FIPRIMA
EUROFARMA
filgrastim
Estimulante de granulócitos.
Apresentações.
Embalagem com 1 seringa preenchida com dispositivo de segurança com 0,5 mL de solução injetável contendo 30 milhões de unidades (MU) ou 300 mg de filgrastim (G-CSF).
VIA INTRAVENOSA OU SUBCUTÂNEA
USO ADULTO
Composição.
Cada seringa preenchida contém: filgrastim 30 MU (300 mg); excipientes* q.s.p. 0,5 mL *Excipientes: ácido acético glacial, acetato de sódio tri-hidratado, sorbitol, polissorbato 80 e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Fiprima (filgrastim) está indicado para redução da duração da neutropenia e da incidência da neutropenia febril em pacientes com neoplasias não mieloides tratados com quimioterapia citotóxica estabelecida. Fiprima (filgrastim) está indicado para redução da duração da neutropenia e suas sequelas clínicas em pacientes submetidos à terapia mieloablativa seguida de transplante de medula óssea.
2. RESULTADOS DE EFICÁCIA
Realizou-se um estudo de fase III, de não inferioridade, randomizado, multicêntrico e aberto, em pacientes portadoras de câncer de mama com indicação de quimioterapia em dose plena (esquemas TAC: docetaxel 75 mg/m2, doxorrubicina 50 mg/m2 e ciclofosfamida 500 mg/m2; ou AT: docetaxel 75 mg/m2 e doxorrubicina 60 mg/m2). O desfecho primário foi taxa de neutropenia de grau 4 após o primeiro ciclo de quimioterapia. Os desfechos secundários foram taxa de neutropenia febril, taxa de neutropenia de qualquer grau, duração da neutropenia de grau 4, frequência de eventos adversos e de alterações laboratoriais. A análise primária de eficácia demonstrou a não inferioridade de Fiprima (filgrastim) em relação ao produto biológico comparador¹. Não houve diferença estatisticamente significativa entre os grupos em relação à taxa de neutropenia de grau 4 após o primeiro ciclo de quimioterapia. A incidência da neutropenia de grau 4 foi de 51,16% no grupo que recebeu Fiprima (filgrastim) e de 51,19% no grupo que recebeu o produto biológico comparador¹ (p=0,9971).
A contagem do número de leucócitos feita entre o primeiro e o décimo dia do estudo demonstra a evolução do comportamento leucocitário. A curva de recuperação dos neutrófilos pode ser visualizada no gráfico da Figura 1.
Em relação aos desfechos secundários e à análise de segurança, os dois tratamentos do estudo mostraram-se semelhantes.
A taxa de neutropenia febril foi definida como a razão entre o número de pacientes que apresentaram neutropenia febril e o número total de pacientes em cada grupo do estudo. A taxa de neutropenia febril foi de 3,67% no grupo que recebeu Fiprima (filgrastim) e de 2,78% no grupo que recebeu o produto biológico comparador¹ (p=0,710).
A taxa de neutropenia de qualquer grau foi definida como a razão entre o número de pacientes que apresentaram neutropenia de qualquer grau [1 a 5] e o número total de pacientes em cada grupo do estudo. A taxa de neutropenia de qualquer grau foi de 90,8% no grupo que recebeu Fiprima (filgrastim) e de 86,1% no grupo que recebeu o produto biológico comparador¹ (p=0,2768).
A comparação dos dois grupos de tratamento em relação à duração da neutropenia de grau 4 não mostrou diferença estatisticamente significativa entre a duração da neutropenia e a filgrastima utilizada (p=0,5436). Considerando duração de neutropenia até 2 dias, a taxa de duração da neutropenia de grau 4 foi de 89,1% no grupo que recebeu Fiprima (filgrastim) e de 92,9% no grupo que recebeu o produto biológico comparador¹. Na análise da taxa de duração da neutropenia de grau 4 entre 2 e 4 dias, os números foram 10,9% no grupo que recebeu Fiprima (filgrastim) e de 7,1% no grupo que recebeu o produto biológico comparador¹. Na população de segurança, 206 pacientes tiveram registro de evento adverso leve e moderado durante o estudo, sendo 94,4% do braço Fiprima (filgrastim) e 95,5% no grupo que recebeu o produto biológico comparador1. Os registros de neutropenia não foram contabilizados como eventos adversos, uma vez que neutropenia era desfecho do estudo e foi, portanto, analisada separadamente nas análises de eficácia.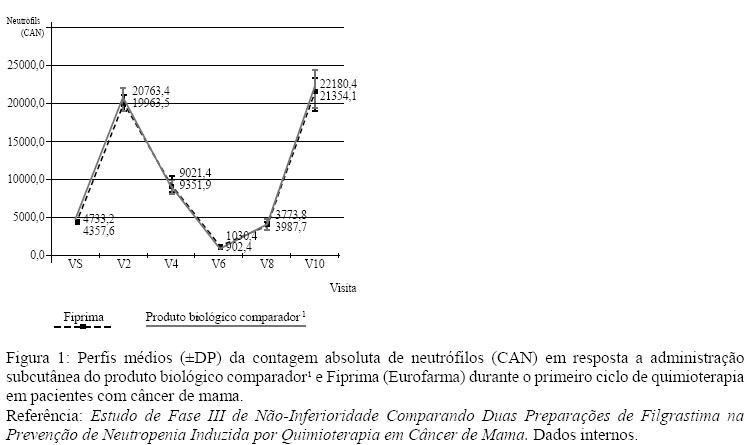
¹Produto biológico comparador (medicamento comparador) -Granulokine® fabricado por Produtos Roche Químicos e Farmacêuticos S.A., registrado na ANVISA sob n° 1.0100.0541.
3. CARACTERÍSTICAS FARMACOLÓGICAS
O fator estimulador de colônias de granulócitos (G-CSF) é uma glicoproteína que regula a produção e liberação dos neutrófilos funcionais da medula óssea. Filgrastim (r-metHuG-CSF), a substância ativa de Fiprima (filgrastim), é a proteína G-CSF recombinante humana altamente purificada, não-glicosilada, produzida em laboratório através de uma cepa de Escherichia coli manipulada geneticamente mediante a inclusão de um gene para o fator estimulador de colônias de granulócitos. Filgrastim provoca aumento evidente na contagem de neutrófilos no sangue periférico em vinte e quatro horas, com elevações mínimas dos monócitos. Em alguns pacientes com neutropenia crônica severa, pode induzir um leve aumento no número de eosinófilos e basófilos circulantes e, alguns destes pacientes podem também apresentar eosinofilia ou basofilia anterior ao tratamento. O aumento da contagem dos neutrófilos é dose-dependente na dose recomendada. Os neutrófilos produzidos em resposta à filgrastim apresentam função normal ou aumentada como demonstrado em testes de função quimiotáxica e fagocitária.
Farmacocinética
Após administração subcutânea, filgrastim é rapidamente absorvido e o pico da concentração sérica é atingido de 2 a 8 horas após a aplicação. A meia-vida de eliminação após administração intravenosa ou subcutânea é de, normalmente, 2 e 4 horas. Depuração e meia-vida são dependentes da dose e da contagem de neutrófilos.
Quando a depuração mediada por neutrófilos está saturada por concentrações elevadas de filgrastim ou diminuída pela neutropenia, a via da depuração linear predomina, e a farmacocinética parece linear. A biodisponibilidade absoluta após administração de filgrastim via subcutânea é estimada em 62% para uma dose de 375 mg e 72% para uma dose de 750 mg. Após descontinuação da dose, concentrações de filgrastim diminuem até chegarem a concentrações endógenas dentro de 24 horas.
Diminuição na concentração sérica de filgrastim é evidenciada depois de aplicações múltiplas em pacientes saudáveis e em pacientes com câncer, antes de quimioterapia. Este aumento da depuração de filgrastim é dose dependente e a magnitude do aumento parece estar intimamente relacionada ao grau de neutrofilia nos receptores, o que é consistente ao aumento da depuração de neutrófilos pelo aumento de sua quantidade. Em pacientes recebendo filgrastim após quimioterapia, a concentração sérica é mantida em um platô até o princípio da recuperação do número de neutrófilos.
Existe uma correlação linear positiva entre a dose e a concentração sérica do filgrastim, seja administrado por via intravenosa ou subcutânea. Após administração subcutânea das doses recomendadas, concentrações séricas acima de 10 ng/mL foram mantidas por 8 a 16 horas, com picos séricos de concentração de 118 ng/mL. O volume de distribuição no sangue é de aproximadamente 150 mL/kg.
A depuração do filgrastim mostrou seguir a farmacocinética de primeira ordem após administração subcutânea e intravenosa. A meia-vida de eliminação sérica de filgrastim é de aproximadamente 3,5 horas com velocidade de depuração de aproximadamente 0,6 mL/kg/min.
A infusão contínua de filgrastim por um período de até 28 dias, em pacientes recuperando-se de transplante autólogo de medula óssea, não resultou em evidência de acúmulo da droga e as meias-vidas de eliminação foram comparáveis.
Segurança pré-clinica:
Carcinogenicidade
O potencial de carcinogenicidade de Fiprima (filgrastim) não foi estudado. Filgrastim não induziu mutações genéticas em bactérias, tanto na presença como na ausência de um sistema enzimático metabolizador do medicamento.
Demonstrou-se que certas células malignas expressam receptores para o fator estimulador de colônias de granulócitos (G-CSF). A possibilidade de que filgrastim possa atuar como fator de crescimento de algum tipo de tumor não pode ser excluída.
Comprometimento da fertilidade
Não foi observado efeito de filgrastim sobre a fertilidade de ratos (machos ou fêmeas) nem sobre a gravidez em doses até 500 mg/kg.
Teratogenicidade
Não há evidências, a partir de estudos realizados em ratos e coelhos, de que filgrastim seja teratogênico. Aumento na incidência da perda embrionária foi observado em coelhos, mas não foi observada nenhuma máformação.
Toxicidade aguda
Fiprima (filgrastim) foi submetido a testes de toxicidade aguda. O objetivo do estudo foi obter informações sobre possíveis efeitos adversos do Fiprima (filgrastim) administrado em dose aguda (10 vezes maior que a maior dose usada para humanos) em duas vias, subcutânea e endovenosa. Foram usados 48 ratos da linhagem Wistar (Rattus novergicus). A administração não causou morte em nenhum dos animais e não foram observadas alterações macroscópicas ou histológicas. Dois estudos de toxicidade crônica foram realizados. Os resultados confirmaram a segurança do Fiprima (filgrastim) em concentrações até 100 vezes maiores que as doses clínicas. A atividade dos neutrófilos produzidos pelo Fiprima (filgrastim) se mostrou normal.
4. CONTRAINDICAÇÕES
Fiprima (filgrastim) não deve ser administrado em pacientes com conhecida hipersensibilidade ao produto ou aos seus componentes.
Fiprima (filgrastim) não deve ser administrado em pacientes portadores de neutropenia congênita grave (Síndrome de Kostman) com citogenética anormal.
Fiprima (filgrastim) não deve ser utilizado para aumentar a dose de terapia citotóxica além do regime estabelecido.
Este medicamento é contraindicado para uso por pacientes pediátricos.
Categoria C de risco na gravidez: Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
5. ADVERTÊNCIAS E PRECAUÇÕES
Não foram estudados os efeitos do Fiprima (filgrastim) em pacientes com redução substancial dos progenitores mieloides. Filgrastim atua primariamente nos precursores neutrofílicos para exercer seu efeito no aumento do número dos neutrófilos. Portanto, em pacientes com redução de precursores (nos casos tratados com radioterapia extensa ou quimioterapia ou aqueles com infiltração na medula óssea por tumor) a resposta neutrofílica pode estar diminuída.
O tamanho do baço deve ser cuidadosamente monitorado (por exemplo, através de exame clínico e ultrassonográfico). Casos de ruptura esplênica foram reportados com pouca frequência depois da administração de filgrastim, sendo que alguns desses casos foram fatais.
Recomenda-se atenção médica especial em pacientes com anemia falciforme uma vez que crises de falcização podem ocorrer em pacientes falcêmicos em uso de filgrastim. Alguns casos foram relatados como fatais. É de extrema importância a avaliação cuidadosa dos potenciais riscos e benefícios.
Trombocitopenia foi frequentemente relatada em pacientes recebendo filgrastim. A contagem de plaquetas deve ser cuidadosamente monitorada.
O efeito do Fiprima (filgrastim) na doença do enxerto versus hospedeiro (GvHD) não foi definido.
O monitoramento da densidade óssea pode ser indicado aos pacientes portadores de doenças osteoporóticas subjacentes submetidos à terapêutica com Fiprima (filgrastim) por mais de 6 meses.
Fiprima (filgrastim) contém sorbitol como excipiente. Nos casos raros de pacientes com intolerância hereditária à frutose (HFI), esse medicamento não deve ser utilizado.
O início de sinais pulmonares (como tosse, febre e dispneia), em associação a sinais radiológicos de infiltrados pulmonares e deterioração da função pulmonar, pode corresponder a sinais preliminares da síndrome da angústia respiratória do adulto (SARA). Em tais circunstâncias, o uso de Fiprima (filgrastim) deve ser descontinuado e tratamento apropriado deve ser instituído.
Crescimento de células malignas
O fator estimulador de colônias de granulócitos pode promover o crescimento de células mieloides in vitro e efeitos semelhantes podem ser observados em algumas células não mieloides in vitro.
A segurança e eficácia da administração de filgrastim em pacientes com mielodisplasia, leucemia mieloide aguda ou leucemia mieloide crônica não foram estabelecidas. Portanto, devido à possibilidade de crescimento tumoral, Fiprima (filgrastim) deve ser administrado com extrema cautela em qualquer condição maligna com características mieloide. Estudos clínicos não estabeleceram ainda se filgrastim influencia a progressão de síndromes mielodisplásicas para leucemia mieloide aguda. Assim, extrema cautela deve ser tomada ao se administrar Fiprima (filgrastim) em qualquer condição mieloide pré-maligna. Deve-se ter particular atenção para distinguir o diagnóstico de transformação blástica da leucemia mieloide crônica do quadro de leucemia mieloide aguda.
Em virtude da existência limitada de dados de eficácia e segurança em pacientes com LMA secundária, Fiprima (filgrastim) deve ser administrado com cautela nesses pacientes. A segurança e a eficácia da administração de Fiprima (filgrastim) em pacientes com LMA de novo, com idade < 55 anos e citogenética favorável [t(8;21), t(15;17) e inv(16)] não foram estabelecidas.
Em pacientes recebendo quimioterapia citotóxica
Leucocitose
Contagem de leucócitos ≥ 100.000/mm3 foi observada em menos de 5% dos pacientes recebendo filgrastim em doses superiores a 0,3 MU/kg/dia (3 mg/kg/dia). Não foram relatados efeitos adversos diretamente atribuíveis a este grau de leucocitose. Contudo, devido aos riscos potenciais associados à leucocitose severa, contagens de leucócitos devem ser realizadas a intervalos regulares durante a terapêutica com Fiprima (filgrastim). Se a contagem leucocitária exceder 50.000/mm3 após o nadir esperado, Fiprima (filgrastim) deve ser imediatamente descontinuado.
Riscos associados com altas doses de quimioterapia
Cuidado especial deve ser tomado ao se tratar pacientes com quimioterapia de altas doses visto que uma melhor resolução tumoral não tem sido demonstrada; doses altas de agentes quimioterápicos podem levar a um aumento de toxicidade, incluindo efeitos cardíacos, pulmonares, neurológicos e dermatológicos (consultar informação sobre prescrição específica dos agentes quimioterápicos utilizados).
O tratamento com Fiprima (filgrastim) não exclui a possibilidade de trombocitopenia e anemia pela quimioterapia mielossupressora. Nestes pacientes, devido à possibilidade de receberem doses mais altas de quimioterapia (por exemplo, doses completas do esquema prescrito), existe maior risco de trombocitopenia e anemia. É recomendado monitoramento periódico do hematócrito e da contagem de plaquetas. Cautela especial deve ser adotada quando da administração de quimioterápicos, conhecidamente trombocitopênicos isolados ou em associação. Não foram realizados estudos com Fiprima (filgrastim) em pacientes com prejuízo severo das funções hepática e renal. Portanto, seu uso em pacientes destes grupos pode não ser recomendado.
Uso em idosos
Os estudos clínicos com filgrastimincluíram pequeno número de pacientes idosos. Estudos especiais não foram realizados neste grupo e, portanto, recomendações específicas de dosagem não podem ser feitas.
Uso em crianças
Não foram estabelecidas segurança e eficácia de Fiprima (filgrastim) em crianças.
Efeitos sobre a capacidade de dirigir e operar máquinas
Não foram relatados efeitos sobre a capacidade de dirigir e operar máquinas.
Gestação e lactação Categoria de risco na gravidez: C Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgiãodentista.
A segurança do Fiprima (filgrastim) não foi estabelecida em gestantes. Há relatos na literatura em que foi demonstrada a passagem transplacentária de filgrastim em gestantes. Estudos em animais mostraram toxicidade reprodutiva. Durante a gestação, o possível risco do uso de Fiprima (filgrastim) para o feto deve ser avaliado com relação aos benefícios terapêuticos esperados. Não se tem conhecimento da excreção do Fiprima (filgrastim) no leite materno. Fiprima (filgrastim) não é recomendado para o uso em lactantes.
Até o momento, não há informações de que Fiprima (filgrastim) possa causar doping.
6. INTERAÇÕES MEDICAMENTOSAS
A segurança e eficácia de Fiprima (filgrastim) administrado no mesmo dia da quimioterapia citotóxica mielossupressora não foram estabelecidas. Tendo em vista a sensibilidade das células mieloides de rápida divisão à quimioterapia citotóxica mielossupressora, o uso de Fiprima (filgrastim) não é recomendado no período de 24 horas antes até 24 horas subsequentes à quimioterapia. Evidência preliminar a partir de um número pequeno de pacientes tratados concomitantemente com filgrastim e 5-fluorouracil indica que a severidade da neutropenia pode ser exacerbada. Não foram investigadas possíveis interações de filgrastim com outros fatores de crescimento hematopoiéticos e com citocinas. Considerando que o lítio promove a liberação de neutrófilos, seu uso pode potencializar o efeito de Fiprima (filgrastim). Embora esta interação não tenha sido formalmente investigada, não há evidências de que seja prejudicial. A atividade hematopoiética aumentada da medula óssea em resposta à terapia com fator de crescimento tem sido associada a alterações temporárias de imagens ósseas, o que deve ser considerado na interpretação dos resultados de exames de imagem.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Fiprima (filgrastim) deve ser armazenado sob refrigeração, entre 2°C e 8°C e protegido da luz. O produto deve ser mantido sempre dentro de sua embalagem secundária até o momento do uso. Fiprima (filgrastim) não pode ser congelado (temperatura de congelamento do produto: -11°C).
Após diluição, manter sob refrigeração entre 2°C e 8°C por até 24 horas. Soluções diluídas de Fiprima (filgrastim) não devem ser preparadas mais de 24 horas antes da administração.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido.
Guarde-o em sua embalagem original.
Fiprima (filgrastim) é uma solução límpida, incolor a levemente amarelada e isenta de partículas visíveis.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Fiprima (filgrastim) deve ser administrado em injeção subcutânea diária ou em infusão intravenosa diária diluída (vide item "diluição").
Antes da administração, a solução deve ser inspecionada quanto à presença de partículas visíveis. A solução deve ser injetada apenas se estiver incolor a levemente amarelada e isenta de partículas visíveis. Evite agitar vigorosamente.
Fiprima (filgrastim) em seringa preenchida é para uso único.
DILUIÇÃO
Se necessário, Fiprima (filgrastim) pode ser diluído em solução glicosada a 5%.
Para aqueles pacientes tratados com Fiprima (filgrastim) diluído a uma concentração entre 0,5 MU/mL (5 mg/mL) e 1,5 MU/mL (15 mg/mL), deve-se adicionar albumina sérica humana para proteção da adsorção de materiais como plásticos até a concentração final de 2 mg/mL. Por exemplo, para o volume de injeção final de 20 mL, doses totais de Fiprima (filgrastim) inferiores a 30 MU (300 mg) devem ser administradas com 0,2 mL de solução de albumina humana a 20%.
Diluições a uma concentração final inferior a 5 mg/mL não são recomendadas em nenhuma eventualidade.
Incompatibilidades
Fiprima (filgrastim) não deve ser diluído em soluções salinas (soro fisiológico).
Fiprima (filgrastim) diluído pode ser adsorvido em materiais plásticos ou vidros. Contudo, quando diluído corretamente, Fiprima (filgrastim) é compatível com vidro e uma variedade de materiais plásticos, incluindo PVC, poliolefina (copolímero do polipropileno e polietileno) e polipropileno.
POSOLOGIA
Quimioterapia citotóxica estabelecida
A dose recomendada de Fiprima (filgrastim) é de 0,5 MU/kg/dia (5 mg/kg/dia). A primeira dose de Fiprima (filgrastim) não deve ser administrada em menos de 24 horas após a quimioterapia citotóxica.
Fiprima (filgrastim) deve ser administrado em injeção subcutânea diária ou em infusão endovenosa diária, diluída em solução glicosada a 5%, durante 30 minutos (vide "Diluição").
A administração diária de Fiprima (filgrastim) deve continuar até que o nadir esperado de neutrófilos seja ultrapassado e a contagem dos neutrófilos tenha retornado a valores normais. Espera-se que a duração do tratamento necessária para preencher estes critérios seja de até 14 dias, dependendo do tipo, dose e esquema quimioterápico citotóxico utilizados.
Em pacientes sob quimioterapia citotóxica, uma elevação transitória da contagem de neutrófilos é tipicamente observada 1 a 2 dias após iniciada a terapêutica com Fiprima (filgrastim). A descontinuação prematura da terapêutica com Fiprima (filgrastim), antes do período do nadir neutrofílico esperado, não é recomendada.
Pacientes tratados com terapia mieloablativa seguida de transplante da medula óssea
A dose inicial recomendada de Fiprima (filgrastim) é de 1,0 MU/kg/dia (10 mg/kg/dia) administrada em 30 minutos ou 24 horas por infusão endovenosa, ou 1,0 MU/kg/dia (10 mg/kg/dia) administrada em 24 horas, de maneira contínua, por via subcutânea. Fiprima (filgrastim) deve ser diluído em 20 mL de solução glicosada a 5% (vide item "diluição").
A primeira dose de Fiprima (filgrastim) não deve ser administrada nas 24 horas seguintes à quimioterapia citotóxica, mas sim dentro das 24 horas após a infusão da medula óssea. A eficácia e a segurança da administração de Fiprima (filgrastim) por mais de 28 dias ainda não foram estabelecidas. Uma vez ultrapassado o nadir neutrofílico, a dose diária de Fiprima (filgrastim) deve ser titulada de acordo com a resposta neutrofílica, como segue: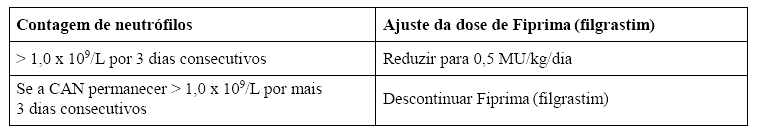
CAN = Contagem absoluta de neutrófilos.
Se a CAN diminuir para < 1,0 x 109/L durante o período de tratamento, a dose de Fiprima (filgrastim) deve ser re-escalonada de acordo com as etapas acima.
Filgrastim tem demonstrado eficácia e boa tolerabilidade neste contexto em doses até 70 mg/kg/dia.
Instruções especiais de dosagens
Idosos: estudos clínicos com filgrastim incluíram número pequeno de pacientes idosos, mas estudos especiais não foram realizados nesse grupo, portanto, recomendações específicas de dosagem não podem ser feitas.
9. REAÇÕES ADVERSAS
Em pacientes com câncer
As reações adversas relatadas a seguir foram obtidas de dados de estudos clínicos publicados e têm sido observadas em pacientes em utilização de filgrastim.
Em estudos clínicos randomizados, placebo controlados, filgrastim não aumentou a incidência dos eventos clínicos adversos associados à quimioterapia citotóxica. Os eventos adversos são relatados com igual frequência em pacientes tratados com filgrastim e quimioterapia ou placebo e quimioterapia e incluíram náusea e vômitos, alopecia, diarreia, fadiga, anorexia, mucosite, cefaleia, tosse, rash cutâneo, dor torácica, fraqueza generalizada, dor de garganta, obstipação e dor inespecífica.
Sintomas sugestivos de reações tipo alérgicas têm sido reportados e aproximadamente metade desses casos estava relacionado com a dose inicial. Em geral, os relatos foram mais comuns após administração intravenosa. Em alguns casos, a retomada da medicação resultou em recorrência dos sintomas.
A administração de filgrastim nas doses recomendadas está frequentemente associada com dor musculoesquelética, especificamente em ossos medulares. Em geral, a dor é discreta ou moderada (10%), mas ocasionalmente severa (3%), e é geralmente controlada com analgésicos comuns.
Efeitos adversos menos frequentes incluíram anormalidades urinárias (predominantemente disúria leve ou moderada).
Hipotensão transitória, sem necessidade de tratamento clínico, foi ocasionalmente relatada.
Distúrbios vasculares (por exemplo, doença veno-oclusiva e distúrbios do volume hídrico) foram ocasionalmente relatados em pacientes submetidos à quimioterapia de altas doses pós-transplante autólogo de medula óssea. A associação causal com filgrastim não foi estabelecida.
Foram relatados casos muito raros de vasculite cutânea em pacientes tratados com filgrastim. O mecanismo de vasculite em pacientes recebendo filgrastim é desconhecido.
A ocorrência de Síndrome de Sweet (dermatite febril aguda) foi ocasionalmente relatada.
Exacerbação de artrite reumatoide foi observada em casos individuais.
Foram relatados eventos adversos pulmonares raros, incluindo pneumonia intersticial, edema pulmonar e infiltrado pulmonar em alguns casos, como resultado de insuficiência respiratória ou síndrome de angústia respiratória do adulto (SARA), que pode ser fatal.
Elevações leves ou moderadas, dose-dependentes e reversíveis de ácido úrico, fosfatase alcalina, gamaglutamiltransferase (GGT) e desidrogenase lática (DHL) podem ocorrer com frequência.
A tabela a seguir apresenta a frequência das reações adversas observadas em pacientes com câncer submetidos à quimioterapia citotóxica: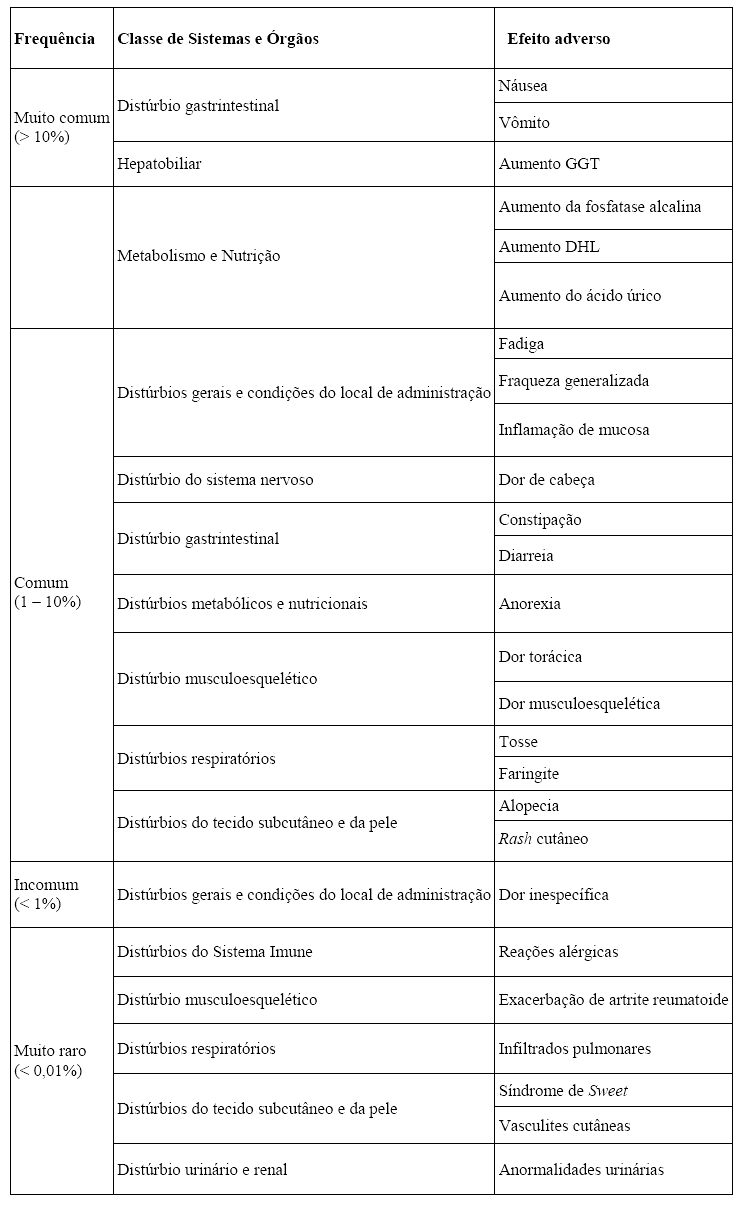
Outros eventos relatados com uso de filgrastim:
Distúrbios do sistema imunológico
- Reações alérgicas incluindo anafilaxia, rash cutâneo e urticária, podem ocorrer no tratamento inicial ou subsequente em pacientes recebendo filgrastim. Em alguns casos, os sintomas ocorreram com a reexposição ao medicamento, sugerindo uma relação causal entre medicamento e efeito.
- Reações alérgicas ao filgrastim foram raramente reportadas. Filgrastim deve ser permanentemente descontinuado em pacientes com experiência de reação alérgica séria.
Distúrbios do sangue e sistema linfático
- Casos isolados de crise de falcização, em alguns casos fatais, foram relatados em pacientes com anemia falciforme.
- Casos de esplenomegalia foram frequentemente relatados (≥ 1% e < 10%) em pacientes tratados com filgrastim.
- Casos de ruptura de baço foram relatados com pouca frequência (≥ 0,1% e < 1%) em doadores normais e pacientes recebendo filgrastim e outros G-CSFs (vide item 5 -"Advertências e precauções").
Distúrbios musculoesqueléticos
- Eventos de pseudogota foram relatados muito raramente (cerca de 0,03 casos por 100.000 exposições ou 0,00003%) em pacientes com câncer tratados com filgrastim.
Distúrbios da pele e tecidos subcutâneos
- Foram relatados raramente (≥ 0,01% e < 0,1%) casos de Síndrome de Sweet (dermatite febril aguda).
- Reações de vasculite cutânea foram relatadas raramente (cerca de 1 caso por 100.000 exposições ou 0,001%) em pacientes com câncer que receberam filgrastim.
Anormalidades laboratoriais
Elevações leves a moderadas e reversíveis de ácido úrico, fosfatase alcalina e desidrogenase lática, sem associação com efeitos clínicos, foram observadas em pacientes recebendo filgrastim após quimioterapia citotóxica.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária -NOTIVISA, disponível em http://www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Os efeitos de doses excessivas de Fiprima (filgrastim) não foram estabelecidos.
Doses de até 138 mg/kg/dia de filgrastim foram administradas aos pacientes em estudos de transplante de medula óssea (BMT) sem efeitos tóxicos.
A descontinuação da terapêutica com Fiprima (filgrastim), em geral, resulta na queda de 50% dos neutrófilos circulantes em 1 a 2 dias, com retorno aos níveis normais em 1 a 7 dias.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
M.S.: 1.0043.1165
Uso restrito a hospitais.
Venda sob prescrição médica.
Esta bula foi aprovada pela ANVISA em 20/10/2015.