EXJADETM
NOVARTIS
deferasirox
Antídoto.
Apresentações.
Comprimidos dispersíveis - via oral. Cada caixa contém 28 comprimidos dispersíveis de ExjadeTM 125 mg, 250 mg e 500 mg.
USO ADULTO E PEDIÁTRICO
Composição.
Cada comprimido contém 125 mg, 250 mg ou 500 mg de deferasirox. Excipientes: lactose, crospovidona, povidona, lauril sulfato de sódio, celulose microcristalina, dióxido de silício e estearato de magnésio.
Indicações.
ExjadeTM é indicado para o tratamento de sobrecarga crônica de ferro devido a transfusões de sangue (hemossiderose transfusional) em pacientes adultos e pediátricos (com 2 anos de idade ou mais).
Resultados de eficácia.
Um estudo de fase III controlado, aberto e randomizado com comparador ativo para comparar ExjadeTM e Desferal (desferroxamina) foi conduzido em pacientes com beta-talassemia e hemosiderose transfusional. Pacientes com idade maior ou igual a 2 anos foram randomizados à razão de 1:1 para receber ExjadeTM oral nas doses iniciais de 5, 10, 20 ou 30 mg/kg uma vez ao dia ou Desferal (desferroxamina) subcutâneo nas doses iniciais de 20 a 60 mg/kg por pelo menos 5 dias por semana, baseados na concentração de ferro hepático (CHF) inicial (2 a 3, > 3 a 7, > 7 a 14 e > 14 mg de Fe/g de peso seco). Foi permitido que pacientes randomizados para desferroxamina que tiveram valores de CHF < 7 mg de Fe/g de peso seco continuassem em sua dose prévia de desferroxamina, mesmo que a dose fosse maior do que aquela estabelecida no protocolo.
A CHF foi avaliada no início da terapia e após 12 meses por biopsia hepática ou não-invasivamente por susceptometria biomagnética. A taxa de sucesso, o desfecho primário de eficácia, foi definido com uma redução na CHF de ≥ 3 mg de Fe/g de peso seco para valores iniciais ≥ 10 mg de Fe/g de peso seco, redução de valores inicias entre 7 e < 10 para < 7 mg de Fe/g de peso seco, ou manutenção ou redução para valores iniciais de < 7 mg de Fe/g de peso seco. ExjadeTM seria declarado como não inferior à desferroxamina se o limite inferior do intervalo de confiança de 95% (two-sided) da diferença nas taxas de sucesso fosse acima de - 15%.
No total, 586 pacientes foram randomizados. As características demográficas foram bem balanceadas. Entre os pacientes, 51% tinham < 16 anos de idade. As taxas globais de sucesso foram de 52,9% para ExjadeTM e 66,4% para desferroxamina com uma diferença de - 13,5 nas taxas de sucesso e um IC de 95% de [- 21,6 a 5,4]. A não-inferioridade à desferroxamina não foi atingida porque o limite inferior do IC foi abaixo de - 15%. Isto foi atribuído ao desequilíbrio da dose estabelecida no protocolo em relação à dose real nas duas coortes de doses mais baixas do braço da desferroxamina (Tabela 1). No entanto, a não-inferioridade foi demonstrada no grupo de pacientes com níveis iniciais de CHF ≥ 7 mg de Fe/g de peso seco que foram alocados para grupos de doses maiores (doses de ExjadeTM de 20 ou 30 mg/kg e doses de desferroxamina ≥ 35 mg/kg). As taxas de sucesso com ExjadeTM e desferroxamina foram de 58,6% e 58,9%, respectivamente, e o limite inferior do IC de 95% (- 10,2%) foi acima do limite de não-inferioridade de - 15%.
Em pacientes com CHF ≥ 7 mg/kg de Fe/g de peso seco que foram tratados com ExjadeTM 20 a 30 mg/kg por dia foi observada uma redução estatisticamente significativa na CHF inicial (- 5,3 ± 8,0 mg de Fe/g de peso seco, p < 0,001, teste t) que não foi estatisticamente diferente da desferroxamina (- 4,3 ± 5,8 mg de Fe/g de peso seco, p = 0,367). Os efeitos doses-dependentes na ferritina sérica e na razão de ferro excretado/recebido nas doses de ExjadeTM de 5 a 30 mg/kg também foram observados (Tabela 1).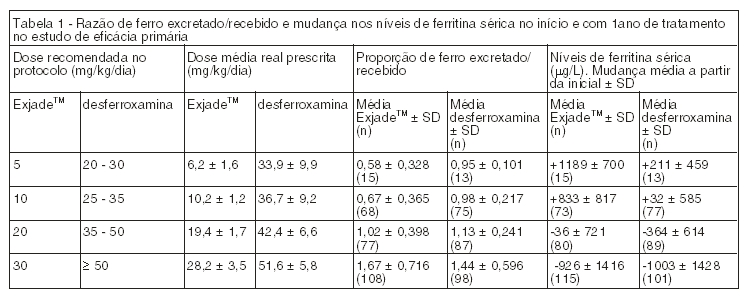
Um segundo estudo de fase II, aberto e não comparativo de eficácia e segurança de ExjadeTM administrado por um ano para pacientes com anemias crônicas e hemosiderose transfusional não tratáveis com desferroxamina também foi conduzido. Os pacientes receberam 5, 10, 20 ou 30 mg/kg por dia de ExjadeTM baseados na CHF inicial. O objetivo principal foi demonstrar uma taxa de sucesso significativamente maior do que 50% com ExjadeTM .
Um total de 184 pacientes foram tratados neste estudo: 85 pacientes com beta-talassemia e 99 pacientes com outras anemias congênitas ou adquiridas (síndromes mielodisplásicas, n = 47; síndrome de Blackfan-Diamond, n = 30; outras, n = 22). Entre os pacientes, 19% tinham < 16 anos e 16% tinham ≥ 65. Trinta e sete pacientes não receberam terapia prévia de quelação. Na população total, a taxa de sucesso (50,5%) não foi estatisticamente maior do que 50%. Isto foi atribuído ao fato que as doses de 5 e 10 mg/kg foram insuficientes para a taxa de ferro que estava sendo recebido por transfusões sanguíneas. No entanto, em pacientes com CHF ≥ 7 mg de Fe/g de peso seco para os quais a CHF inicial e ao final do estudo estavam disponíveis e que receberam ExjadeTM 20 a 30 mg/kg por dia, a taxa de sucesso foi 58,5% [p = 0,022 (50,3; 66,6)] e houve uma redução estatisticamente significativa na CHF absoluta no final do estudo (- 5,5 ± 7,4 mg de Fe/g de peso seco, p < 0,001, teste t). Houve também um efeito dose-dependente na ferritina sérica e na razão de ferro excretado/recebido nas doses de 5 a 30 mg/kg por dia.
Um terceiro estudo foi conduzido em pacientes com doença falciforme e hemosiderose transfusional. Este estudo foi um estudo de fase II, aberto, randomizado, de segurança e eficácia de ExjadeTM comparado à desferroxamina administrados por um ano. Os pacientes foram randomizados para ExjadeTM nas doses de 5, 10, 20 ou 30 mg/kg por dia ou desferroxamina subcutânea nas doses de 20 a 60 mg/kg por dia por 5 dias por semana de acordo com a CHF inicial.
Um total de 195 pacientes foram tratados no estudo: 132 com ExjadeTM e 63 com desferroxamina. 44% dos pacientes tinham < 16 anos e 91% eram negros. No final do estudo, a mudança média na CHF na população per protocolo-1 (PP-1), que consistiu de pacientes que tiveram pelo menos uma avaliação de CHF após o início do estudo, foi - 1,3 mg de Fe/g de peso seco para pacientes recebendo ExjadeTM (n = 113) e - 0,7 mg de Fe/g de peso seco para pacientes recebendo desferroxamina (n = 54). Um subestudo cardíaco foi conduzido como parte de um estudo de fase IV.
O subestudo cardíaco foi de um ano, de braço único, aberto e prospectivo, e incluíram duas coortes de pacientes beta-talassêmicos com sobrecarga de ferro grave com valores de fração de ejeção de ventrículo esquerdo (LVEF) ≥ 56%. Foram estudados 114 pacientes com valores iniciais de T2* > 5 a < 20 ms, indicando siderose miocárdica (coorte de tratamento) e 78 pacientes com T2* ≥ 20 ms, indicando depósito de ferro cardíaco sem significância clínica (coorte de prevenção). Na coorte de tratamento, a dose inicial de deferasirox foi 30 mg/kg/dia, com escalonamento até o máximo de 40 mg/kg/dia. Na coorte de prevenção, a dose inicial foi 20-30 mg/kg/dia, com escalonamento até o máximo de 40 mg/kg/dia. O objetivo principal do subestudo cardíaco foi a mudança no T2* em um ano. Na coorte de tratamento, o T2* (média geométrica coeficiente de variação) aumentou significativamente do valor inicial de 11,2 ms ± 40,5% a 12,9 ms ± 49,5%, representando uma melhora significativa de 16% (p < 0,0001). Na coorte de tratamento, foi observada melhora no T2* em 69,5% dos pacientes e estabilização de T2* em 14,3% dos pacientes. A LVEF permaneceu estável e dentro da variação normal: 67,4 ± 5,7% para 67,1 ± 6,0%. Na coorte de prevenção,o T2* permaneceu dentro da faixa normal e não mudou do valor inicial de 32,0 ms ± 25,6% para 32,5 ms ± 25,1% (+ 2%; p = 0,565), indicando que o tratamento diário com deferasirox pode prevenir sobrecarga de ferro cardíaca em pacientes com beta-talassemia com história de exposição transfusional alta e regular, e com transfusões em andamento.
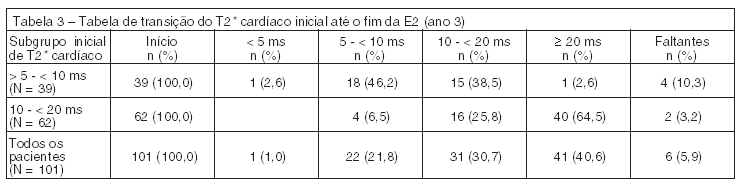
Pacientes na coorte de tratamento do estudo de 1 ano tiveram a opção de participar em duas extensões de 1 ano. Durante o período de 3 anos de tratamento, houve melhora estatisticamente significativa (p < 0,0001), progressiva e clinicamente relevante na média geométrica do T2* cardíaco a partir do inicial no geral, no subgrupo com sobrecarga de ferro grave, o qual está associado a alto risco de insuficiência cardíaca (T2* > 5 a < 10 ms), e no subgrupo com sobrecarga de ferro leve a moderada (T2* 10 a < 20 ms) (Tabela 2). Usando a taxa da média geométrica, o aumento de T2* foi de 43% acima do inicial em todos os pacientes, 37% acima do inicial no subgrupo de T2* > 5 a < 10 ms, e 46% acima do inicial no subgrupo de T2* 10 a < 20 ms). O tratamento contínuo com ExjadeTM por até 3 anos em doses > 30 mg/kg/dia reduziu efetivamente o ferro cardíaco em pacientes com talassemia maior com siderose miocárdica, como mostrado pelo número de pacientes que tiveram seus T2* normalizados ou melhorados a uma categoria associada a menor risco de insuficiência cardíaca (Tabela 3).
Caract. farmacológicas.
Grupo farmacoterapêutico: agente quelante de ferro, código ATC V03AC03.
Mecanismo de ação
O deferasirox é um quelante ativo oral que é altamente seletivo para ferro (III). É um agente tridentado que se liga ao ferro com alta afinidade na proporção 2:1. O deferasirox promove excreção de ferro, principalmente nas fezes. O deferasirox tem uma baixa afinidade por zinco e cobre, não alterando os níveis séricos destes.
Farmacodinâmica
Em um estudo de balanço metabólico de ferro em pacientes adultos talassêmicos com sobrecarga de ferro, ExjadeTM em doses diárias de 10, 20 e 40 mg/kg induziu média de excreção líquida de 0,119; 0,329 e 0,445 mg de Fe/kg de peso corpóreo por dia, respectivamente. ExjadeTM foi investigado em pacientes adultos e pediátricos (com 2 anos de idade ou mais) com sobrecarga crônica de ferro devido a transfusões sanguíneas. As condições clínicas que requeriam transfusão foram beta-talassemia, anemia falciforme e outras anemias congênitas e adquiridas (síndromes mielodisplásticas, síndrome de Blackfan-Diamond, anemia aplástica e outras anemias raras). O tratamento diário com ExjadeTM nas doses de 20 e 30 mg/kg por um ano em pacientes adultos e pediátricos politransfundidos com beta-talassemia levou a reduções em indicadores de ferro corpóreo total; a concentração de ferro hepático foi reduzida em aproximadamente - 0,4 e - 8,9 mg de Fe/g de tecido hepático (peso seco da biópsia) em média, respectivamente, e a ferritina sérica foi reduzida em aproximadamente - 36 e - 926 micrograma/L em média, respectivamente. Nestas mesmas doses, as razões de excreção de ferro: consumo de ferro foram de 1,02 (indicando balanço líquido de ferro) e 1,67 (indicando remoção líquida de ferro), respectivamente. ExjadeTM induziu respostas similares em pacientes com sobrecarga crônica de ferro devido a outras anemias. Doses diárias de 10 mg/kg por um ano mantiveram os níveis de ferro hepático e de ferritina sérica e induziram o balanço líquido de ferro em pacientes recebendo transfusões eventuais ou exsanguíneo-transfusões. A ferritina sérica avaliada mensalmente refletiu as mudanças na concentração de ferro hepático, indicando que tendências na ferritina sérica podem ser usadas para monitorar a resposta à terapia. Em pacientes com acúmulo cardíaco de ferro (MRI T2* < 20 ms), o tratamento com ExjadeTM mostrou remover o ferro cardíaco, demonstrado por melhora progressiva nos valores de T2* durante 3 anos de observação. Em pacientes sem acúmulo cardíaco, ExjadeTM mostrou prevenir o acúmulo cardíaco de ferro de modo clinicamente relevante (manutenção de T2* > 20 ms) durante 1 ano de observação, embora houvesse significante exposição a transfusões sanguíneas.
Farmacocinética
Absorção
O deferasirox é absorvido após administração oral com um tempo mediano para a concentração plasmática máxima (tmáx) de aproximadamente 1,5 a 4 horas. A biodisponibilidade absoluta (AUC) do deferasirox de ExjadeTM comprimidos é de aproximadamente 70% comparada a uma dose intravenosa. A exposição total (AUC) foi aproximadamente dobrada quando administrado junto com um desjejum gorduroso (conteúdo de gordura > 50% das calorias), e por aproximadamente 50% quando administrado junto com um desjejum convencional. A biodisponibilidade (AUC) do deferasirox foi moderadamente elevada (aproximadamente 13 a 25%) quando administrado 30 minutos antes das refeições com conteúdo normal ou alto de gordura. A biodisponibilidade (AUC) do deferasirox foi moderadamente elevada (aproximadamente 13 a 25%) quando administrado 30 minutos antes de refeições normais ou de alto teor de gordura. A exposição total (AUC) do deferasirox após administração dos comprimidos dispersos em suco de laranja ou maçã foi equivalente à exposição total (AUC) obtida após administração dos comprimidos de deferasirox dispersos na água (razões relativas de AUC foram de 103% e 90%, respectivamente).
Distribuição
O deferasirox tem alta afinidade por proteínas plasmáticas (99%), quase exclusivamente pela albumina sérica, e tem um pequeno volume de distribuição, de aproximadamente 14 L em adultos.
Biotransformação
A glucuronidação é a principal via de metabolização do deferasirox, com subsequente excreção biliar. Parece ocorrer desconjungação de glucuronidatos no intestino e subsequente reabsorção (ciclo enterohepático). O deferasirox é glucuronizado principalmente por UGT1A1 e, em um menor grau, por UGT1A3. O metabolismo oxidativo via CYP450 parece ter importância menor no metabolismo do deferasirox em humanos (8%). Não foi observada inibição do metabolismo in vitro do deferasirox por hidroxiureia. O deferasirox sofre reciclagem entero-hepática. Em estudo com voluntários sadios, a administração de colestiramina após uma dose única de deferasirox resultou em diminuição de 45% na exposição (AUC) de deferasirox.
Eliminação
O deferasirox e seus metabólitos são excretados principalmente nas fezes (84% da dose). A excreção renal do deferasirox e de seus metabólitos é mínima (8% da dose). A média da meia-vida de eliminação (t1/2) variou de 8 a 16 horas.
Linearidade/não-linearidade
A Cmáx e a AUC0-24h do deferasirox aumentam de forma linear com doses abaixo das condições em estado de equilíbrio. A exposição em múltiplas dosagens aumentou linearmente a Cmáx com um fator de acúmulo de 1,3 a 2,3.
Populações especiais
Pacientes pediátricos
Aparentemente, o clearance do deferasirox em adolescentes (12 a ≤ 17 anos) e crianças (2 a < 12 anos) expostos à droga, após doses únicas ou múltiplas, foi maior, levando a uma menor exposição à droga. Em crianças menores de 6 anos, a exposição à droga é aproximadamente 50% menor do que em adultos. Como a dosagem é individualmente ajustada de acordo com a resposta à terapia, não são esperadas consequências clínicas.
Sexo
Mulheres têm um clearance discretamente menor (aproximadamente 17,5%) para deferasirox comparadas aos homens. Como a dosagem é individualmente ajustada de acordo com a resposta à terapia, não são esperadas consequências clínicas.
Pacientes idosos
A farmacocinética do deferasirox não foi estudada em pacientes idosos (com 65 anos ou mais).
Insuficiência renal e hepática
A farmacocinética do deferasirox não foi estudada em pacientes com insuficiência renal. A AUC média de deferasirox em 6 indivíduos com insuficiência hepática leve (Child-Pugh A) aumentou 16% em relação àquela encontrada em 6 indivíduos com função hepática normal, enquanto que a AUC média de deferasirox em 6 indivíduos com insuficiência hepática moderada (Child-Pugh B) aumentou 76% em relação ao encontrado em 6 indivíduos com função hepática normal. A Cmax média de deferasirox em indivíduos com disfunção hepática leve ou moderada aumentou 22% em relação ao encontrado em indivíduos com função hepática normal. O impacto da insuficiência hepática grave (Child-Pugh C) foi avaliado em apenas um indivíduo (vide "Posologia" e "Advertências"). A farmacocinética do deferasirox não foi influenciada por níveis de transaminases hepáticas até 5 vezes o limite superior para a idade.
Dados de segurança pré-clínicos
Os dados pré-clínicos não revelam riscos especiais para pacientes com sobrecarga de ferro, baseados em estudos convencionais de segurança farmacológica, toxicidade de dose repetida, genotoxicidade ou potencial carcinogênico. Os principais achados foram toxicidade renal e opacidade do cristalino (catarata). Achados similares foram observados em animais neonatos e jovens. A toxicidade renal é considerada principalmente devido à privação de ferro em animais que não tinham, previamente, sobrecarga de ferro.
O potencial de toxicidade para reprodução foi avaliado em ratos e coelhos. Em ratos sem sobrecarga de ferro e submetidos a altas doses de deferasirox, a medicação não mostrou efeito teratogênico, mas causou frequência aumentada de prematuridade e variações esqueléticas em ratos recém-natos. O deferasirox não causou outros efeitos na fertilidade ou reprodução.
Contraindicações.
ExjadeTM é contraindicado quando o clearance de creatinina é < 40 mL/min ou a creatinina sérica > 2 vezes o limite superior da normalidade. Em pacientes com síndrome mielodisplásica (SMD) de alto risco e pacientes com outras malignidades hematológicas e não-hematológicas, nos quais não se esperam benefícios da terapia de quelação devido à rápida progressão da doença.
ExjadeTM é contraindicado em pacientes com hipersensibilidade à substância ativa ou a qualquer dos excipientes da fórmula.
Este medicamento é contraindicado na faixa etária abaixo de 2 anos de idade.
Advertências e precauções.
A decisão de remover a sobrecarga de ferro deve ser individualizada e baseada nos riscos e benefícios clínicos antecipados da terapia de quelação (vide "Posologia"). Deve-se ter cautela ao ser usado em pacientes idosos devido à maior frequência de reações adversas.
Renal
Aumentos não-progressivos na creatinina sérica foram observados em alguns pacientes tratados com ExjadeTM, geralmente dentro da variação normal. Casos de insuficiência renal aguda foram relatados após a comercialização de ExjadeTM (vide "Reações adversas"). Houve casos raros de insuficiência renal aguda que necessitaram de diálise.
É recomendado que a creatinina sérica e/ou o clearance de creatinina sejam avaliados em duplicata antes de iniciar a terapia e seguida mensalmente durante o tratamento.
Pacientes com condições renais preexistentes, ou pacientes que estejam recebendo medicamentos que possam deprimir a função renal podem ter maior risco de complicações e recomenda-se o seguimento semanal da creatinina sérica e/ou do clearance de creatinina no primeiro mês após início ou modificação da terapia, e mensalmente durante o tratamento. Deve-se ter cautela ao ser usado em pacientes com clearance de creatinina entre 40 e 60 mL/min, particularmente nos casos em que há fatores de risco adicionais que possam comprometer a função renal, tais como medicações concomitantes, desidratação, ou infecções graves.
Tubulopatia renal foi relatada em pacientes tratados com ExjadeTM . A maioria destes pacientes eram crianças e adolescentes com beta-talassemia e níveis de ferritina sérica < 1.500 micrograma/L. Exames de proteinúria devem ser realizados mensalmente.
Deve-se ter cuidado para manter hidratação adequada em pacientes que apresentem diarreia ou vômito. Para pacientes adultos, a dose diária de ExjadeTM pode ser reduzida para 10 mg/kg se um aumento não progressivo na creatinina sérica maior que 33% acima da média das medidas pré-tratamento for detectado em duas consultas consecutivas, e não puder ser atribuído a outras causas (vide "Posologia"). Para pacientes pediátricos, a dose deve ser reduzida para 10 mg/kg se os níveis da creatinina sérica aumentarem acima do limite superior da normalidade para a idade em duas consultas consecutivas. Se houver um aumento progressivo na creatinina sérica, além do limite superior para a idade, ExjadeTM deve ser interrompido. A terapia com ExjadeTM deve ser reiniciada dependendo das circunstâncias clínicas individuais.
Hepático
ExjadeTM não é recomendado em pacientes com insuficiência hepática grave (Child-Pugh C) (vide "Posologia" e "Farmacocinética -Populações Especiais").
O tratamento com ExjadeTM foi iniciado somente em pacientes com níveis de transaminases hepáticas até 5 vezes o limite superior da normalidade para a idade. A farmacocinética do deferasirox não foi influenciada por tais níveis de transaminase. O deferasirox é eliminado principalmente por glucuronidação e é minimamente (aproximadamente 8%) metabolizado pelas enzimas oxidativas do citocromo P450 (vide "Farmacocinética").
Embora incomuns (0,3%), foram observadas, em estudos clínicos, elevações de transaminases maiores que 10 vezes o limite superior da normalidade, sugerindo hepatite. Houve relatos, pós-comercialização, de falência hepática em pacientes tratados com ExjadeTM. A maioria dos relatos de falência hepática envolveram pacientes com comorbidades significativas incluindo cirrose hepática e falência múltipla de órgãos; casos fatais foram relatados em alguns destes pacientes (vide "Reações adversas"). É recomendado que as transaminases séricas, bilirrubina e fosfatase alcalina sejam monitoradas antes do início do tratamento, a cada 2 semanas durante o primeiro mês e depois mensalmente. Se houver um aumento persistente e progressivo nos níveis de transaminases séricas que não possa ser atribuído a outras causas, ExjadeTM deve ser interrompido. Uma vez que a causa dos testes anormais de função hepática sejam esclarecidos ou após retorno aos níveis normais, deve-se ter cautela ao reiniciar o tratamento com ExjadeTM em uma dose menor, seguida de um escalonamento gradual de dose.
Distúrbios sanguíneos
Após o início da comercialização, houve relatos (ambos espontâneos e de estudos clínicos) de citopenias em pacientes tratados com ExjadeTM . A maioria destes pacientes tinham distúrbios hematológicos preexistentes, que são frequentemente associados à falência medular (vide "Reações adversas"). A relação destes episódios ao tratamento com ExjadeTM é incerta. De acordo com a prática clínica e o padrão de tais distúrbios hematológicos, contagens sanguíneas devem ser avaliadas regularmente. Em pacientes que desenvolvam citopenia de forma inexplicada, deve-se considerar a interrupção do tratamento com ExjadeTM. A reintrodução da terapia com ExjadeTM pode ser considerada, uma vez que a causa da citopenia seja elucidada.
Gastrintestinal
Podem ocorrer irritações gastrintestinais (GI) durante o tratamento com ExjadeTM . Foram relatadas ulcerações gastrintestinais superiores e hemorragia em pacientes recebendo ExjadeTM , inclusive em crianças e adolescentes. Houve raros relatos de hemorragias GI fatais, especialmente em pacientes idosos que tinham malignidades hematológicas avançadas e/ou contagem baixa de plaquetas. Em alguns pacientes foram observadas úlceras múltiplas (vide "Reações adversas"). Médicos e pacientes devem ficar atentos aos sinais e sintomas de ulcerações GI e hemorragias durante a terapia com ExjadeTM e, se houver suspeita de um evento adverso GI grave, deve-se iniciar avaliação adicional e tratamento prontamente.
Deve-se ter cautela em pacientes que estejam tomando ExjadeTM em combinação com drogas que são conhecidas como ulcerogênicas potenciais, tais como antiinflamatórios não-esteroidais, corticosteroides, ou bisfosfonatos orais, em pacientes que estejam recebendo anticoagulantes (vide "Interações medicamentosas"), e em pacientes com contagem plaquetária < 50 x 109/L.
Reações de hipersensibilidade
Casos raros de reações graves de hipersensibilidade (tais como anafilaxia e angioedema) foram relatados em pacientes recebendo ExjadeTM, com o começo da reação ocorrendo, na maioria dos casos, dentro do primeiro mês de tratamento (vide "Reações adversas"). No caso de reações graves, ExjadeTM deve ser descontinuado e intervenções médicas apropriadas devem ser instituídas.
Distúrbios de pele
Erupções de pele (rash) podem aparecer durante o tratamento com ExjadeTM . Para os casos de erupções (rash) de severidade leve ou moderada, ExjadeTM deve ser continuado sem ajuste de dose, uma vez que a erupção (rash) frequentemente se resolve espontaneamente. Para erupções (rash) mais graves, onde a interrupção do tratamento pode ser necessária, ExjadeTM pode ser reintroduzido após resolução da erupção (rash) em uma dose menor, seguida por escalonamento gradual de dose. Em casos graves, esta reintrodução pode ser conduzida em combinação com administração oral de esteroides por um período curto. Casos raros de eritema multiforme foram relatados durante o tratamento com ExjadeTM .
Visão e audição
Distúrbios auditivos (diminuição de audição) e oculares (opacidade de cristalino) foram reportados com o tratamento com ExjadeTM (vide "Reações adversas"). Testes auditivos e oftalmológicos (incluindo fundoscopia) são recomendados antes do início do tratamento com ExjadeTM e em intervalos regulares durante a terapia (a cada 12 meses). A redução ou interrupção da dose pode ser considerada, se estes distúrbios forem observados.
Outras considerações
É recomendado que a ferritina sérica seja dosada todo mês para avaliação da resposta do paciente à terapia (vide "Posologia"). Em pacientes nos quais o nível de ferritina sérica atingiu o valor desejado (normalmente entre 500 e 1.000 microgramas/L), reduções de dose em etapas de 5 a 10 mg/kg devem ser consideradas para manter os níveis de ferritina sérica dentro da faixa alvo. Se a ferritina sérica cair consistentemente abaixo de 500 mg/L, a interrupção do tratamento deve ser considerada. Assim como com outros quelantes de ferro, o risco de toxicidade de ExjadeTM pode ser aumentado quando doses mais altas são administradas inapropriadamente a pacientes com baixa sobrecarga de ferro ou com níveis de ferritina sérica que estejam levemente elevados.
ExjadeTM não foi associado com retardo no crescimento de crianças seguidas por até 5 anos em estudos clínicos. Entretanto, como uma medida geral de precaução, o peso corpóreo e o crescimento longitudinal em pacientes pediátricos podem ser monitorados em intervalos regulares (a cada 12 meses). ExjadeTM não deve ser combinado com outras terapias quelantes de ferro, pois a segurança de tais combinações não foi estabelecida.
Os comprimidos contêm lactose (1,1 mg de lactose para cada mg de deferasirox). Este medicamento não é recomendado para pacientes com problemas hereditários raros de intolerância à galactose, de deficiência grave de lactase ou de má absorção de glicose-galactose.
Usos específicos
Gravidez
Não estão disponíveis dados clínicos em mulheres grávidas expostas a deferasirox. Estudos em animais têm demonstrado alguma toxicidade reprodutiva em doses tóxicas (vide "Dados de segurança pré-clínica"). O risco potencial para humanos não é conhecido.
Como uma precaução, é recomendado que ExjadeTM não seja usado durante a gravidez, a menos que claramente necessário.
ExjadeTM enquadra-se na categoria B de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Lactação
Em estudos em animais, deferasirox foi demonstrado ser rápido e extensivamente secretado no leite materno. Não foram observados efeitos na ninhada. Não se sabe se o deferasirox é secretado no leite humano. A lactação não é recomendada durante o tratamento com ExjadeTM .
Efeitos sobre a habilidade de dirigir veículos e/ou operar máquinas
Não foram realizados estudos para avaliação dos efeitos de ExjadeTM sobre a habilidade de dirigir veículos e/ou operar máquinas. Pacientes que apresentaram tontura, uma reação adversa incomum, devem ter cuidado ao dirigir veículos ou operar máquinas.
Incompatibilidade
A dispersão em bebidas gaseificadas ou leite não é recomendada devido à formação de espuma e lenta dispersão, respectivamente.
Interações medicamentosas.
Agentes que podem diminuir a exposição sistêmica ao ExjadeTM
Em estudo com voluntários sadios, a administração concomitante de ExjadeTM (dose única de 30 mg/kg) e rifampicina, potente indutor de UDP-glucorosiltransferase (UGT) (dose repetida de 600 mg/dia) resultou em diminuição da exposição de deferasirox de 44% (90% IC: 37% -51%). Desta forma, o uso concomitante de ExjadeTM com indutores potentes de UGT (ex.: rifampicina, fenitoína, fenobarbital, ritonavir) pode resultar na diminuição da eficácia de ExjadeTM. Se ExjadeTM e um indutor potente de UGT forem usados concomitantemente, aumentos na dose de ExjadeTM devem ser considerados baseando-se na resposta clínica à terapia.
Interação com alimentos
A biodisponibilidade do deferasirox foi aumentada para uma extensão variável quando tomada ao longo da refeição. Embora a biodisponibilidade tenha sido quase dobrada quando ExjadeTM foi ingerido com refeições contendo alto teor de gordura, este aumento também variou de forma importante com outros teores de gordura e horários de ingestão de ExjadeTM com relação às refeições (vide "Farmacocinética: Absorção"). Como nos estudos pivotais, ExjadeTM foi administrado em pacientes com estômago vazio, recomenda-se que ExjadeTM deva ser tomado com o estômago vazio, pelo menos 30 minutos antes da refeição, e de preferência no mesmo horário todos os dias (vide "Posologia").
A dispersão dos comprimidos de ExjadeTM em suco de laranja ou maçã não afeta a biodisponibilidade.
Interação com midazolam e outros agentes metabolizados pelo CYP3A4
Em um estudo com voluntários sadios, a administração concomitante de ExjadeTM e midazolam (um substrato do CYP3A4) resultou na diminuição da exposição de midazolam em 17% (90% IC: 8% - 26%). Na prática clínica, este efeito pode ser mais pronunciado. Portanto, devido à possível diminuição na eficácia, deve-se ter cautela quando deferasirox for combinado com substâncias metabolizadas pelo CYP3A4 (como ciclosporina, sinvastatina, contraceptivos hormonais).
Interação com repaglinida e outros agentes metabolizados pelo CYP2C8
Em estudo com voluntários sadios, a administração concomitante de ExjadeTM (dose repetida de 30 mg/kg/dia) com repaglinida, um substrato da CYP2C8 (dose única de 0,5 mg), resultou em aumento na AUC e Cmáx de repaglinida de 131% (90% IC: 103% -164%) e 62% (90% IC: 42% -84%), respectivamente. Quando ExjadeTM e repaglinida são usados concomitantemente, deve ser feito monitoramento cuidadoso dos níveis de glicose. A interação entre ExjadeTM e outros substratos como paclitaxel não podem ser excluídos.
Interação com teofilina e outros agentes metabolizados pelo CYP1A2
Em um estudo com voluntários sadios, a administração concomitante de ExjadeTM (dose repetida de 30 mg/kg/dia) com teofilina, um substrato do CYP1A2 (dose única de 120 mg), resultou em aumento da AUC da teofilina em 84% (90% IC: 73% a 95%). A Cmáx da dose única não foi afetada, mas um aumento da Cmáx de teofilina deverá ocorrer com administração crônica. Quando ExjadeTM e teofilina são utilizados concomitantemente, o monitoramento da concentração de teofilina e uma possível redução da dose de teofilina devem ser considerados. Uma interação entre ExjadeTM e outros substratos de CYP1A2 pode ser possível.
Outras informações
Não foram observadas interações entre ExjadeTM e digoxina em voluntários sadios. A administração concomitante de ExjadeTM e vitamina C não foi estudada formalmente. As doses de vitamina C até 200 mg por dia não foram associadas com reações adversas.
Interações previstas resultantes do uso de medicação concomitante não recomendada
A administração concomitante de ExjadeTM e preparações antiácidas contendo alumínio não foi estudada formalmente. Embora deferasirox tenha uma afinidade mais baixa por alumínio que por ferro, comprimidos de ExjadeTM não devem ser tomados com preparações antiácidas contendo alumínio. A administração concomitante de ExjadeTM com drogas que são conhecidas como ulcerogênicas potenciais, tais como anti-inflamatórios não-esteroidais, corticosteroides, ou bisfosfonatos orais, e o uso de ExjadeTM em pacientes recebendo anticoagulantes podem aumentar o risco de irritação gastrintetinal (vide "Precauções e advertências").
Cuidados de armazenamento.
Manter em temperatura ambiente (entre 15 e 30°C).
Posologia e modo de usar.
Modo de usar e cuidados de conservação depois de aberto
Via oral. Após aberto, manter na embalagem original.
Posologia
Recomenda-se que a terapia com ExjadeTM seja iniciada após a transfusão de aproximadamente 20 unidades (aproximadamente 100 mL/kg) de bolsas de hemácias ou quando há evidência de sobrecarga crônica de ferro por avaliação clínica (p. ex. ferritina sérica > 1000 mg/L). As doses (em mg/kg) devem ser calculadas e arredondadas para um número mais próximo de comprimidos inteiros. A terapia com agentes quelantes de ferro tem o objetivo de remover a quantidade de ferro administrada nas transfusões e, quando necessário, reduzir a carga de ferro existente. A decisão de remover o acúmulo de ferro deve ser individualizada com base no benefício clínico já comprovado e nos riscos da terapia de quelação.
População alvo geral
Dose inicial
A dose diária inicial de ExjadeTM é de 20 mg/kg de peso corpóreo.
Uma dose diária inicial de 30 mg/kg pode ser considerada para pacientes recebendo mais que 14 mL/kg/mês de hemácias (aproximadamente mais de 4 unidades/mês para um adulto) e para aqueles cujo objetivo é reduzir a sobrecarga de ferro.
Uma dose inicial de 10 mg/kg pode ser considerada para pacientes recebendo menos que 7 mL/kg/mês de hemácias (aproximadamente menos de 2 unidades/mês para um adulto) e para aqueles cujo objetivo é a manutenção do nível de ferro no organismo.
Para pacientes que já estão bem controlados com o tratamento com desferroxamina, uma dose inicial de ExjadeTM equivale numericamente à metade da dose de desferroxamina administrada [por ex.: um paciente recebendo 40 mg/kg/dia de desferroxamina, por 5 dias na semana (ou equivalente), pode ser transferido para o tratamento com ExjadeTM utilizando uma dose inicial de 20 mg/kg/dia].
Se o paciente esquecer-se de tomar uma dose, ele deve tomá-la no dia ou assim que se lembrar. A próxima dose deve ser tomada como de costume. O paciente não deve tomar uma dose dobrada no dia seguinte para completar os comprimidos que faltaram.
Dose de manutenção
Recomenda-se que a ferritina sérica seja monitorada todo mês e que a dose de ExjadeTM seja ajustada, se necessário, a cada 3 a 6 meses, baseando-se na tendência da ferritina sérica. Ajustes de dose podem ser feitos por etapas de 5 a 10 mg/kg e de acordo com as respostas individuais dos pacientes e seus objetivos terapêuticos (manutenção ou redução de sobrecarga de ferro). Em pacientes não controlados adequadamente com doses de 30 mg/kg (por exemplo, níveis de ferritina sérica persistentemente acima de 2.500 microgramas/L, e não mostrando uma tendência decrescente ao longo do tempo) doses de até 40 mg/kg podem ser consideradas. As doses acima de 40 mg/kg não são recomendadas porque há somente experiências limitadas com doses acima deste nível.
Em pacientes nos quais o nível de ferritina sérica atinge o valor desejado (geralmente entre 500 e 1.000 microgramas/L), reduções de doses em etapas de 5 a 10 mg/kg devem ser consideradas até a manutenção do nível de ferritina sérico dentro do intervalo desejado. Se a ferritina sérica cair consistentemente abaixo de 500 microgramas/L, deve ser considerada a interrupção do tratamento. Como ocorre com outros quelantes de ferro, o risco de toxicidade de ExjadeTM pode ser aumentado quando pacientes com baixa carga de ferro ou com níveis de ferritina sérica ligeiramente elevados recebem doses mais altas inapropriadamente (vide "Advertências").
Método de administração
ExjadeTM deve ser tomado uma vez por dia com o estômago vazio pelo menos 30 minutos antes da refeição e, de preferência, no mesmo horário todos os dias. Os comprimidos são dispersíveis por agitação em um copo de água ou suco de laranja ou maçã (100 - 200 mL) até que uma fina suspensão seja obtida. Após a ingestão da suspensão, qualquer resíduo deve ser novamente disperso em um pequeno volume de água ou suco de laranja ou maçã e ingerido. Os comprimidos não devem ser mastigados ou engolidos inteiros. A dispersão em bebidas gaseificadas ou leite não é recomendada devido à formação de espuma e à demora para dispersão, respectivamente.
Populações especiais
Pacientes idosos
As recomendações de dosagem para pacientes idosos são as mesmas descritas acima. Em estudos clínicos, pacientes idosos tiveram uma frequência maior de reações adversas do que pacientes mais jovens e devem ser monitorados cautelosamente para reações adversas que podem requerer o ajuste de dose.
Pacientes pediátricos
As recomendações de dosagem para pacientes pediátricos são as mesmas para pacientes adultos. Mudanças no peso de pacientes pediátricos ao longo do tempo devem ser consideradas no cálculo da dose.
Pacientes com insuficiência renal
O tratamento com ExjadeTM deve ser usado com cautela em pacientes com níveis de creatinina sérica acima do limite para a idade. Deve-se ter cautela especialmente ao ser usado em pacientes com clearance de creatinina entre 40 e 60 mL/min, particularmente nos casos em que há fatores de risco adicionais que podem comprometer a função renal, tais como medicações concomitantes, desidratação, ou infecções graves. As recomendações de dosagem inicial para pacientes com insuficiência renal são as mesmas descritas acima. A creatinina sérica deve ser monitorada mensalmente em todos os pacientes e, se necessário, doses diárias podem ser reduzidas em 10 mg/kg (vide "Advertências").
Pacientes com i