EVRYSDI
ROCHE
risdiplam
Tratamento de atrofia muscular espinhal (AME).
Apresentações.
Pó para solução oral
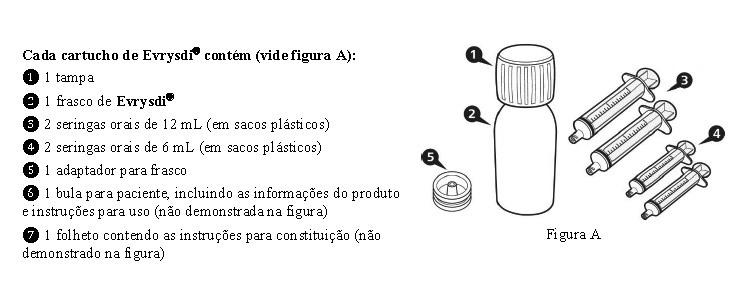
Cada cartucho contém 1 frasco de vidro âmbar contendo 2,0 g de pó para solução oral, 2 seringas reutilizáveis de 6 mL, 2 seringas reutilizáveis de 12 mL e um adaptador.
VIA ORAL
USO ADULTO E PEDIÁTRICO
Composição.
Princípio ativo: cada 2,0 g de pó para solução oral contém 60 mg de risdiplam. Cada mL da solução constituída contém 0,75 mg de risdiplam.
Excipientes: manitol, isomalte, aroma de morango (composto por maltodextrina de milho, amido de cera de milho modificado e ingredientes aromatizantes), ácido tartárico, benzoato de sódio, macrogol, sucralose, ácido ascórbico, edetato dissódico di-hidratado.
Informações técnicas.
1. INDICAÇÕES
Evrysdi® é indicado para o tratamento de atrofia muscular espinhal (AME).
2. RESULTADOS DE EFICÁCIA
A eficácia de Evrysdi® no tratamento de pacientes com AME de início na infância e AME de início tardio foi avaliada em 2 estudos clínicos pivotais, o FIREFISH e o SUNFISH, e é suportada por dados adicionais do estudo JEWELFISH. Os achados globais desses estudos embasam a eficácia de Evrysdi® em pacientes com AME.
AME de início na infância1, 2
O estudo BP39056 (FIREFISH) é um estudo aberto de 2 partes para investigar a eficácia, segurança, farmacocinética e farmacodinâmica (PD) de Evrysdi® em pacientes sintomáticos com AME tipo 1 (todos os pacientes tinham doença geneticamente confirmada com 2 cópias do gene SMN2). A parte 1 do FIREFISH foi projetada como a parte do estudo para determinação da dose. A parte 2 confirmatória do estudo FIREFISH avaliou a eficácia de Evrysdi® na dose terapêutica selecionada com base nos resultados da parte 1 (vide item "8. Posologia e Modo de Usar"). Os pacientes da parte 1 não participaram da parte 2.
Nas partes 1 e 2, o principal desfecho de eficácia foi a capacidade de se sentar sem apoio por ao menos 5 segundos, conforme medido pelo item 22 das escalas de desenvolvimento infantil de Bayley - terceira edição (Bayley III Scales of Infant and Toddler Development - BSID-III), escala da função motora grossa, após tratamento de 12 meses com Evrysdi®.
FIREFISH - parte 2
Na parte 2 do FIREFISH, foram incluídos 41 pacientes com AME tipo 1. A mediana de idade de início dos sinais e sintomas clínicos da AME tipo 1 foi de 1,5 meses (faixa de 1,0 - 3,0 meses), 54% eram mulheres, 54% eram caucasianos e 34% eram asiáticos. A idade mediana na inclusão foi de 5,3 meses (faixa de 2,2 - 6,9 meses) e o tempo mediano entre o início dos sintomas e a primeira dose foi de 3,4 meses (faixa de 1,0 - 6,0 meses). No início do estudo, a pontuação mediana de CHOP-INTEND foi de 22,0 pontos (faixa de 8,0 - 37,0) e a pontuação mediana de HINE-2 foi de 1,0 ponto (faixa de 0,0 - 5,0).
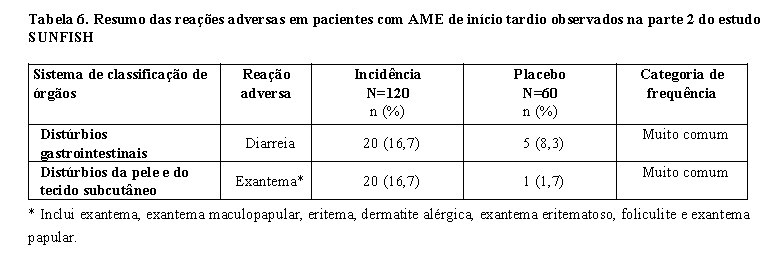
O desfecho primário foi a proporção de pacientes com a capacidade de sentar sem apoio por ao menos 5 segundos após 12 meses de tratamento (escala da função motora grossa BSID-III, item 22). Os desfechos de eficácia dos pacientes tratados com Evrysdi® foram comparados a coortes semelhantes de história natural de pacientes não tratados com AME de início na infância (definido como critérios de desempenho), conforme mostrado na tabela 1.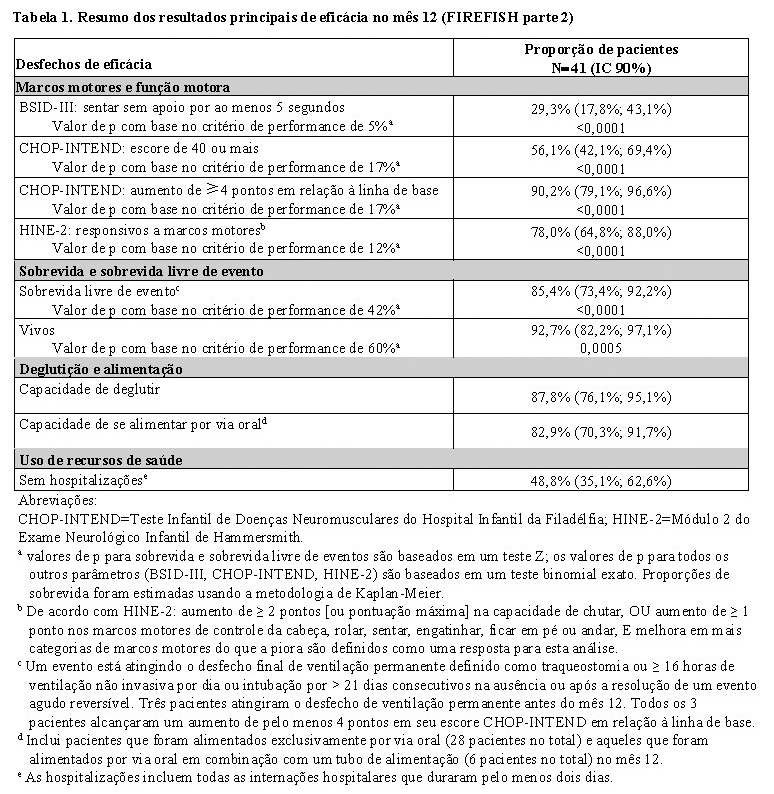
Após 12 meses de tratamento com Evrysdi®, 29% (12/41) dos pacientes atingiram os critérios para sentar sem apoio (BSID-III, item 22), 93% (38/41) dos pacientes estavam vivos e 85% (35/41) dos pacientes estavam vivos e livre de eventos (sem ventilação permanente), conforme demonstrado na Figura 1. Esses resultados indicam um desvio clinicamente significativo da história natural da AME com início na infância não tratada. Pacientes não tratados com AME de início na infância nunca seriam capazes de se sentar sem apoio e apenas 25% sobreviveriam sem ventilação permanente além dos 14 meses de idade.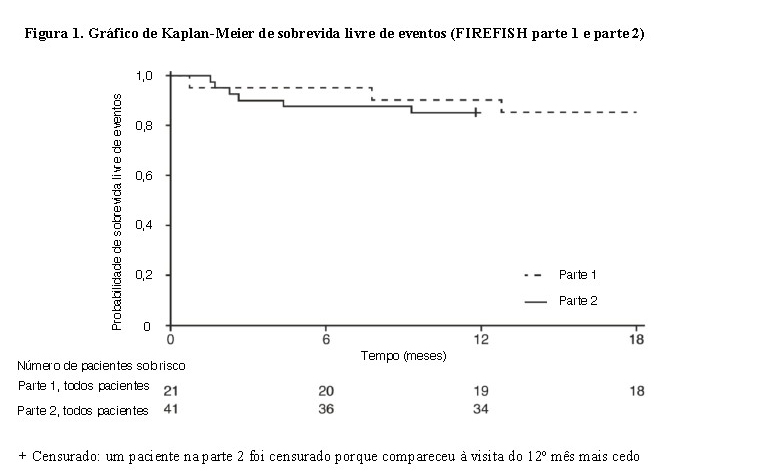
A maioria dos pacientes atingiu uma resposta nas categorias de marcos motores de HINE-2, incluindo algum nível de controle da cabeça (76%, 31/41), sentar (61%, 25/41), rolar (56%, 23/41) e ficar em pé (22%, 9/41). A melhoria da função motora também foi observada conforme medido pelo escore CHOP-INTEND total, conforme demonstrado na Figura 2.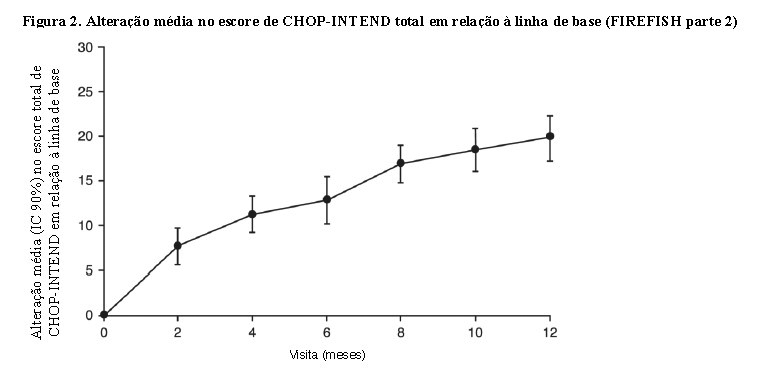
FIREFISH - parte 1
A eficácia de Evrysdi® em pacientes com AME tipo 1 também é suportada pelos resultados da parte 1 do estudo FIREFISH. Para os 21 pacientes da parte 1, as características na linha de base foram consistentes com pacientes sintomáticos com AME do tipo 1. A idade mediana na inclusão foi de 6,7 meses (faixa de 3,3 - 6,9 meses) e o tempo médio entre o início dos sintomas e a primeira dose foi de 4,0 meses (faixa de 2,0 - 5,8 meses). Um total de 17 pacientes receberam a dose terapêutica (dose selecionada para a parte 2) durante os primeiros 12 meses de tratamento. Após 12 meses de tratamento, 41% (7/17) dos pacientes conseguiam sentar-se independentemente por ao menos 5 segundos (BSID-III, item 22). Após 18 meses de tratamento, 88% (15/17) dos pacientes estavam vivos e livres de eventos (sem ventilação permanente), conforme demonstrado na Figura 1. Esses resultados na sobrevida e no desenvolvimento do marco motor foram consistentes com o FIREFISH parte 2.
AME de início tardio 2, 3, 4,5
O BP39055 (SUNFISH), é um estudo clínico de 2 partes, multicêntrico, para investigar a eficácia, segurança, farmacocinética (PK) e farmacodinâmica (PD) de Evrysdi® em pacientes com AME tipo 2 ou tipo 3 entre 2 e 25 anos de idade. A parte 1 compreendeu uma análise de determinação da dose, enquanto a parte 2 foi a porção confirmatória, randomizada, duplo-cega e controlada por placebo. Os pacientes da parte 1 não foram incluídos na parte 2.
O desfecho primário foi a alteração na escala de medida da função motora 32 (Motor Function Measure - MFM32) no mês 12 em relação à linha de base. A MFM32 avalia uma ampla variedade de funções motoras em uma ampla gama de pacientes com AME. O escore total da MFM32 é expresso em percentual (faixa de 0 - 100) do escore máximo possível, com as pontuações mais altas indicando maior função motora. A MFM32 avalia habilidades motoras que se relacionam a funções cotidianas importantes. Pequenas mudanças na função motora podem resultar em ganho ou perda significativa de funções da vida cotidiana.
SUNFISH - parte 2
A parte 2 é porção randomizada, duplo-cega e controlada por placebo do estudo SUNFISH, que incluiu 180 pacientes não-deambuladores com AME tipo 2 (71%) ou tipo 3 (29%). Os pacientes foram randomizados em uma proporção de 2:1 para receber Evrysdi® na dose terapêutica (vide item "8. Posologia e Modo de Usar") ou placebo. A randomização foi estratificada por faixa etária (2 a 5 anos, 6 a 11 anos, 12 a 17 anos, 18 a 25 anos).
A idade mediana dos pacientes no início do tratamento foi de 9,0 anos (faixa de 2 - 25 anos), o tempo mediano entre o início dos sintomas iniciais da AME e o primeiro tratamento foi de 102,6 (1 - 275) meses. Dos 180 pacientes incluídos no estudo, 51% eram do sexo feminino, 67% eram caucasianos e 19% eram asiáticos. No início do estudo, 67% dos pacientes tinham escoliose (32% deles com escoliose grave). Os pacientes tiveram um escore MFM32 médio na linha de base de 46,1 e RULM de 20,1. As características demográficas gerais na linha de base foram bem equilibradas entre os grupos com Evrysdi® e placebo, com exceção de um desequilíbrio de pacientes com escoliose (63,3% dos pacientes no braço Evrysdi® e 73,3% dos pacientes no braço placebo).
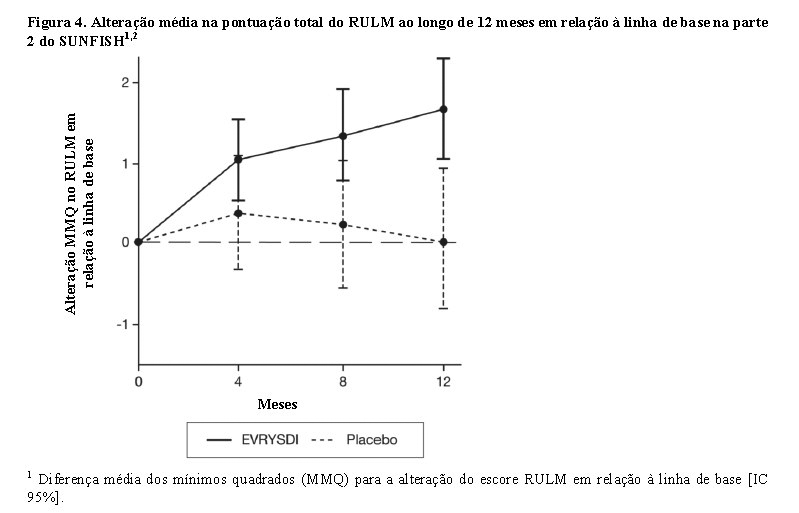
A análise primária da parte 2 do SUNFISH mostrou uma diferença clínica e estatisticamente significativa na alteração da pontuação total do MFM32 no mês 12 em comparação com a linha de base entre os pacientes tratados com Evrysdi® e placebo. Os resultados da análise primária e dos principais objetivos secundários são mostrados na tabela 2 e nas figuras 3 e 4.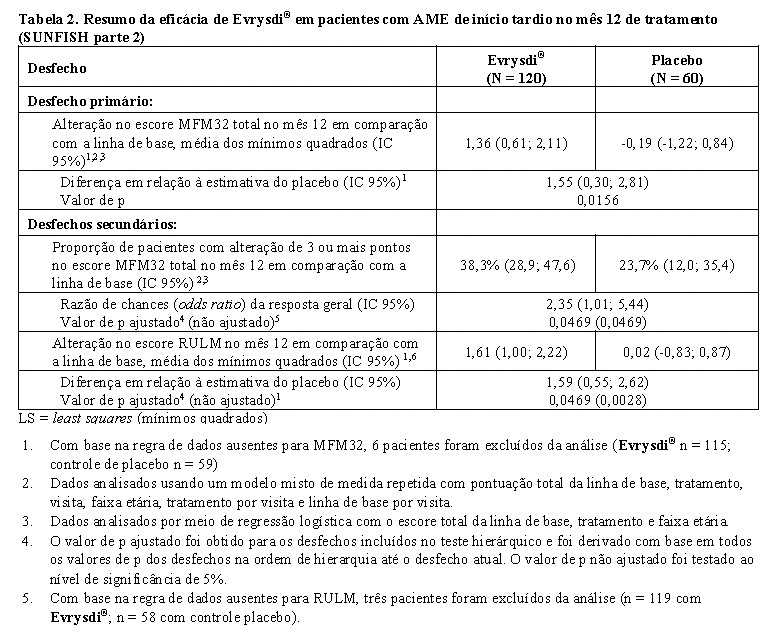
Em comparação ao placebo, os pacientes tratados com Evrysdi® demonstraram melhora significativa na função motora avaliada por MFM32 (diferença média de 1,55 ponto; p = 0,0156) após 12 meses de tratamento. Os pacientes com 2 - 5 anos de idade tratados com Evrysdi® demonstraram a maior melhora no MFM32 em comparação ao controle placebo (aumento ≥ 3 pontos: 78,1% vs. 52,9%). Pacientes com idade igual ou superior a 18 anos tratados com Evrysdi® alcançaram estabilização da doença (alteração ≥ 0 ponto(s) do escore MFM32 total em relação à linha de base: 57,1% vs. 37,5%). Observou-se melhora consistente no MFM32 em relação à linha de base em pacientes com AME tipo 2 e 3 (1,54 pontos [IC 95%: 0,06; 3,02]; 1,49 pontos [IC 95%: -0,94; 3,93], respectivamente) tratados com Evrysdi® em comparação com o placebo.
O estudo também atingiu um desfecho secundário independente da função motora, o RULM. Nessa escala foi observada melhora estatística e clinicamente significativa na função motora após 12 meses de tratamento em comparação à linha de base. Os pacientes de 2 - 5 anos de idade tratados com Evrysdi® demonstraram a maior melhora no RULM (3,41 pontos [IC 95%: 1,55; 5,26]) e também foi observada melhora nos pacientes com idade ≥18 anos (1,74 pontos [IC 95%: -1,06; 4,53]).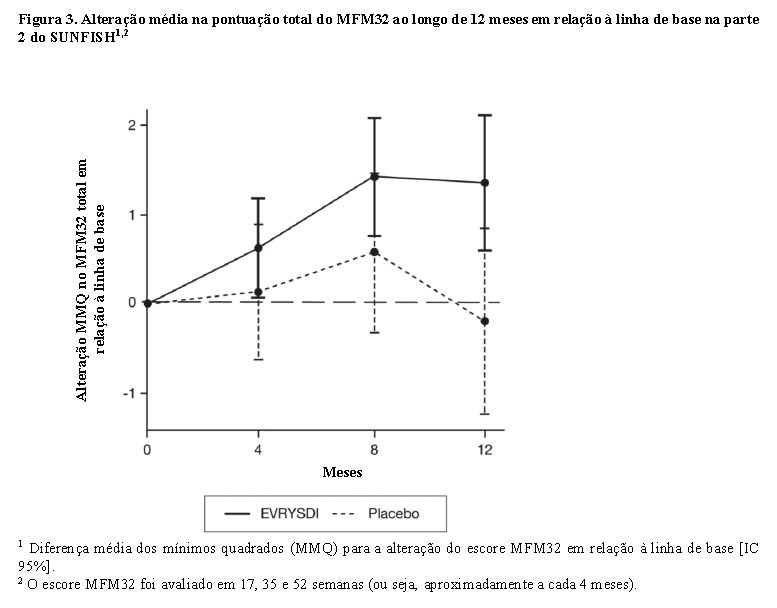
SUNFISH - parte 1
A eficácia do Evrysdi® em pacientes com AME de início tardio também foi suportada pelos resultados da parte 1 do SUNFISH que determina a dose do estudo. Na parte 1, 51 pacientes com AME tipos 2 e 3 (incluindo 7 pacientes ambulantes) entre 2 e 25 anos de idade foram incluídos. Após 1 ano de tratamento com a dose terapêutica (a dose selecionada para a parte 2), houve uma melhora clinicamente significativa na função motora medida por MFM32 com uma mudança média em relação à linha de base de 2,7 pontos (IC 95%: 1,5; 3,8). A melhora em MFM32 foi mantida até 2 anos sob o tratamento com Evrysdi® (alteração média de 2,7 pontos [IC 95%: 1,2; 4,2]).
Em uma análise exploratória, a função motora avaliada por MFM foi comparada entre a parte 1 do SUNFISH e uma coorte de história natural (ponderada com base nos principais fatores prognósticos). A mudança em relação à linha de base de MFM total após 1 ano e 2 anos foi maior em pacientes que receberam Evrysdi® em comparação com a coorte de história natural (após 1 ano: diferença de 2,7 pontos e p < 0,0001; após dois anos: diferença de 4,0 pontos e p < 0,0001). A coorte de história natural apresentou um declínio na função motora conforme esperado com base na progressão natural da AME (após 1 ano: -0,6 de alteração média; após 2 anos: -2,0 de alteração média).
Uso em pacientes com AME previamente tratados 6,7
O BP39054 (JEWELFISH) é um estudo aberto de braço único, para investigar a segurança, tolerabilidade, farmacocinética e farmacodinâmica de Evrysdi® em pacientes com AME de início na infância e de início tardio com idade entre 6 meses a 60 anos, que receberam tratamento farmacológico anterior para AME (incluindo nusinersena e onasemnogeno abeparvoveque). Dos 174 pacientes incluídos, 76 pacientes foram tratados anteriormente com nusinersena (9 pacientes com AME tipo 1, 43 com AME tipo 2 e 24 com AME tipo 3) e 14 pacientes foram tratados anteriormente com onasemnogeno abeparvoveque (4 pacientes com AME tipo 1 e 10 com AME tipo 2).
Pacientes tiveram, em média, um aumento superior a 2 vezes nos níveis da proteína SMN no sangue em comparação ao período basal após 4 semanas de tratamento com Evrysdi®.
Referências bibliográficas:
1 - Primary CSR Study BP39056 (FIREFISH): A two part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of risdiplam in infants with type 1 Spinal Muscular Atrophy. Report No. 1100385. April 2020.
2 - Risdiplam CTD Module 2, section 2.7.3 Summary of Clinical Efficacy. August 2019.
3 - Interim Clinical Study Report, Study BP39055 (SUNFISH): A two-part seamless, multi-center, randomized, placebo-controlled, double-blind study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of RO7034067 in Type 2 and 3 spinal muscular atrophy patients. Report No. 1088216. July 2019.
4 - Risdiplam CTD. Module 2, section 2.7.3 Summary of Clinical Efficacy. August 2019.
5 - Primary CSR Study BP39055, (SUNFISH): A two-part seamless, multicenter randomized, placebo-controlled, double-blind study to investigate the safety, tolerability, pharmacokinetics, pharmacodynamics and efficacy of ro7034067 in type 2 and 3 Spinal Muscular Atrophy Patients. Report No. 1099250, Feb 2020.
6 - Interim Clinical Study Report, Study BP39054 (Jewelfish): Open-Label Study to Investigate the Safety, Tolerability, and Pharmacokinetics/Pharmacodynamics of RO7034067 in Adult and Pediatric Patients With Spinal Muscular Atrophy. July 2019.
7 - Interim CSR BP39054 (JEWELFISH): An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of RO7034067 in adult and pediatric patients with spinal muscular atrophy. Report No. 1100549. June, 2020.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Mecanismo de ação
Risdiplam é um modificador do splicing (maturação) do pré-mRNA de sobrevivência do neurônio motor 2 (SMN2) desenvolvido para tratar a AME causada por mutações no gene SMN1 presente no cromossomo 5q que levam à deficiência na síntese da proteína SMN. A deficiência na proteína SMN funcional é o mecanismo fisiopatológico de todos os tipos de AME. Risdiplam corrige o splicing de SMN2 para deslocar o equilíbrio da exclusão do éxon 7 para a inclusão desse éxon no mRNA transcrito, promovendo um aumento na produção da proteína SMN funcional e estável. Assim, risdiplam trata a AME aumentando e mantendo os níveis funcionais da proteína SMN.
Risdiplam se distribui de modo uniforme em todas as partes do corpo, incluindo o sistema nervoso central (SNC), atravessando a barreira hematoencefálica e levando, assim, ao aumento da proteína SMN no SNC e em todo o corpo. As concentrações de risdiplam no plasma e da proteína SMN no sangue refletem sua distribuição e seus efeitos farmacodinâmicos em tecidos como o cerebral e o muscular.
Em todos os estudos clínicos, risdiplam levou a um aumento consistente e persistente na proteína SMN, com uma alteração mediana superior a 2 vezes em relação ao período basal até 4 semanas do início do tratamento, medida no sangue. Esse aumento no nível de proteína SMN foi sustentado ao longo do período de tratamento de até 2 anos para pacientes com AME de início na infância e pacientes com AME de início tardio (vide item "2. Resultados de Eficácia").
Farmacocinética
Os parâmetros farmacocinéticos de Evrysdi® foram caracterizados em indivíduos adultos saudáveis e em pacientes com AME.
Após a administração de Evrysdi® como solução oral, a farmacocinética de risdiplam foi aproximadamente linear entre 0,6 e 18 mg. A melhor descrição da farmacocinética (PK) de risdiplam foi feita por um modelo de farmacocinética populacional com absorção tricompartimental de trânsitos, distribuição bicompartimental e eliminação de primeira ordem. Verificou-se que o peso corporal e a idade têm um efeito significativo na farmacocinética.
A exposição estimada (ASC0-24h média) para pacientes com AME de início na infância (idade de 2 a 7 meses no momento da inclusão) na dose terapêutica de 0,2 mg/kg uma vez ao dia foi de 1930 ng.h/mL. A exposição estimada para pacientes com AME de início tardio (idade de 2 a 25 anos no momento da inclusão) no estudo SUNFISH (parte 2) na dose terapêutica foi de 2070 ng.h/mL (0,25 mg/kg uma vez ao dia para pacientes com peso corporal < 20kg) e 1680 ng.h/mL (5 mg uma vez ao dia para pacientes com peso corporal ≥ 20 kg). A concentração máxima observada (Cmáx média) foi 194 ng/mL a 0,2 mg/kg no FIREFISH e 120 ng/mL no SUNFISH parte 2.
Absorção
Risdiplam foi rapidamente absorvido em jejum, com tmáx plasmático variando de 1 a 4 horas após a administração oral. A ingestão de alimento (café da manhã com alto teor de gorduras e calorias) não teve efeito relevante na exposição de risdiplam.
Distribuição
As estimativas de parâmetros farmacocinéticos populacionais foram de 98 L para o volume de distribuição central aparente, 93 L para o volume periférico e 0,68 L/hora para depuração intercompartimental.
Risdiplam se liga predominantemente à albumina sérica, sem nenhuma ligação a alfa-1-glicoproteína ácida, com uma fração livre de 11%.
Metabolismo
Risdiplam é metabolizado principalmente por flavina monooxigenase 1 e 3 (FMO1 e FMO3) e também pelas CYPs 1A1, 2J2, 3A4 e 3A7.
A administração concomitante de 200 mg de itraconazol duas vezes ao dia, um forte inibidor de CYP3A, com uma dose oral única de 6 mg de risdiplam não mostrou efeito clinicamente relevante na PK de risdiplam (aumento de 11% na ASC, redução de 9% na Cmáx).
Eliminação
As análises de PK populacional estimaram uma depuração aparente (Cl/F) de 2,6 L/h para risdiplam.
A meia-vida efetiva de risdiplam foi de aproximadamente 50 horas em pacientes com AME.
Risdiplam não é um substrato da proteína humana tipo 1 de resistência a múltiplos medicamentos (MDR1).
Aproximadamente 53% da dose (14% de risdiplam inalterado) foram excretados nas fezes e 28% na urina (8% de risdiplam inalterado). O fármaco original foi o principal componente encontrado no plasma, totalizando 83% do material relacionado ao fármaco na circulação. O metabólito farmacologicamente inativo M1 foi identificado como o principal metabólito circulante.
Farmacocinética em populações especiais
População pediátrica
O peso corporal e a idade foram identificados como covariáveis na análise de PK populacional. Desse modo, a dose é ajustada com base na idade (abaixo e acima de 2 anos) e peso corporal (até 20 kg) para obter uma exposição semelhante entre as faixas de idade e peso corporal. Não há dados disponíveis em pacientes com menos de 2 meses de idade.
População geriátrica
Não foram realizados estudos específicos para investigar a farmacocinética de Evrysdi® em pacientes com AME acima de 60 anos de idade. Pacientes com AME até 60 anos de idade foram incluídos no estudo JEWELFISH. Indivíduos sem AME até 69 anos de idade foram incluídos em estudos clínicos de PK, o que indica que nenhum ajuste de dose é necessário para pacientes até 69 anos de idade.
Insuficiência renal
Nenhum estudo foi realizado para investigar a farmacocinética de risdiplam em pacientes com insuficiência renal. A eliminação de risdiplam como composto inalterado via excreção renal é insignificante (8%).
Insuficiência hepática
A insuficiência hepática leve e moderada não teve impacto na farmacocinética de risdiplam. Após a administração de 5 mg de risdiplam, as taxas médias para Cmáx e ASC foram de 0,95 e 0,80 em indivíduos com insuficiência hepática leve (n = 8) e 1,20 e 1,08 em indivíduos com insuficiência hepática moderada (n = 8) em comparação com controles saudáveis compatíveis (n = 10). A segurança e a farmacocinética em pacientes com insuficiência hepática grave não foram estudadas.
Etnia
A farmacocinética de risdiplam não é diferente em indivíduos japoneses e caucasianos.
Segurança não clínica
Carcinogenicidade
Um estudo de carcinogenicidade realizado com risdiplam em camundongos rasH2 transgênicos não forneceu nenhuma evidência de potencial tumorigênico de risdiplam com animais expostos até 7 vezes a exposição em humanos na dose terapêutica.
Genotoxicidade
Risdiplam não é mutagênico em um ensaio de mutação reversa bacteriana. Em células de mamíferos in vitro e na medula óssea de ratos, risdiplam aumenta a frequência de células micronucleadas. A indução de micronúcleo na medula óssea foi observada em diversos estudos de toxicidade em ratos (animais adultos e jovens). O nível sem efeitos adversos observáveis (NOAEL) entre os estudos está associado a uma exposição de aproximadamente 1,5 vezes a exposição em humanos na dose terapêutica. Dados indicaram que esse efeito é indireto e secundário a uma interferência de risdiplam no ciclo celular de células de divisão. Esses efeitos também se manifestam em outros tecidos com elevada regeneração celular, com alterações na pele, trato gastrointestinal (GI), células germinativas masculinas, na toxicidade embrionária e na medula óssea. Risdiplam não possui potencial para danificar o DNA diretamente.
Comprometimento da fertilidade
O tratamento com risdiplam foi associado a uma suspensão das células germinativas masculinas em ratos e macacos. Esses efeitos levaram a uma degeneração de espermatócitos, degeneração/ necrose do epitélio seminífero e oligo/aspermia no epidídimo. Além disso, foram observadas reduções nas concentrações de esperma e na motilidade de espermatozoides, associadas a um aumento no número de anormalidades na morfologia dos espermatozoides. Em ratos jovens, os efeitos foram observados nos níveis de exposição atingidos na dose terapêutica de risdiplam nos pacientes. No entanto, não houve comprometimento da fertilidade masculina observado em um estudo respectivo em ratos. Os efeitos de risdiplam nos espermatozoides provavelmente estão relacionados a uma interferência de risdiplam no ciclo celular das células de divisão, sendo específicos do estágio e reversíveis. Não foram observados efeitos nos órgãos reprodutores femininos em ratos e macacos após o tratamento com risdiplam.
Toxicidade reprodutiva
Em estudos em ratas prenhes tratadas com risdiplam, a toxicidade embriofetal com peso mais baixo do feto e atraso de desenvolvimento foram evidentes. O NOAEL para este efeito foi aproximadamente duas vezes acima dos níveis de exposição alcançados na dose terapêutica de risdiplam nos pacientes. Em estudos com coelhas prenhes, efeitos dismorfogênicos foram observados a exposições também associadas à toxicidade materna. Tais efeitos consistiram em quatro fetos (4%) de 4 ninhadas (22%) com hidrocefalia. O NOAEL foi de aproximadamente quatro vezes os níveis de exposição alcançados na dose terapêutica de risdiplam nos pacientes.
Em um estudo pré e pós-natal em ratas tratadas diariamente com risdiplam, observou-se que risdiplam causou um leve prolongamento do tempo de gestação. Nenhum efeito adverso foi registrado na sobrevida, crescimento, desempenho funcional (comportamental ou reprodutivo) dos filhotes. Não houve efeitos nas células germinativas femininas, conforme avaliado por contagens do folículo primordial e por histopatologia ovariana.
Estudos em ratas prenhes e lactantes mostraram que risdiplam atravessa a barreira placentária e é excretado no leite.
Outros
Efeito na estrutura da retina
O tratamento crônico de macacos com risdiplam produziu evidências de efeito na retina em termos de degeneração de fotorreceptores a partir da periferia da retina. Mediante a interrupção do tratamento, os efeitos na retinografia foram parcialmente reversíveis, mas a degeneração de fotorreceptores não se reverteu. Os efeitos foram monitorados por tomografia de coerência óptica (OCT) e na eletrorretinografia (ERG). Alguns dados experimentais indicam que o efeito pode ser causado por um comprometimento da reciclagem dos fotorreceptores no epitélio pigmentar da retina. O efeito tem um NOAEL evidente na dose clínica utilizada para risdiplam. Os efeitos foram observados com exposições superiores a 2 vezes a exposição em humanos na dose terapêutica. Nenhum achado semelhante foi observado em ratos albinos ou pigmentados que receberam risdiplam de forma crônica a exposições que excederam às exposições em macacos. Esses achados não foram observados em estudos clínicos em pacientes com AME com monitoramento oftalmológico regular (incluindo OCT de domínio espectral [SD OTC] e avaliação da função visual).
Efeito nos tecidos epiteliais
Os efeitos na histologia da pele, laringe e pálpebra e no trato gastrointestinal (GI) foram evidentes em ratos e macacos tratados com risdiplam. As alterações começaram a ser notadas em doses elevadas no tratamento de 2 semanas ou mais. Com o tratamento crônico por 39 semanas em macacos, o NOAEL ocorreu a uma exposição superior a 2 vezes a exposição média em humanos na dose terapêutica. Os efeitos no tecido epitelial cutâneo observados em estudos em animais não foram observados em estudos clínicos em pacientes com AME.
Efeito nos parâmetros hematológicos
No teste de micronúcleo em medula óssea após exposição aguda em ratos, uma redução de mais de 50% na proporção de eritrócitos policromáticos (jovens) para normocromáticos (adultos), indicativa de toxicidade substancial na medula óssea, foi observada no nível de dose alta a uma exposição superior a 15 vezes a exposição média em humanos na dose terapêutica. Com o tratamento de ratos por 4 semanas, tais efeitos não foram observados até a dose mais alta a uma exposição de aproximadamente 7 vezes a exposição média em humanos na dose terapêutica, embora mortes e sacrifícios prematuros provavelmente com base nos efeitos hematológicos tenham sido observados com o tratamento crônico de ratos durante 26 semanas na mesma exposição. O NOAEL para efeitos hematológicos em ratos tratados por 26 semanas foi atingido em aproximadamente 3,5 vezes acima da exposição alcançada em humanos na dose terapêutica. A indução de micronúcleos na medula óssea foi observada em diversos estudos de toxicidade em ratos (animais adultos e jovens), com uma exposição no NOAEL de aproximadamente 1,5 vez a exposição média em humanos na dose terapêutica. Os parâmetros hematológicos permaneceram inalterados durante o tratamento com Evrysdi® nos estudos clínicos em pacientes com AME.
Estudos em animais jovens
A toxicidade de risdiplam foi estudada com a administração crônica em ratos e macacos, incluindo estudos em animais jovens. Os estudos em animais jovens não indicaram efeito específico do tratamento com risdiplam nos sistemas de órgãos em desenvolvimento. Em relação à toxicidade observada após o tratamento com risdiplam em vários sistemas de órgãos com alta regeneração celular (pele, trato GI, medula óssea), os estudos em animais não indicaram nenhuma diferença na sensibilidade entre animais jovens, adolescentes ou adultos.
4. CONTRAINDICAÇÕES
Evrysdi® é contraindicado para pacientes com hipersensibilidade conhecida a risdiplam ou a qualquer um dos excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Geral
Toxicidade embriofetal
Toxicidade embriofetal foi observada em estudos em animais (vide item "3. Características Farmacológicas - Segurança não clínica). Os pacientes com potencial reprodutivo devem ser informados dos riscos e devem utilizar contracepção altamente eficaz durante o tratamento e até pelo menos 1 mês após a última dose de Evrysdi® para pacientes do sexo feminino e 4 meses após a última dose de Evrysdi® para pacientes do sexo masculino.
Possíveis efeitos na fertilidade masculina
Devido aos efeitos reversíveis de Evrysdi® na fertilidade masculina com base nas observações de estudos em animais, os pacientes do sexo masculino não devem doar esperma durante o tratamento e por 4 meses após a última dose de Evrysdi®.
Uso em populações especiais
Homens e mulheres com potencial reprodutivo
Fertilidade em pacientes do sexo masculino
A fertilidade masculina pode ficar comprometida durante o tratamento com Evrysdi® com base nos achados não clínicos. Em órgãos reprodutores de ratos e macacos, observou-se degeneração do esperma e números reduzidos de esperma (vide item "3. Características Farmacológicas - Segurança não clínica"). Os efeitos nos espermatozoides são reversíveis mediante a descontinuação de risdiplam. Antes de iniciar o tratamento com Evrysdi®, estratégias para preservar a fertilidade devem ser discutidas com os pacientes do sexo masculino que receberem Evrysdi®. Os pacientes do sexo masculino podem considerar armazenar esperma antes do início do tratamento ou após um período sem tratamento de no mínimo 4 meses. Os pacientes do sexo masculino que desejarem ter filhos devem interromper o tratamento com Evrysdi® por no mínimo 4 meses. O tratamento pode ser reiniciado após a concepção.
Fertilidade em pacientes do sexo feminino
Com base em dados não clínicos, não é esperado impacto de Evrysdi® na fertilidade feminina (item "3. Características Farmacológicas - Segurança não clínica").
Teste de gravidez
O status de gravidez de mulheres com potencial reprodutivo deve ser verificado antes de iniciar a terapia com Evrysdi®. As mulheres grávidas devem ser claramente alertadas sobre o possível risco ao feto.
Contracepção
Pacientes do sexo masculino e do sexo feminino com potencial reprodutivo devem aderir às seguintes exigências de contracepção:
• As pacientes do sexo feminino com potencial para engravidar devem utilizar contracepção altamente eficaz durante o tratamento com Evrysdi® e por no mínimo 1 mês após a última dose.
• Tanto os pacientes do sexo masculino quanto suas parceiras com potencial para engravidar devem utilizar contracepção altamente eficaz durante o tratamento com Evrysdi® e por no mínimo 4 meses após a última dose.
Gravidez e lactação
Categoria de risco na gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Gravidez
Não há dados clínicos sobre o uso de Evrysdi® em mulheres grávidas. Risdiplam demonstrou ser embriofetotóxico e teratogênico em animais. Com base nos achados de estudos em animais, risdiplam atravessa a barreira placentária e pode causar dano fetal (vide item "3. Características Farmacológicas - Segurança não clínica").
Evrysdi® não deve ser utilizado durante a gravidez, a menos que o benefício para a mãe supere os possíveis riscos ao feto. Se uma gestante precisar ser tratada com Evrysdi®, ela deve ser claramente alertada sobre o possível risco ao feto.
O uso seguro de Evrysdi® durante o parto e trabalho de parto não foi estabelecido.
Lactação
Não se sabe se Evrysdi® é excretado no leite materno humano. Estudos em ratos demostram que risdiplam é excretado no leite (vide item "3. Características Farmacológicas - Segurança não clínica"). Uma vez que o potencial para causar dano ao lactente é desconhecido, a decisão deve ser tomada com o médico responsável pelo tratamento do paciente. Recomenda-se não amamentar durante o tratamento com Evrysdi®.
Uso pediátrico
Vide itens "1. Indicações", "8. Posologia e Modo de Usar", "2. Resultados de Eficácia", "3. Características Farmacológicas", "9. Reações Adversas".
Uso geriátrico
A farmacocinética e a segurança de Evrysdi® foram avaliadas em indivíduos sem AME até 69 anos de idade. Evrysdi® não foi estudado em pacientes com AME acima de 60 anos de idade (vide itens "3. Características Farmacológicas" e "2. Resultados de Eficácia").
Insuficiência renal
A segurança e a eficácia de Evrysdi® em pacientes com insuficiência renal não foi estudada. Não se espera que uma mudança da dose seja necessária em pacientes com insuficiência renal (vide itens "8. Posologia e Modo de Usar" e "3. Características Farmacológicas").
Insuficiência hepática
A farmacocinética, segurança e tolerabilidade de uma dose única de 5 mg de risdiplam foram avaliadas em indivíduos com insuficiência hepática leve ou moderada em um estudo clínico dedicado. A insuficiência hepática leve ou moderada não impactou a farmacocinética de risdiplam. Portanto, não é necessário realizar o ajuste de dose em pacientes com insuficiência hepática leve ou moderada. Evrysdi® não foi estudado em pacientes com insuficiência hepática grave (vide itens "8. Posologia e Modo de Usar" e "3. Características Farmacológicas").
Abuso e dependência do medicamento
Evrysdi® não tem potencial de causar abuso e dependência.
Efeitos sobre a capacidade de conduzir veículos ou operar máquinas
Evrysdi® não tem influência na capacidade de dirigir e operar máquinas.
Outras
Até o momento, não há informações de que Evrysdi® (risdiplam) possa causar doping.
6. INTERAÇÕES MEDICAMENTOSAS
Risdiplam é metabolizado principalmente pela flavina monooxigenase 1 e 3 (FMO1 e 3) e também por CYPs 1A1, 2J2, 3A4 e 3A7. Risdiplam não é um substrato da proteína humana de resistência a múltiplos medicamentos tipo 1 (MDR1).
Efeitos de outros medicamentos em Evrysdi®.
A coadministração de 200 mg de itraconazol duas vezes ao dia, um forte inibidor de CYP3A, com uma dose oral única de 6 mg de risdiplam não demonstrou um efeito clinicamente relevante na farmacocinética (PK) de risdiplam (aumento de 11% na ASC, redução de 9% na Cmáx). Nenhum ajuste da dose é necessário quando Evrysdi® é administrado concomitantemente com um inibidor de CYP3A.
Nenhuma interação medicamentosa é esperada pela via FMO1 e FMO3.
Efeitos de Evrysdi® em outros medicamentos
Risdiplam in vitro e seu principal metabólito circulante M1 não induziram CYP1A2, 2B6, 2C8, 2C9, 2C19 ou 3A4. Risdiplam e M1 in vitro não inibiram (inibição reversível ou dependente do tempo) nenhuma das enzimas CYP testadas (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6) com exceção de CYP3A.
Evrysdi® é um inibidor fraco da CYP3A. Em indivíduos adultos saudáveis, a administração de Evrysdi® uma vez por dia durante 2 semanas aumentou ligeiramente a exposição a midazolam, um substrato sensível da CYP3A (ASC 11%; Cmáx 16%). A extensão da interação não é considerada clinicamente relevante e, portanto, nenhum ajuste de dose é necessário para substratos de CYP3A. Com base em modelagem farmacocinética baseada na fisiologia, uma magnitude semelhante deste efeito é esperada em crianças e bebês a partir dos 2 meses de idade.
Estudos in vitro demonstraram que risdiplam e seu principal metabólito não são inibidores significativos da MDR1 humana, do polipeptídeo transportador de ânion orgânico (OATP)1B1, OATP1B3, e dos transportadores de ânions orgânicos 1 e 3 (OAT 1 e 3). No entanto, risdiplam e seu metabólito são inibidores in vitro do transportador de cátion orgânico humano 2 (OCT2) e dos transportadores de extrusão de múltiplos fármacos e toxinas (MATE)1 e MATE2-K. Em concentrações terapêuticas do medicamento, não se espera nenhuma interação com substratos de OCT2. Com base em dados in vitro, Evrysdi® pode aumentar as concentrações plasmáticas de medicamentos eliminados por MATE1 ou MATE2-K. A relevância clínica da administração concomitante com substratos de MATE1/2-K é desconhecida.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenamento
Pó para solução: conservar sob refrigeração (entre 2 e 8°C). Manter no cartucho.
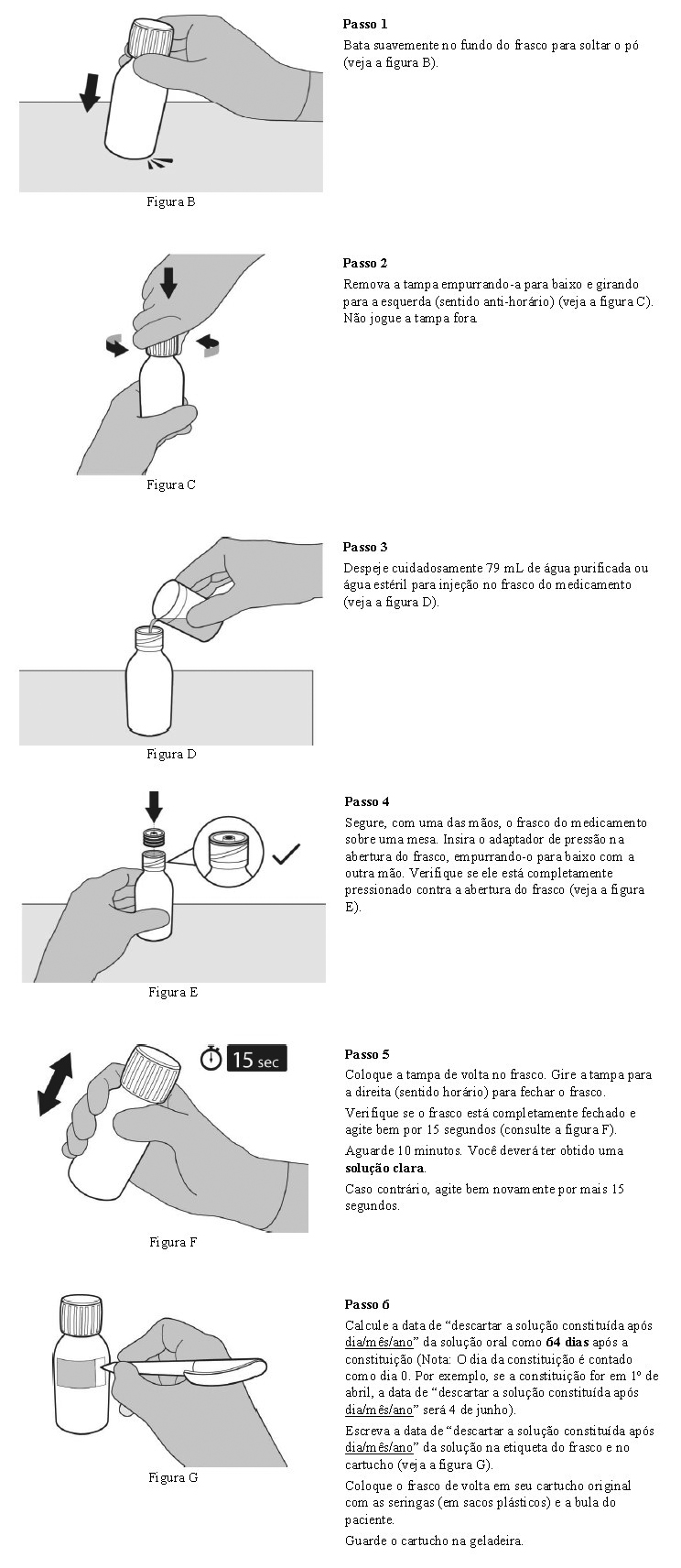
Solução constituída: após constituição, conservar sob refrigeração (entre 2 e 8°C) por até 64 dias. Não congelar. Manter a solução oral no frasco original, sempre em posição vertical e com a tampa bem fechada. Manter no frasco âmbar original para proteger da luz.
Prazo de validade
Este medicamento não deve ser utilizado após a data de validade ("Val:" para o pó e "Descartar a solução constituída após dia/mês/ano" para a solução oral constituída) na embalagem e no frasco.
Esse medicamento em pó possui prazo de validade de 24 meses a partir da data de fabricação.
Após o preparo, manter a solução oral sob refrigeração (entre 2 e 8°C) por 64 dias.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Descarte de medicamentos não utilizados/ vencidos
O descarte de produtos farmacêuticos no meio-ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto e o descarte em lixo doméstico deve ser evitado.
As exigências locais devem ser seguidas para o processo de descarte de medicamentos não utilizados / vencidos.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
Evrysdi® é um pó amarelo esverdeado a amarelo e, após constituição, é uma solução oral amarelo esverdeada a amarelada.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
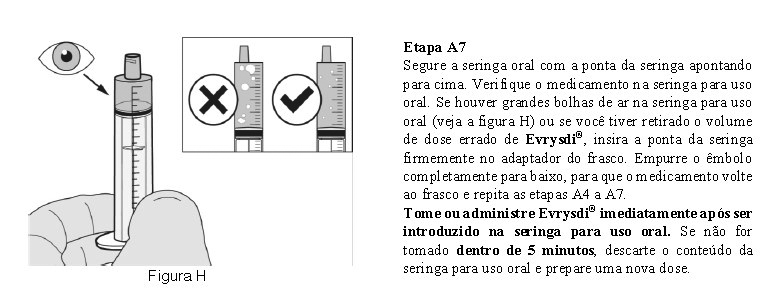
8. POSOLOGIA E MODO DE USAR
Evrysdi® pó para solução oral deve ser constituído para uma solução oral por um profissional de saúde antes de ser dispensado.
Instruções gerais
O tratamento para AME deve ser iniciado assim que possível após o diagnóstico.
Evrysdi® é administrado por via oral uma vez ao dia, utilizando a seringa oral fornecida, aproximadamente no mesmo horário todos os dias.
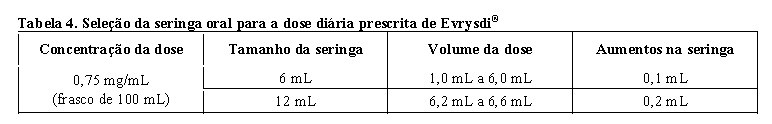
A dose diária recomendada de Evrysdi® para pacientes com AME é determinada pela idade e peso corporal (vide tabela 3) a seguir.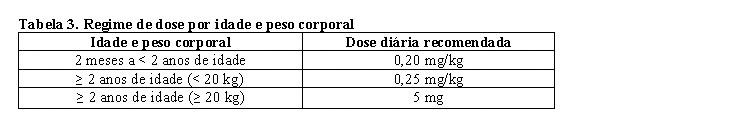
Mudanças na dose devem ser realizadas sob a supervisão de um profissional de saúde. O tratamento com dose diária acima de 5 mg não foi estudado. Não há dados disponíveis em bebês com menos de 2 meses de idade.
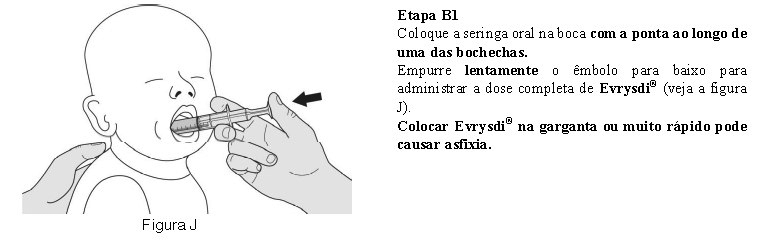
Método de administração
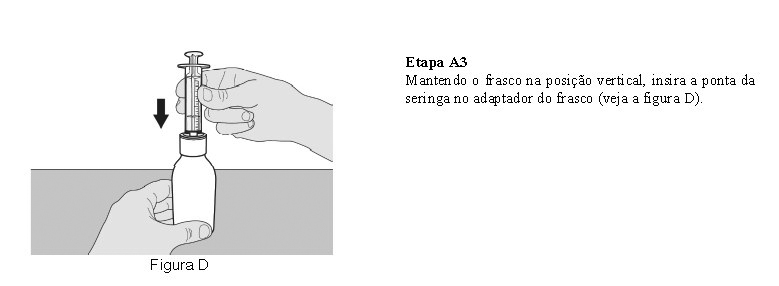
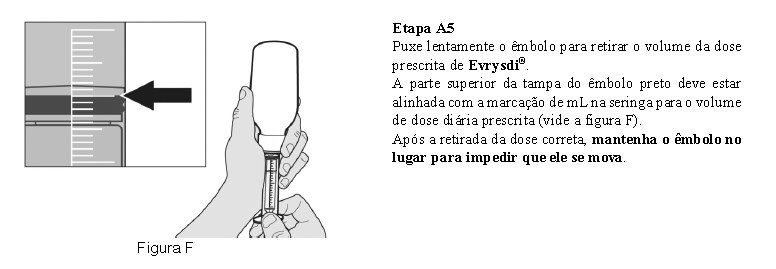
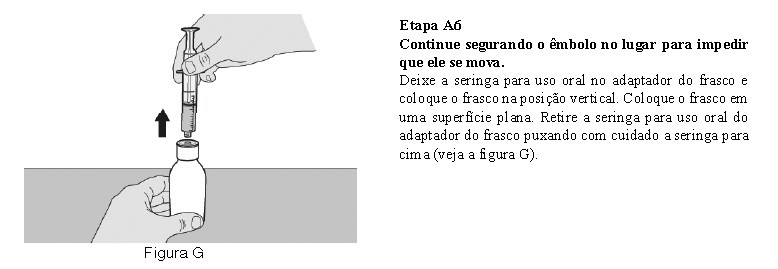
Utilize a seringa oral reutilizável fornecida para administrar a dose diária de Evrysdi®. Recomenda-se que o profissional de saúde discuta com o paciente ou cuidador sobre como preparar a dose diária prescrita antes da administração da primeira dose.
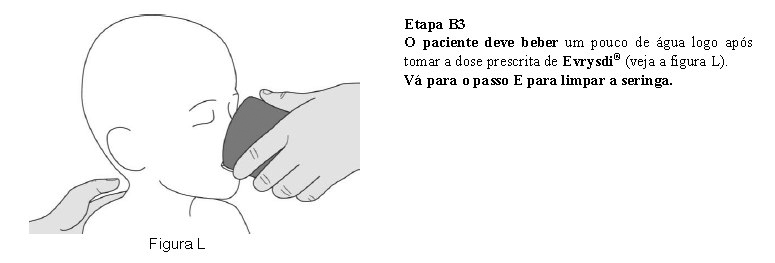
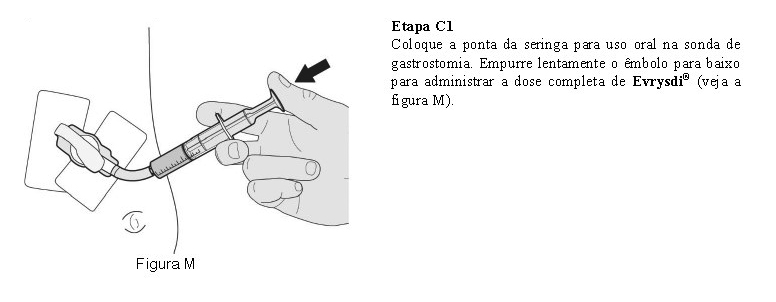
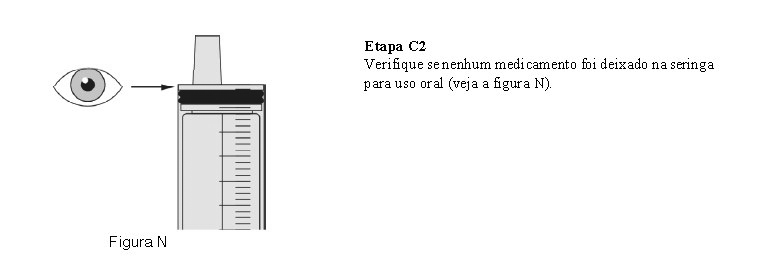
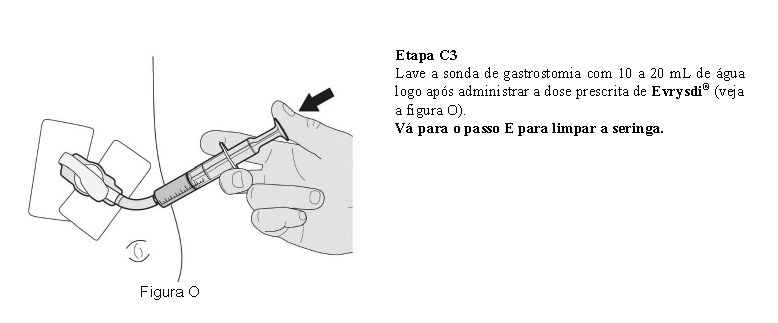
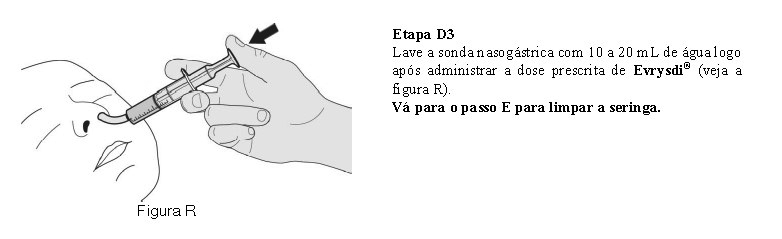
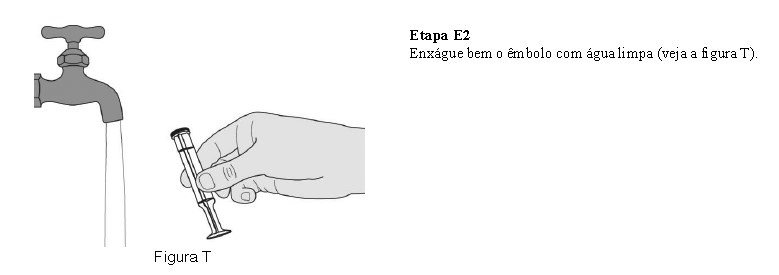
O paciente deve beber água após tomar Evrysdi® para garantir que o medicamento foi engolido completamente. Se o paciente não conseguir engolir e tiver uma sonda nasogástrica ou de gastrostomia, administre Evrysdi® pela sonda. A sonda deve ser lavada com água após a administração de Evrysdi®.
Doses atrasadas ou esquecidas
Evrysdi® é administrado via oral uma vez ao dia, aproximadamente no mesmo horário todos os dias. Se alguma dose de Evrysdi® for esquecida, administre-a assim que possível se ainda estiver dentro de 6 horas da dose programa