FABHALTA
NOVARTIS
iptacopana, cloridrato monoidratado
Trat. da hemoglobinúria paroxística noturna.
Apresentações.
Fabhalta® 200 mg - embalagens contendo 56 cápsulas duras.
VIA ORAL
USO ADULTO
Composição.
Cada cápsula dura de Fabhalta® contém 200 mg de iptacopana (na forma de 225,8 mg de cloridrato de iptacopana monoidratada). Excipientes:
Gelatina dura, óxido de ferro vermelho, dióxido de titânio, óxido de ferro amarelo, óxido de ferro preto, solução concentrada de amônia, hidróxido de potássio, propilenoglicol e goma-laca.
Informações técnicas.
1. INDICAÇÕES
Fabhalta® é indicado para o tratamento de pacientes adultos com hemoglobinúria paroxística noturna (HPN).
2. RESULTADOS DE EFICÁCIA
A eficácia e a segurança de Fabhalta® em pacientes adultos com HPN foram avaliadas em dois estudos de Fase 3 multicêntricos, abertos, de 24 semanas: um estudo controlado por comparador ativo (APPLY-PNH; NCT04558918) e um estudo de braço único (APPOINT-PNH; NCT04820530)[4].
APPLY-PNH: tratamento com anti-C5 em pacientes com HPN
O APPLY-PNH incluiu pacientes adultos com HPN e anemia residual (hemoglobina < 10 g/dL) que receberam tratamento anterior com um regime estável de tratamento com anti-C5 (eculizumabe ou ravulizumabe) por pelo menos 6 meses antes da randomização.[4]
Noventa e sete pacientes foram randomizados na proporção de 8:5 para receber Fabhalta® 200 mg por via oral duas vezes ao dia (n=62) ou para continuar o tratamento com anti-C5 (eculizumabe n=23 ou ravulizumabe n=12) durante todo o período controlado randomizado (RCP) de 24 semanas. A randomização foi estratificada com base no tratamento anterior com anti-C5 e no histórico de transfusões nos últimos 6 meses. Após a conclusão do RCP de 24 semanas, todos os pacientes eram elegíveis para inclusão em um período de extensão de tratamento de 24 semanas e para receber Fabhalta® em monoterapia. Posteriormente, os pacientes foram elegíveis para entrar em um estudo de extensão de longo prazo separado. [4]
Os pacientes precisavam ser vacinados contra Neisseria meningitidis e recomendou-se que fossem vacinados contra Streptococcus pneumoniae e Haemophilus influenzae tipo B. Se o paciente não tivesse sido vacinado anteriormente ou se fosse necessário um reforço, a vacina era administrada pelo menos 2 semanas antes da primeira dose. Se o tratamento com Fabhalta® fosse iniciado antes de 2 semanas após a vacinação, a profilaxia com medicamentos antibacterianos era administrada.[5]
Os dados demográficos e as características basais da doença foram bem equilibradas entre os grupos de tratamento (vide Tabela 1). [5] O tempo médio de tratamento anterior com anti-C5 foi de 3,8 e 4,2 anos para os grupos com Fabhalta® e anti-C5, respectivamente. O tamanho médio basal do clone HPN em hemácias (Tipo II + III) foi de 64,6% para o grupo com Fabhalta® e 57,4% para o grupo com anti-C5. A hemoglobina média basal foi de 8,9 g/dL para ambos os grupos, sendo que aproximadamente 57% e 60% dos pacientes necessitaram de transfusão nos 6 meses anteriores à randomização, nos grupos com Fabhalta® e anti-C5, respectivamente. O nível médio de lactato desidrogenase (LDH) basal foi de 269,1 U/L para o grupo com Fabhalta® e 272,7 U/L para o grupo com anti-C5. Havia 19,4% e 28,6% de pacientes com histórico de eventos adversos vasculares graves (MAVEs) nos grupos com Fabhalta® e anti-C5, respectivamente. [4,5,8]
Durante o RCP, uma paciente no grupo Fabhalta® descontinuou o tratamento devido à gravidez; nenhum paciente no grupo anti-C5 descontinuou.[5]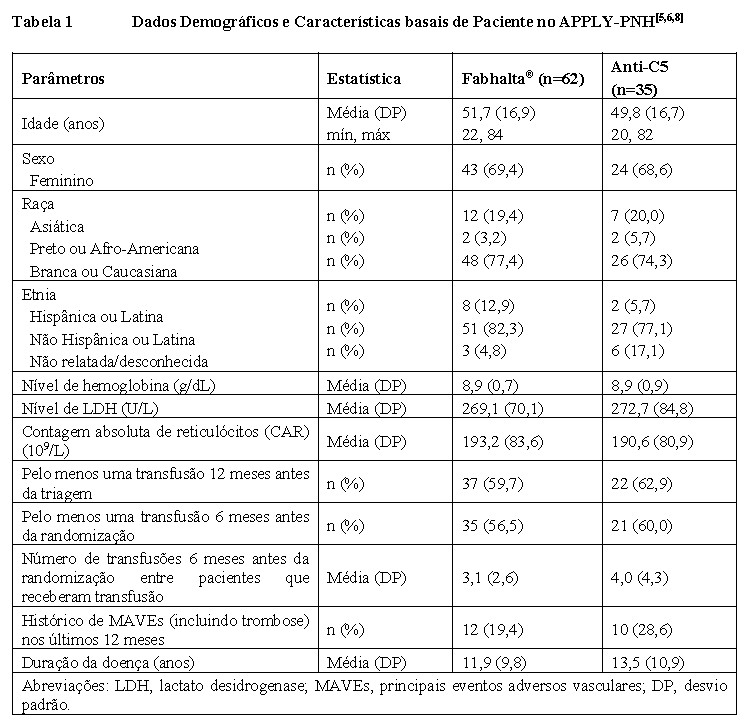
A eficácia baseou-se em dois desfechos primários para demonstrar a superioridade de Fabhalta® em relação ao anti-C5 na obtenção de resposta hematológica após 24 semanas de tratamento, sem necessidade de transfusão, avaliando a proporção de pacientes que demonstraram: 1) aumento sustentado de ≥ 2 g/dL nos níveis de hemoglobina basal (melhora da hemoglobina) e/ou 2) níveis sustentados de hemoglobina ≥12 g/dL. Os desfechos secundários incluíram prevenção de transfusões, alteração em relação a linha de base nos níveis de hemoglobina, alteração em relação a linha de base nas pontuações da Avaliação Funcional da Terapia de Doenças Crônicas (FACIT)-Fadiga, ocorrência de hemólise de escape (breakthrough) clínica e alteração em relação a linha de base nas contagens absolutas de reticulócitos.[4]
Fabhalta® foi superior ao tratamento com anti-C5, com uma diferença significativa na taxa de resposta de 80,2% (82,2% vs 2%) para melhora da hemoglobina (aumento sustentado dos níveis de hemoglobina ≥ 2 g/dL em relação a linha de base) e 67% (68,8% vs 1,8%) para nível sustentado de hemoglobina ≥12 g/dL sem necessidade de transfusão de hemácias para ambos os desfechos primários, após 24 semanas de tratamento (p < 0,0001) (vide Tabela 2).[1,8]
No geral, mais pacientes alcançaram melhora da hemoglobina no grupo com Fabhalta® (51/60) em comparação com o grupo com anti-C5 (0/35) e hemoglobina sustentada ≥12 g/dL (42/60 no grupo com Fabhalta® em comparação com 0/35 no grupo com anti-C5) sem necessidade de transfusão de hemácias (vide Tabela 2).[1]
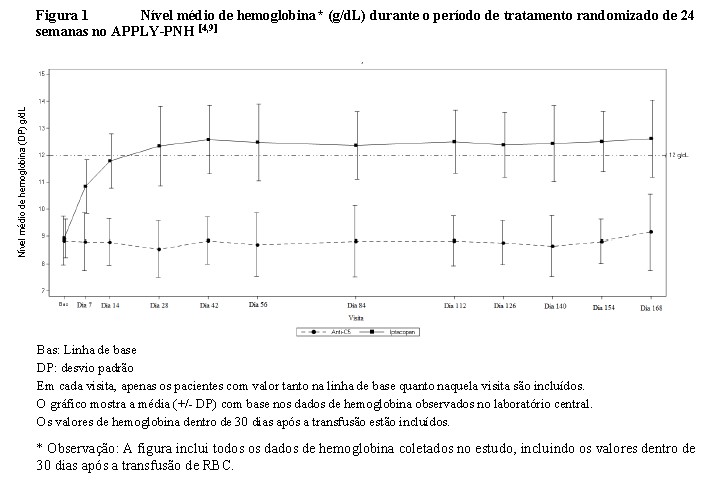
Fabhalta® também foi superior ao tratamento com anti-C5 em relação à taxa de prevenção de transfusão com uma diferença de tratamento de 68,9% (94,8% vs 25,9% (p < 0,0001)) e alteração em relação a linha de base no nível de hemoglobina (diferença de tratamento de +3,66 g/dL; p < 0,0001). O efeito do tratamento de Fabhalta® na hemoglobina foi observado logo no Dia 7 e mantido durante o estudo (consulte a Figura 1).[4,8]
Fabhalta® foi superior ao tratamento com anti-C5 na melhora da fadiga conforme avaliado pela FACIT-Fadiga (diferença de tratamento de +8,29 pontos; p < 0,001), e os pacientes tratados com Fabhalta® apresentaram melhoras clinicamente significativas na fadiga relatada pelo paciente em relação a linha de base (+8,59 pontos). Fabhalta® também foi superior ao tratamento com anti-C5 na taxa anualizada de hemólise de escape clínica (diferença de tratamento de 90%; p=0,01) e redução na contagem absoluta de reticulócitos em relação a linha de base (diferença de tratamento de -116,2x109/L; p < 0,0001) consistente com a inibição de hemólise extravascular (EVH).[4]
A proporção de LDH em relação a linha de base foi semelhante para ambos os grupos de tratamento, demonstrando que Fabhalta® manteve o controle da hemólise intravascular (IVH) após a descontinuação do tratamento com anti-C5 (vide Tabela 2).[4,8]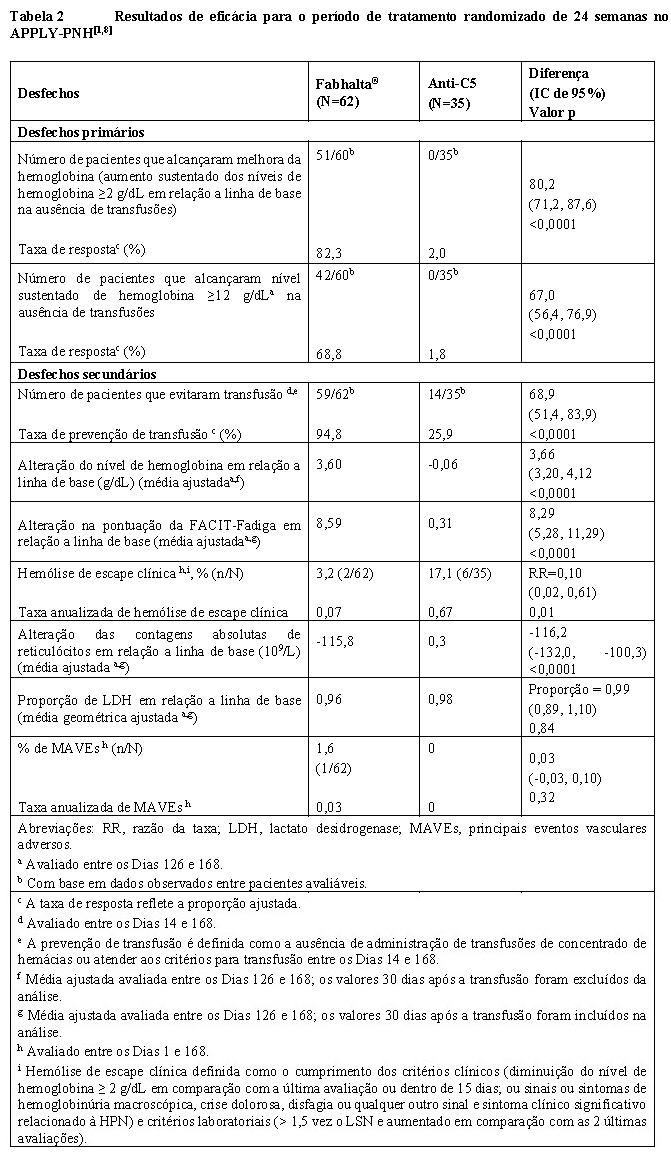
Os resultados para os desfechos primários foram consistentes nos subgrupos predefinidos estudados, incluindo duração da doença, idade, sexo, hemoglobina na linha de base, histórico de MAVEs, tratamento anterior com anti-C5 (eculizumabe ou ravulizumabe), necessidade de transfusão nos últimos 6 meses, número de transfusões nos últimos 6 meses ( < 2 ou ≥2), nível de LDH na linha de base e duração do tratamento anterior com anti-C5.[4]
Extensão do tratamento
Um total de 95 pacientes participantes do APPLY-PNH entraram no período de extensão do tratamento de 24 semanas, no qual todos os pacientes receberam Fabhalta®, resultando em uma exposição total de até 48 semanas. Os resultados de eficácia na semana 48 foram consistentes com os da semana 24 e demonstraram a eficácia sustentada do tratamento com Fabhalta®.
APPOINT-PNH: Estudo em pacientes virgens de tratamento com inibidor de complemento
O APPOINT-PNH estudou 40 pacientes adultos com HPN (tamanho do clone HPN ≥10%) com hemoglobina < 10 g/dL e LDH > 1,5 LSN, que não foram previamente tratados com um inibidor de complemento. Todos os 40 pacientes receberam Fabhalta® 200 mg por via oral duas vezes ao dia durante o período de tratamento principal aberto de 24 semanas. Posteriormente, os pacientes foram elegíveis para inclusão em um período de extensão de tratamento de 24 semanas e continuar a receber o Fabhalta®, seguido por um estudo de extensão de longo prazo separado.[4]
Os pacientes precisavam ser vacinados contra Neisseria meningitidis e recomendou-se que fossem vacinados contra Streptococcus pneumoniae e Haemophilus influenzae Tipo B. Se o paciente não tivesse sido vacinado anteriormente ou se fosse necessário um reforço, a vacina era administrada pelo menos 2 semanas antes ou até 2 semanas após a primeira dose. Se o tratamento com Fabhalta® fosse iniciado antes de 2 semanas após a vacinação, a profilaxia com medicamentos antibacterianos era administrada.[5]
A Tabela 3 mostra os dados demográficos e as características da doença na linha de base. Nenhum paciente descontinuou o período de tratamento principal do estudo.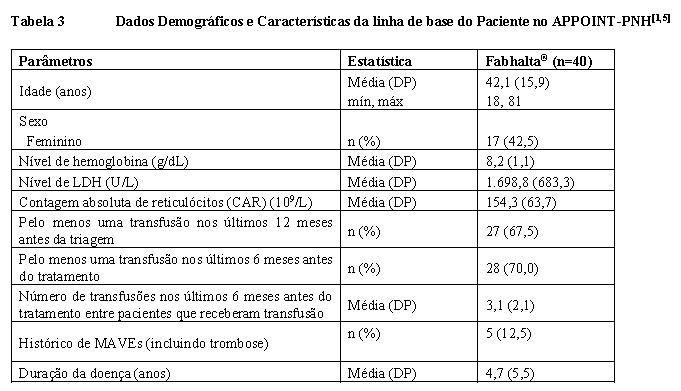
A eficácia baseou-se no desfecho primário que avalia o efeito do tratamento com Fabhalta® na proporção de pacientes que alcançaram melhora da hemoglobina (aumento sustentado de ≥2 g/dL nos níveis de hemoglobina em relação a linha de base, sem necessidade de transfusão de hemácias, após 24 semanas). Os desfechos secundários incluíam: hemoglobina sustentada ≥12 g/dL (sem necessidade de transfusão de hemácias) após 24 semanas, prevenção de transfusão, alteração em relação a linha de base nos níveis de hemoglobina, alteração em relação a linha de base nas pontuações da FACIT-Fadiga, ocorrência de hemólise de escape clínica e alteração em relação a linha de base nas contagens absolutas de reticulócitos.[4]
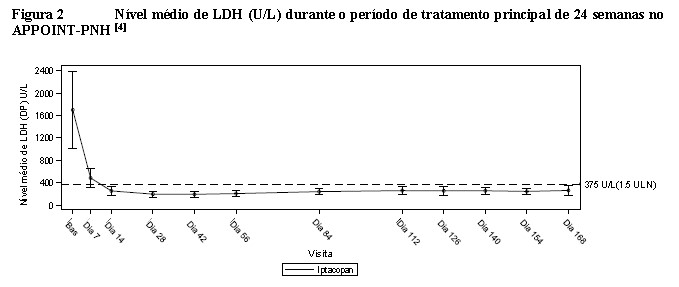
O tratamento com Fabhalta® resultou em uma taxa de resposta de 92,2% (IC de 95%: 82,5, 100,0) para melhora da hemoglobina, sem necessidade de transfusão de hemácias, após 24 semanas.[1] A taxa de resposta para pacientes que alcançaram hemoglobina ≥12 g/dL, sem necessidade de transfusão de hemácias, foi de 62,8% (IC de 95%: 47,5, 77,5). O tratamento com Fabhalta® levou a uma taxa de prevenção de transfusão de 97,6% (IC de 95%: 92,5, 100,0). Os pacientes tratados com Fabhalta® apresentaram melhora clinicamente significativa na fadiga relatada pelo paciente (alteração na pontuação da FACIT-Fadiga em relação a linha de base de +10,8; IC de 95%: 8,7, 12,8). Nenhum paciente apresentou hemólise de escape clínica ou MAVEs. Quando comparado com a linha de base, em pacientes tratados com Fabhalta®, os níveis de hemoglobina aumentaram 4,3 g/dL (IC de 95%: 3,9, 4,7), as contagens absolutas de reticulócitos alteraram -82,5 x 109/L (IC de 95%: -89,3; -75,6) e a mudança na porcentagem de LDH foi de -83,6% (IC de 95%: -84,9, -82,1) após 24 semanas.[1] O efeito do tratamento de Fabhalta® no LDH foi observado logo no Dia 7 e atingiu < 1,5 LSN no Dia 14, o que foi mantido durante o estudo. (Consulte a Tabela 4 e a Figura 2).[4]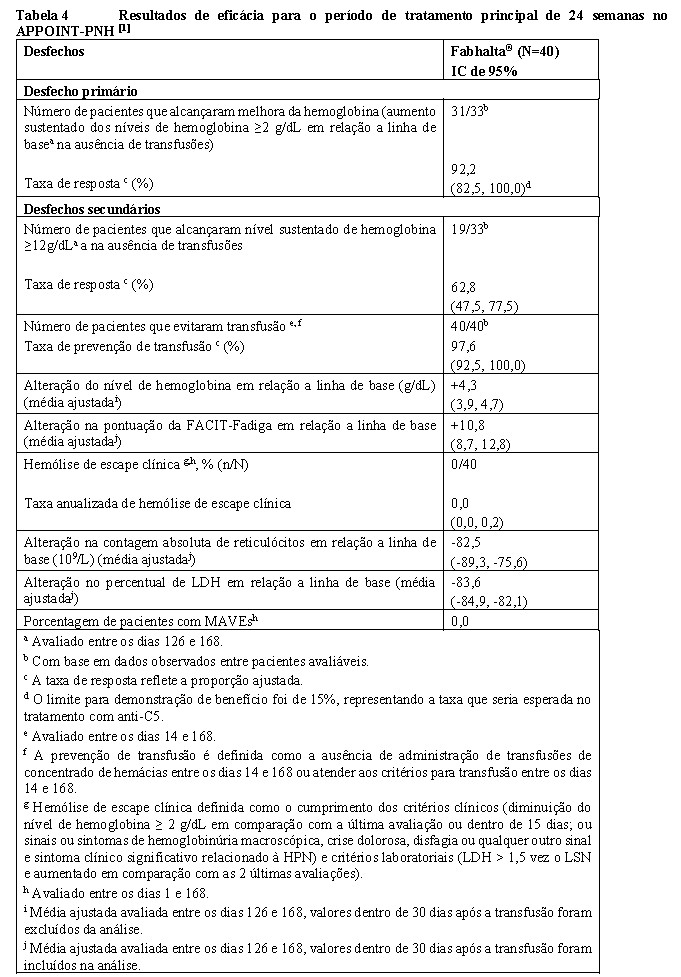
Os resultados para o desfecho primário foram consistentes nos subgrupos predefinidos examinados, incluindo duração da doença, idade, sexo, hemoglobina basal, histórico de MAVEs, necessidade de transfusão nos últimos 6 meses, número de transfusões nos últimos 6 meses ( < 2 ou ≥2). [1]
Extensão do tratamento
Todos os 40 pacientes participantes do estudo APPOINT-PNH entraram no período de extensão do tratamento de 24 semanas, onde todos os pacientes continuaram o tratamento com Fabhalta®, resultando em uma exposição total de até 48 semanas. Os resultados de eficácia semana 48 foram consistentes com os da semana 24 demonstrando a eficácia sustentada do tratamento com Fabhalta®.
Referências bibliográficas
1. [2.5 Visão Geral Clínica]. LNP023C1 (CTD 2.5 Visão geral clínica na hemoglobinúria paroxística noturna). Novartis. 29-Mar-2023.
2. [2.4 Visão Geral Não Clínica]. LNP023C1 (Iptacopana) - 2.4 Visão geral não clínica.Novartis. 21-Fev-2023.
3. [2.7.2 Resumo dos Estudos de Farmacologia Clínica] LNP023 (Iptacopana- CLPN023)- 2.7.2 Resumo dos Estudos de Farmacologia Clínica. Novartis. 02-Mar-2023.
4. [2.7.3 Resumo de Eficácia Clínica] LNP023C1 (Iptacopana) - 2.7.3 Resumo de Eficácia Clínica na hemoglobinúria paroxística noturna. Novartis. 10-Mar-2023.
5. [2.7.4 Resumo de Segurança Clínica] LNP023C1 (2.7.4 Resumo de Segurança Clínica na hemoglobinúria paroxística noturna. Novartis. 10-Mar-2023.
6.[Estudo CLNP023C12302]. Estudo aberto randomizado, multicêntrico, controlado por Compara dor ativo para avaliar a eficácia e a segurança do LNP023 oral, duas vezes ao dia, em pacientes adultos com HPN e anemia residual, apesar do tratamento com um anticorpo anti-C5 intravenoso. Novartis. 27-Fev-2023.
7. 2.5 Clinical Overview, Rationale for changes to Core Data Sheet (CDS) / Product Information - ADR section to remove PNH-REP data from frequency calculation. Novartis. 2023.
8. CLNP023C12302. Supplementary Clinical Study Report, v1.0 A randomized,
multicenter, active-comparator controlled, open-label trial to evaluate efficacy and safety of oral, twice daily LNP023 in adult patients with PNH and residual anemia, despite treatment with an intravenous anti-C5 antibody. Novartis. 06-Mar-2023
9. 2.5 Clinical Overview Rationale for changes to Core Data Sheet (CDS) / Product Information - Updates to APPLY-PNH (CLNP023C12302) analyses. Novartis. 14-Feb-2024
3. CARACTERÍSTICAS FARMACOLÓGICAS
Grupo farmacoterapêutico: Inibidores do complemento.
Código ATC: L04AJ08
Mecanismo de ação
Iptacopana é um inibidor do complemento proximal que tem como alvo o Fator B (FB) para inibir seletivamente a via alternativa, deixando intacta a sinalização direta da lectina e das vias clássicas. A inibição do FB impede a atividade da C3 convertase relacionada à via alternativa e a subsequente formação de C5 convertase.
Na HPN, a hemólise intravascular (IVH) é mediada pelo complexo de ataque à membrana (MAC) a jusante, enquanto a hemólise extravascular (EVH) é facilitada pela opsonização com fragmentos de C3. Iptacopana atua proximalmente na via alternativa da cascata do complemento para controlar tanto a EVH mediada por C3 quanto a IVH mediada por complexo terminal do complemento.
Farmacodinâmica
O início da inibição de biomarcadores da via alternativa do complemento, ensaio de via alternativa ex vivo e Bb plasmático (fragmento Bb de FB), foi ≤2 horas após uma dose única de iptacopana em voluntários saudáveis.
Em pacientes com HPN que recebem tratamento concomitante com anti-C5 e iptacopana 200 mg duas vezes ao dia, o ensaio de via alternativa ex vivo e o Bb plasmático diminuíram em relação ao valor de linha de base em 54,1% e 56,1%, respectivamente, na primeira observação no Dia 8. Em pacientes com HPN virgens de tratamento, esses mesmos biomarcadores diminuíram em relação ao valor de linha de base em 78,4% e 58,9%, respectivamente, na primeira observação após 4 semanas de tratamento com iptacopana 200 mg duas vezes ao dia.
Em pacientes com HPN em tratamento concomitante com anti-C5 e iptacopana 200 mg duas vezes ao dia, o tamanho médio do clone HPN de hemácias (RBC) foi de 54,8% na linha de base e aumentou para 89,2% após 13 semanas; a proporção de RBCs de HPN Tipo II + III com depósito de C3 foi de 12,4% na linha de base e diminuiu para 0,2% após 13 semanas. Em pacientes com HPN virgens de tratamento, o tamanho médio do clone HPN de hemácias foi de 49,1% na linha de base e aumentou para 91,1% após 12 semanas; havia RBCs de HPN Tipo II + III insignificantes com depósito de C3 nesta população devido à predominância de IVH.
Iptacopana reduz os níveis séricos de LDH. Em pacientes com HPN previamente tratados com eculizumabe, todos os pacientes tratados com iptacopana 200 mg duas vezes ao dia alcançaram uma redução dos níveis de LDH para < 1,5 vezes o limite superior de normalidade (LSN) após 13 semanas e mantiveram o efeito até o final do estudo. Em pacientes com HPN virgens de tratamento, iptacopana 200 mg duas vezes ao dia reduziu o LDH em > 60% em comparação com a linha de base após 12 semanas e manteve o efeito até o final do estudo.
Eletrofisiologia cardíaca
Em um estudo clínico do intervalo QTc em voluntários saudáveis, doses únicas supraterapêuticas de iptacopana de até 1200 mg (que forneceram pico de concentração maior que 4 vezes da dose máxima recomendada a humanos (DMRH)) não mostraram efeito na repolarização cardíaca ou no intervalo QT.
Farmacocinética
Absorção
Após a administração oral, a iptacopana atingiu o pico de concentração plasmática aproximadamente 2 horas após a dose. No regime posológico recomendado de 200 mg duas vezes ao dia, o estado de equilíbrio é alcançado em aproximadamente 5 dias com pequena acumulação (1,4 vez). Os dados de Cmáx e AUC de um estudo do efeito dos alimentos que envolve a administração de iptacopana a voluntários saudáveis em jejum ou com uma refeição com alto teor de gordura indicaram que a exposição à iptacopana não é afetada pelos alimentos. Portanto, Fabhalta® pode ser tomado com ou sem alimentos.
Distribuição
Iptacopana mostrou ligação às proteínas plasmáticas dependente da concentração devido à ligação ao FB alvo na circulação sistêmica. Iptacopana apresentou 75% a 93% de ligação às proteínas in vitro nas concentrações plasmáticas clínicas relevantes. Após administração de iptacopana 200 mg duas vezes ao dia, o volume aparente de distribuição no estado de equilíbrio foi de aproximadamente 288 L.
Biotransformação/metabolismo
O metabolismo é uma via de eliminação predominante para iptacopana com aproximadamente 50% da dose atribuída às vias oxidativas. O metabolismo da iptacopana inclui N-desalquilação, O-desetilação, oxidação e desidrogenação, principalmente impulsionada pelo CYP2C8 (98%) com uma pequena contribuição do CYP2D6 (2%). Glicuronidação (UGT1A1, UGT1A3, UGT1A8) é uma via secundária. No plasma, a iptacopana foi o principal componente respondendo por 83% da AUC 0-48hr. Dois acil glicuronídeos foram os únicos metabólitos detectados no plasma e foram menores, respondendo por 8% e 5% da AUC 0-48hr. Os metabólitos de Iptacopana não são considerados farmacologicamente ativos.
Eliminação
Em um estudo em humanos, após uma dose oral única de 100 mg de [14C] iptacopana, a excreção total média de radioatividade (iptacopana e metabólitos) foi de 71,5% nas fezes e 24,8% na urina, resultando em excreção média total de > 96% da dose. Especificamente, 17,9% da dose foi excretada como iptacopana original na urina e 16,8% nas fezes. A meia-vida (t1/2) de iptacopana no estado de equilíbrio é de aproximadamente 25 horas após a administração de Fabhalta® 200 mg duas vezes ao dia.
Linearidade/não linearidade
Em doses entre 25 mg e 200 mg duas vezes ao dia, a iptacopana, em geral, era menos do que proporcional à dose. No entanto, as doses orais de 100 mg e 200 mg foram aproximadamente proporcionais à dose.
Estudos de Fase 2
A eficácia de Fabhalta® em adultos com HPN também foi corroborada por dois estudos abertos de Fase 2 (CLNP023X2201 como terapia adicional a eculizumabe e CLNP023X2204 como monoterapia) em 29 pacientes com hemólise ativa e um tamanho de clone HPN de hemácias de pelo menos 10%, nível médio de hemoglobina < 10,5 g/dL e um nível de LDH de pelo menos 1,25 X LSN. Os pacientes que receberam Fabhalta® como terapia adjuvante no estudo CLNP023X2201 descontinuaram o eculizumabe durante o estudo e continuaram com Fabhalta® em monoterapia. Esses 29 pacientes foram tratados por até 3,4 anos com Fabhalta® em monoterapia e apresentaram aumento sustentado nos níveis de hemoglobina e redução nos níveis de LDH durante o tratamento.[4]
Dados de segurança pré-clinicos
Dados pré-clinicos não revelam risco especial para humanos com base nos estudos convencionais da farmacologia de segurança, da toxicidade de doses repetidas, da genotoxicidade, do potencial carcinogênico e da toxicidade reprodutiva.
Farmacologia de segurança
Os estudos de segurança cardiovascular de iptacopana foram realizados em ratos, cães e macacos após administração oral. Nenhum efeito cardiovascular foi observado em ratos após uma dose oral única de 1000 mg/kg (equivalente a aproximadamente 10 vezes a dose máxima recomendada a humanos (DMRH) com base na Cmáx). Em cães, após o início do tratamento, observou-se aumento da frequência cardíaca dependente da dose e diminuição da pressão arterial. A magnitude das alterações da frequência cardíaca diminuiu com o tempo e o efeito não foi considerado adverso até 150 mg/kg/dia (equivalente a aproximadamente 14 vezes a DMRH com base na AUC e aproximadamente 19 vezes com base na Cmáx) e sem nível de efeito no QTc a 100 mg/kg (equivalente a aproximadamente 9 vezes a DMRH com base na Cmáx). Em macacos cynomolgus, foi observado prolongamento do QTc após administração única de iptacopana ≥300 mg/kg (equivalente a > 21 vezes a DMRH com base em Cmáx).[2]
Nenhum efeito relacionado a iptacopana no sistema respiratório ou nervoso foi identificado em estudos pré-clínicos de farmacologia de segurança.[2]
Toxicidade de doses repetidas
O perfil de segurança pré-clínica de iptacopana foi avaliado em ratos com doses orais de até 750 mg/kg/dia (aproximadamente 7 vezes a DMRH com base na AUC) por 26 semanas e em cães com doses orais de até 150 mg/kg/dia por via oral (aproximadamente 14 vezes a DMRH com base na AUC) por 39 semanas. Achados adversos e irreversíveis nos estudos de toxicidade crônica limitaram-se à fibrose da medula óssea e diseritropoiese em um cão na dose mais alta. Achados reversíveis e não graves incluíram hipertrofia das células foliculares da tireoide e degeneração tubular testicular. [2]
Efeitos cardíacos adversos (por exemplo, degeneração celular e fibrose) foram observados apenas em estudos em cães de curta duração de até 4 semanas de duração em doses ≥300 mg/kg/dia (equivalente a > 39 vezes a DMRH com base na AUC).[2]
Mutagenicidade e carcinogenicidade
Iptacopana não foi genotóxica ou mutagênica em uma série de testes in vitro e in vivo. Estudos de carcinogenicidade realizados com iptacopana em camundongos e ratos por meio da administração oral não identificaram nenhum potencial carcinogênico. As doses mais altas de iptacopana estudadas em camundongos (1000 mg/kg/dia) e ratos (750 mg/kg/dia) foram aproximadamente 4 e 12 vezes a DMRH com base na AUC, respectivamente. [2]
Toxicidade reprodutiva
Para informações sobre toxicidade reprodutiva, veja a seção "Gravidez, lactação, mulheres e homens com potencial reprodutivo".
Populações especiais
Uma análise farmacocinética populacional foi realizada em dados de 234 pacientes. Idade, peso corporal, TFGe, raça e sexo não influenciaram significativamente a farmacocinética de iptacopana. Estudos que incluíram indivíduos asiáticos mostraram que a farmacocinética de iptacopana foi semelhante a indivíduos caucasianos (brancos).
Comprometimento renal
Apenas 17,9% de iptacopana foram excretados na urina como medicamento original. Portanto, o rim é uma via de eliminação secundária. O efeito do comprometimento renal no clearance da iptacopana foi avaliado por meio de uma análise farmacocinética populacional. Não houve diferenças clinicamente relevantes no clearance de iptacopana entre pacientes com função renal normal e pacientes com comprometimento renal leve (TFGe 60 a < 90 mL/min/1,73m2) ou moderado (TFGe 30 a < 60 mL/min/1,73m2), e nenhum ajuste de dose é necessário (vide seção 8. POSOLOGIA E MODO DE USAR). Pacientes com comprometimento renal grave ou em diálise não foram estudados.
Comprometimento hepático
Com base em um estudo em pacientes com comprometimento hepático leve, moderado ou grave, foi observado um efeito insignificante na exposição de iptacopana. Um aumento de aproximadamente 1,04 vez na Cmáx de iptacopana foi observado em pacientes com comprometimento hepático leve (n=8) e nenhuma alteração foi observada em pacientes com comprometimento hepático moderado (n=8) ou grave(n=6). O aumento na AUCinf em pacientes com comprometimento hepático leve e grave foi de 1,03 vez, enquanto não houve alteração em pacientes com comprometimento hepático moderado.
Nenhum ajuste de dose é necessário para pacientes com comprometimento hepático leve, moderado ou grave (vide seção 8. POSOLOGIA E MODO DE USAR).
4. CONTRAINDICAÇÕES
Fabhalta® é contraindicado:
• Em pacientes com hipersensibilidade a iptacopana ou a qualquer um dos excipientes.
• Em pacientes não vacinados atualmente contra Neisseria meningitidis e Streptococcus pneumoniae a menos que o risco de adiar o tratamento com Fabhalta® supere o risco de desenvolver uma infecção por essas bactérias encapsuladas (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
• Para iniciação em pacientes com infecção grave não solucionada causada por bactérias encapsuladas, incluindo Streptococcus pneumoniae, Neisseria meningitidis ou Haemophilus influenzae tipo B.
5. ADVERTÊNCIAS E PRECAUÇÕES
Infecções graves causadas por bactérias encapsuladas
O uso de inibidores do complemento, como Fabhalta®, pode predispor os indivíduos a infecções graves, com risco à vida ou fatais causadas por bactérias encapsuladas. Para reduzir o risco de infecção, os pacientes devem ser vacinados contra bactérias encapsuladas, incluindo Neisseria meningitidis e Streptococcus pneumoniae. Recomenda-se vacinar os pacientes contra Haemophilus influenzae tipo B, se disponível. Consulte as recomendações das diretrizes locais de vacinação.
As vacinas devem ser administradas pelo menos 2 semanas antes da administração da primeira dose de Fabhalta®. Caso Fabhalta® deva ser iniciado antes da vacinação, os pacientes devem ser vacinados o mais rápido possível e receber profilaxia antibacteriana até 2 semanas após a administração da vacina.
Se necessário, os pacientes podem ser revacinados de acordo com as recomendações das diretrizes locais de vacinação.
A vacinação reduz, mas não elimina, o risco de infecção grave. Infecções graves podem tornar-se rapidamente de risco à vida ou fatais se não forem reconhecidas e tratadas precocemente. Os pacientes devem ser informados e monitorados quanto aos primeiros sinais e sintomas de infecção grave. Os pacientes devem ser imediatamente avaliados e tratados se houver suspeita de infecção. O uso de Fabhalta® durante o tratamento de infecções graves pode ser considerado após uma avaliação dos riscos e benefícios (vide seção 9. REAÇÕES ADVERSAS).
Monitoramento das manifestações de HPN após a descontinuação do Fabhalta®
Se o tratamento com Fabhalta® precisar ser descontinuado, os pacientes devem ser rigorosamente monitorados quanto a sinais e sintomas de hemólise por pelo menos 2 semanas após a última dose. Esses sinais incluem níveis elevados de lactato desidrogenase (LDH), juntamente com diminuição súbita na hemoglobina ou no tamanho do clone de HPN, fadiga, hemoglobinúria, dor abdominal, dispneia, principais eventos adversos vasculares (incluindo trombose), disfagia ou disfunção erétil. Se a descontinuação de Fabhalta® for necessária, considerar terapia alternativa.
Se ocorrer hemólise após a descontinuação de Fabhalta®, deve ser considerado o reinício do tratamento com Fabhalta®, se apropriado, ou o início de outro tratamento para HPN.
Gravidez, lactação, mulheres e homens com potencial reprodutivo
Gravidez
Resumo de riscos
Não há dados suficientes sobre o uso do Fabhalta® em mulheres grávidas para informar um risco associado ao medicamento de defeitos congênitos graves, aborto espontâneo ou outros resultados maternos ou fetais adversos. Existem riscos para a mãe e para o feto associados à HPN não tratada na gravidez (vide Considerações Clínicas). O uso de Fabhalta® em mulheres grávidas ou mulheres que planejam engravidar pode ser considerado após uma avaliação dos riscos e benefícios.
Estudos de reprodução animal em ratos e coelhos demonstraram que a administração oral de Fabhalta® durante a organogênese não induziu toxicidade embrionária ou fetal adversa até as doses mais altas testadas. Isso corresponde a 5 vezes (ratos) e 8 vezes (coelhos) a dose máxima recomendada em humanos (DMRH) de 200 mg duas vezes ao dia com base na AUC (vide Dados em animais).
Considerações clínicas
Risco fetal/embrionário e/ou materno associado à doença
A hemoglobinúria paroxística noturna na gravidez está associada a resultados maternos adversos, incluindo piora da citopenia, eventos trombóticos, infecções, sangramento, abortos espontâneos e aumento da mortalidade materna, bem como resultados fetais adversos, incluindo morte fetal e parto prematuro.
Dados
Dados em animais
No estudo de desenvolvimento embriofetal em ratos, a iptacopana administrado por via oral durante a organogênese não induziu toxicidade materna, embrionária ou fetal adversa até a dose mais alta de 1.000 mg/kg/dia, que corresponde a 5 vezes a DMRH com base na AUC. Achados não adversos em ratos incluíram atrasos na ossificação do crânio fetal e cistos benignos no lado esquerdo da região parietal da cabeça, sem impacto no crânio, cérebro ou qualquer outra estrutura da cabeça, observados em apenas dois fetos em uma de 22 ninhadas a 1.000 mg/kg/dia.
No estudo de desenvolvimento embriofetal em coelhos, a iptacopana não induziu toxicidade embrionária ou fetal adversa em qualquer dose administrada por via oral, enquanto a toxicidade materna foi observada devido à perda de peso corporal adversa e consumo reduzido de alimentos em animais prenhes na dose mais alta de 450 mg/kg/dia, o que corresponde a 8 vezes a MRHD com base na AUC.
No estudo de desenvolvimento pré e pós-natal em ratos, com iptacopana administrada por via oral a fêmeas durante a gestação, parto e lactação (desde o 6° dia de gestação até o 21° dia de lactação), não houve efeitos adversos em fêmeas prenhas e filhotes até a dose mais alta testada de 1.000 mg/kg/dia (5 vezes a DMRH estimada com base na AUC).
Lactação
Resumo de riscos
Não se sabe se a iptacopana é transferida para o leite materno após a administração oral de Fabhalta®. Não há dados sobre os efeitos de Fabhalta® na criança amamentada ou na produção de leite.
Os benefícios da amamentação na saúde e no desenvolvimento devem ser considerados em conjunto com a necessidade clínica da mãe para o Fabhalta® e quaisquer efeitos adversos em potencial (por ex., infecções graves decorrentes de bactérias encapsuladas) no lactente a partir do Fabhalta® ou da condição subjacente materna.
Dado que muitos medicamentos são excretados no leite humano e devido ao potencial de reações adversas graves na criança que está sendo amamentada, a amamentação deve ser interrompida durante o tratamento e durante 5 dias após a dose final.
Mulheres e homens com potencial reprodutivo
Infertilidade
Não existem dados humanos sobre o efeito de Fabhalta® na fertilidade. Em estudos de fertilidade animal de dose oral, a iptacopana não afetou a fertilidade em ratos machos até a dose mais alta testada (750 mg/kg/dia), que corresponde a 6 vezes a DMRH com base na AUC. Efeitos reversíveis no sistema reprodutor de machos (degeneração tubular testicular e hipoespermatogênese) foram observados em estudos de toxicidade de dose repetida após administração oral em ratos e cães em doses > 3 vezes a DMRH com base na AUC, sem efeitos aparentes no número, morfologia ou motilidade de espermatozoides ou fertilidade.
No estudo de fertilidade de fêmeas e do desenvolvimento embrionário inicial em ratos, os achados relacionados a iptacopana foram limitados ao aumento das perdas pré e pós-implantação e, consequentemente, diminuição do número de embriões vivos apenas na dose mais alta de 1.000 mg/kg/dia por via oral, que corresponde a aproximadamente 5 vezes a DMRH com base na AUC. A dose de 300 mg/kg/dia é o nível sem efeito adverso observado (NOAEL) que corresponde a aproximadamente 2 vezes a DMRH com base na AUC.
Este medicamento pertence à categoria B de risco à gravidez, portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Atenção: Contém os corantes dióxido de titânio, óxido de ferro vermelho, óxido de ferro amarelo e óxido de ferro preto que podem, eventualmente, causar reações alérgicas.
6.INTERAÇÕES MEDICAMENTOSAS
Não se espera que Fabhalta® tenha interações clinicamente relevantes com outros medicamentos com base nosresultados abaixo.
Iptacopana não inibe as enzimas comuns do citocromo P450 (CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 ou3A4/5) ou induz CYP1A2, 2B6, 2C8, 2C9 ou 2C19 em concentrações clinicamente relevantes. Iptacopana nãoinibe os transportadores MATE1, MATE2-K, OAT1, OAT3, OCT1, OCT2. Iptacopana é um substrato paraCYP2C8 e OATP1B1/1B3 (transportadores). A administração concomitante com clopidogrel (um inibidormoderado de CYP2C8) ou ciclosporina (um potente inibidor de OATP1B1/1B3) não levou a aumentosclinicamente relevantes na Cmáx ou na área sob a curva (AUC)inf da iptacopana. Iptacopana não afetou a exposiçãoda digoxina (um substrato da glicoproteína P) ou da rosuvastatina (um substrato do OATP). Vide seção 3.CARACTERÍSTICAS FARMACOLÓGICAS.
Um estudo específico de interação medicamentosa no qual iptacopana foi administrada concomitantemente comoutros medicamentos foi conduzido em voluntários saudáveis e não demonstrou nenhuma interação clinicamenterelevante:
Quando administrado concomitantemente com clopidogrel (um inibidor moderado de CYP2C8), a Cmáx e a AUCda iptacopana aumentaram 5% e 36%, respectivamente.
Quando administrada concomitantemente com ciclosporina (um potente inibidor de OATP 1B1/1B3), a Cmáx e aAUC de iptacopana aumentaram 41% e 50%, respectivamente.
Na presença de iptacopana, a Cmáx da digoxina (um substrato da PgP) aumentou 8% enquanto a AUC permaneceuinalterada.
Na presença de iptacopana, a Cmáx e a AUC da rosuvastatina (um substrato de OATP) permaneceram inalteradas.
7.CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar o medicamento em temperatura ambiente (entre 15°C e 30°C). Armazenar em sua embalagem original.
O prazo de validade a partir da data de fabricação é de 36 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características do medicamento
Cápsulas duras de 200 mg: amarelas claras, opacas, com "LNP200" impresso no corpo e "NVR" na tampa, contendo pó branco a quase branco a rosa-arroxeado claro.
Antes de usar, observe o aspecto do medicamento.
TODO MEDICAMENTO DEVE SER MANTIDO FORA DO ALCANCE DAS CRIANÇAS.
8.POSOLOGIA E MODO DE USAR
A dose recomendada é de 200 mg duas vezes ao dia.
Caso uma dose ou doses tenha(m) sido esquecida(s), o paciente deve ser aconselhado a tomar uma dose deFabhalta® o mais rápido possível (mesmo que seja um pouco antes da próxima dose programada) e depois retomarocronograma posológico regular.
A HPN é uma doença que requer tratamento crônico. A descontinuação deste medicamento não é recomendada amenos que clinicamente indicado (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Pacientes que trocam de anti-C5 (eculizumabe, ravulizumabe) ou outras terapias para HPN para o Fabhalta®
Para reduzir o possível risco de hemólise com a descontinuação abrupta do tratamento:
•Para pacientes que trocam de eculizumabe, Fabhalta® deve ser iniciado no máximo 1 semana após a últimadose de eculizumabe.
•Para pacientes que trocam de ravulizumabe, Fabhalta® deve ser iniciado no máximo 6 semanas após aúltima dose de ravulizumabe.
•Ao trocar de outras terapias de HPN para Fabhalta®, o intervalo de administração da dose e o modo deação dos medicamentos anteriores devem ser considerados.
Aderência ao cronograma posológico
Os profissionais de saúde devem orientar os pacientes com HPN sobre a importância da aderência ao cronograma posológico a fim de minimizar o risco de hemólise (vide seção 5. ADVERTÊNCIAS E PRECAUÇÕES).
Populações especiais
Comprometimento renal
Não é necessário nenhum ajuste da dose em pacientes com comprometimento renal leve (taxa de filtração glomerular estimada [TFGe] de 60 a < 90 mL/min/1,73 m2) a moderado (TFGe de 30 a < 60 mL/min/1,73 m2). Não há dados atualmente disponíveis em pacientes com comprometimento renal grave ou em diálise. O uso de Fabhalta® não é recomendado em pacientes com insuficiência renal grave (taxa de filtração glomerular estimada (TFGe) < 30 mL/min/1,73 m2) com ou sem hemodiálise (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Comprometimento hepático
Não é necessário ajuste de dose para pacientes com comprometimento hepático leve (Child-Pugh classe A), moderado (Child-Pugh classe B) ou grave (Child-Pugh classe C) (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Pacientes pediátricos
A segurança e a eficácia de Fabhalta® em pacientes com menos de 18 anos de idade não foram estabelecidas.
Pacientes geriátricos (65 anos ou mais)
Nenhum ajuste de dose é necessário para pacientes com 65 anos ou mais (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Método de administração
Para uso oral. Fabhalta® pode ser tomado com ou sem alimentos (vide seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Este medicamento não deve ser partido, aberto ou mastigado.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
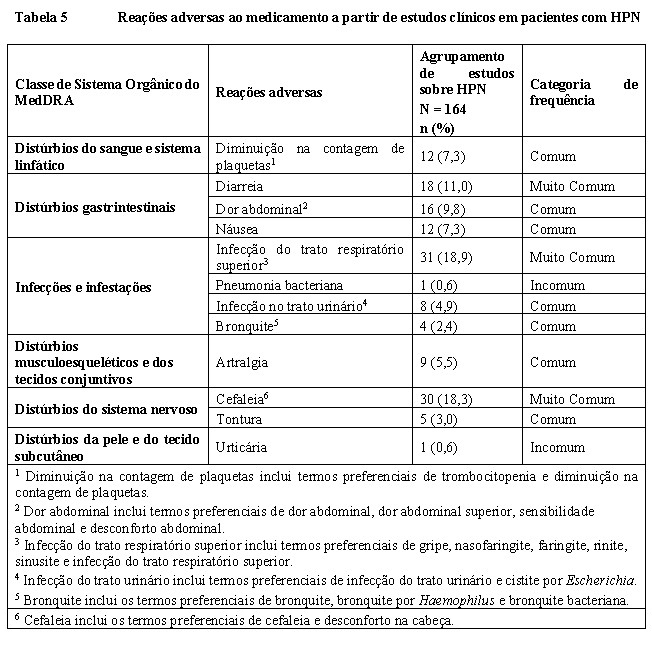
O perfil de segurança de Fabhalta® é baseado na análise de dados de segurança agrupados de 164 pacientes com HPN tratados com Fabhalta® 200 mg duas vezes ao dia em vários estudos. A duração mediana da exposição ao Fabhalta® foi de 10,2 meses. As reações adversas mais comumente relatadas em pacientes tratados com Fabhalta® foram infecção do trato respiratório superior (18,9%), cefaleia (18,3%) e diarreia (11%%).
Reações adversas a partir de estudos clínicos
As reações adversas ao medicamento a partir de estudos clínicos (Tabela 5) estão relacionadas de acordo com a classe de sistema orgânico do MedDRA. Em cada classe de sistema orgânico, as reações adversas ao medicamento são classificadas por frequência, relacionando primeiramente as reações mais frequentes. Dentro de cada agrupamento de frequência, as reações adversas ao medicamento são apresentadas em ordem de gravidade decrescente. Além disso, a categoria de frequência correspondente para cada reação adversa ao medicamento baseia-se na convenção a seguir (CIOMS III): muito comum (≥1/10); comum (≥1/100 a < 1/10); incomum (≥1/1.000 a < 1/100); raro