monitoramento do tratamento em pacientes com MSA, MS-ADHNM e LCM
A avaliação de resposta pode ser feita a partir de exame físico, radiografia de tórax, tomografia computadorizada ou ressonância magnética de tórax/abdome/pelve, inventário ósseo, hemograma, bioquímica e coagulograma.