EVOBRIG
TAKEDA
brigatinibe
Antineoplásico.
Apresentações.
Comprimidos revestidos contendo 30 mg de brigatinibe - Embalagem com 28 comprimidos
Comprimidos revestidos contendo 90 mg de brigatinibe - Embalagem com 28 comprimidos
Comprimidos revestidos contendo 180 mg de brigatinibe - Embalagem com 28 comprimidos.
Embalagem contendo 28 comprimidos revestidos - 7 comprimidos revestidos de 90 mg + 21 comprimidos revestidos de 180 mg.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém 30 mg, 90 mg ou 180 mg de brigatinibe. Excipientes: lactose monoidratada, celulose microcristalina, amidoglicolato de sódio, sílica coloidal hidrofóbica, estearato de magnésio, talco, macrogol, álcool polivinílico, dióxido de titânio.
Informações técnicas.
1. INDICAÇÕES
EVOBRIG (brigatinibe) é indicado para o tratamento de pacientes com câncer de pulmão não pequenas células (CPNPC) localmente avançado ou metastático que seja positivo para quinase de linfoma anaplásico (ALK), previamente tratados com crizotinibe.
2. RESULTADOS DE EFICÁCIA
• ALTA (Estudo 201) - CPNPC avançado ou metastático ALK-positivo previamente tratado com Crizotinibe2
A segurança e a eficácia de brigatinibe foram avaliadas em um estudo randomizado (1:1), aberto, multicêntrico (ALTA) em 222 pacientes adultos com câncer de pulmão de células não pequenas, metastático ou localmente avançado, positivo para ALK que progrediram com crizotinibe. Os critérios de elegibilidade permitiram a inclusão de pacientes com rearranjo de ALK documentado, escala de performance ECOG de 0-2, quimioterapia anterior e metástases no sistema nervoso central (SNC), desde que fossem neurologicamente estáveis e não exigissem o aumento de dose de corticosteroides. Pacientes com histórico de doença pulmonar intersticial ou pneumonite relacionada ao medicamento foram excluídos.
Os pacientes foram randomizados na razão 1:1 para receber 90 mg de brigatinibe uma vez ao dia (regime de 90 mg, n=112) ou 180 mg de brigatinibe uma vez ao dia, com uma dose inicial de 90 mg por 7 dias (regime de 180 mg, n=110). A duração média do acompanhamento foi de 22,9 meses (faixa: 0,1 - 39,2).
A randomização foi estratificada por metástases cerebrais (presente, ausente) e melhor resposta anterior à terapia com crizotinibe (resposta completa ou parcial, qualquer outra resposta/desconhecido).
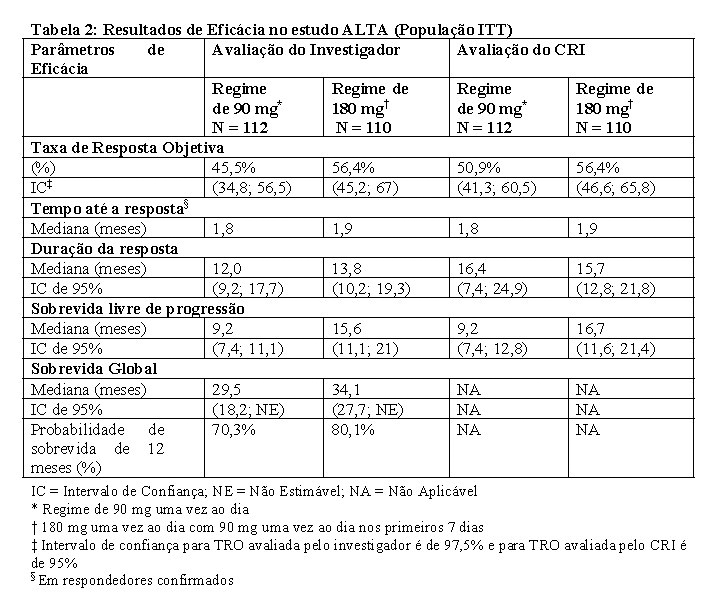
O principal desfecho foi a taxa de resposta objetiva (TRO) confirmada, de acordo com os Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST v1.1), conforme avaliado pelo investigador. Desfechos adicionais incluíram taxa de resposta objetiva confirmada, conforme avaliado por um Comitê de Revisão Independente (CRI); tempo até a resposta; sobrevida livre de progressão (SLP); duração da resposta (DR); sobrevida global (SG); qualidade de vida (QoL); e taxa de resposta objetiva intracraniana, duração da resposta intracraniana e sobrevida livre de progressão intracraniana, conforme avaliado pelo CRI. A análise dos resultados obtidos no estudo em ambos os braços informou a dose recomendada.
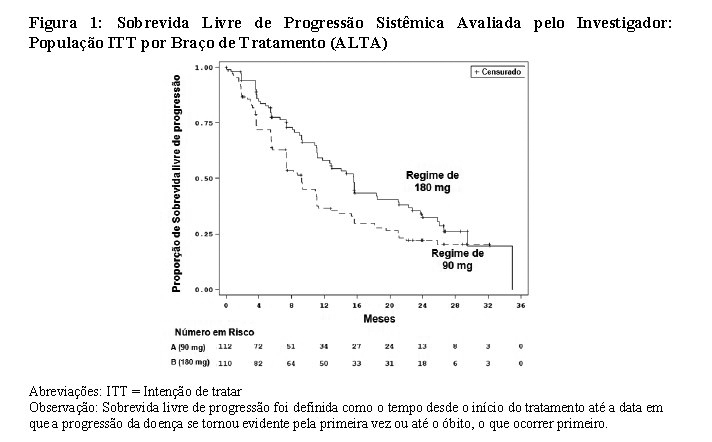
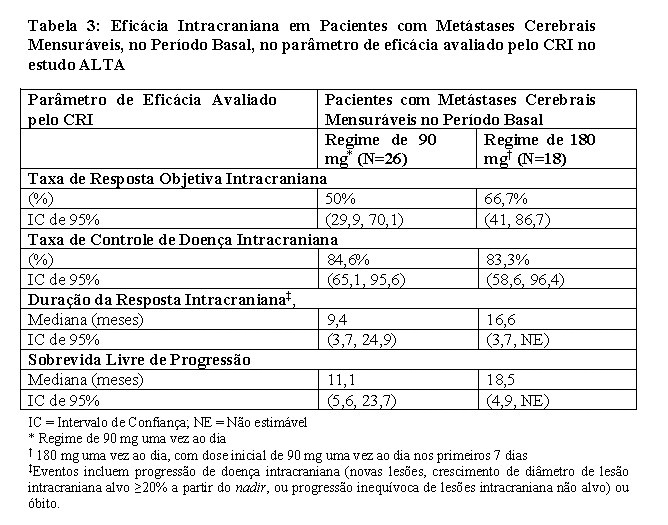
Os dados demográficos basais e características da doença no estudo ALTA (Tabela 1) foram idade mediana de 54 anos (faixa de 18 a 82 anos; 23% com 65 anos ou mais), 67% de brancos e 31% de asiáticos, 57% do sexo feminino, 36% de ECOG PS 0 e 57% de ECOG PS 1, 95% nunca fumaram ou são ex-fumantes, 98% no Estádio IV, 97% com adenocarcinoma e 74% com quimioterapia anterior. Os locais mais comuns de metástase extratorácica incluíram: cérebro 69,4% (dos quais 62,3% haviam recebido radiação anterior no cérebro), osso 39,6% e fígado 25,2%. Os resultados de eficácia da análise de ALTA são resumidos na Tabela 2 e as curvas de Kaplan-Meier (KM) para SLP sistêmica avaliada pelo investigador e pelo CRI são apresentadas na Figura 1 e Figura 2, respectivamente.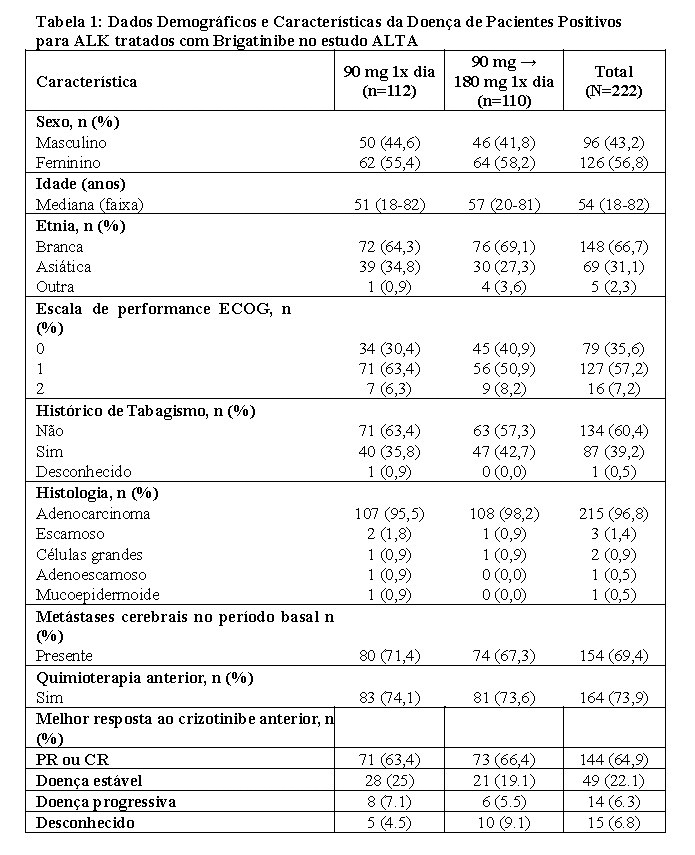
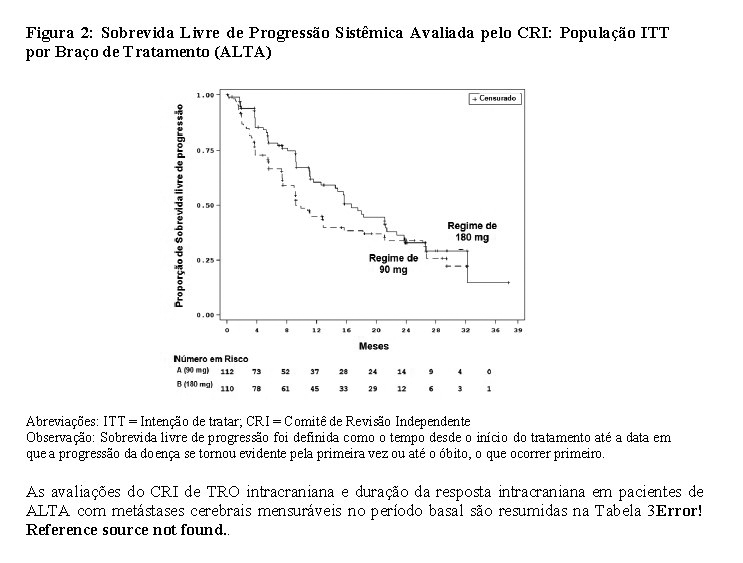
Em geral, os pacientes apresentaram alterações positivas em relação ao período basal na QoL durante o tratamento com brigatinibe, no estudo ALTA. O IC 95% da média da alteração (em relação à linha de base) da pontuação transformada da escala de Status de Saúde Global/Qualidade de Vida, do instrumento EORTC QLQC-30, foi positivo durante grande parte do tratamento em ambos os grupos de dose.
Os dados clínicos atualmente disponíveis não são conclusivos para informar sobre quais mutações secundárias específicas são susceptíveis ou conferem resistência ao brigatinibe
Estudo 101
No Estudo 101, 25 pacientes com CPNPC positivo para ALK que progrediram com crizotinibe receberam 180 mg de brigatinibe uma vez ao dia com uma dose inicial de 90 mg uma vez ao dia nos primeiros 7 dias. Destes, 19 pacientes apresentaram uma resposta objetiva confirmada avaliada pelo investigador (76%; IC de 95%: 55, 91) e a SLP mediana de KM foi de 16,3 meses (IC de 95%: 9,2, NE) e a probabilidade de sobrevida global de 12 meses foi de 84,0% (IC de 95%: 62,8, 93,7).
Nos estudos clínicos de brigatinibe, para os pacientes que reduziram a dose para 60 mg e não voltaram a aumentar para mais de 60 mg, o tempo mediano de administração de 60 mg foi de 7,08 meses.
Referências Bibliográficas
1. Clinical Study Report AP26113-13-301
2. Clinical Study Report AP26113-13-201
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Eletrofisiologia Cardíaca
No Estudo 101, o potencial de prolongamento do intervalo QT do brigatinibe foi avaliado em 123 pacientes com doenças malignas avançadas, após doses diárias de brigatinibe de 30 mg a 240 mg. A média máxima do QTcF (QT corrigido pelo método de Fridericia) alterada em relação ao valor basal foi inferior a 10 mseg. Uma análise QT de exposição sugeriu que não há prolongamento do intervalo QTc dependente da concentração. Na análise conjunta dos estudos clínicos pivotais, o aumento QTcF de mais de 60 mseg ocorreu em 8,1% (20/246) dos pacientes, e para os pacientes com QTcF basal < 500 mseg, 1,6% (4/246) dos pacientes apresentaram um aumento do QTcF > 500 mseg.
Mecanismo de Ação
O brigatinibe é um inibidor de tirosina quinase que tem como alvos ALK, ROS1 e o receptor do fator de crescimento semelhante à insulina 1 (IGF 1R). Entre eles, o brigatinibe é mais ativo contra ALK. O brigatinibe inibiu a autofosforilação de ALK e a fosforilação mediada por ALK da proteína STAT3 de sinalização descendente ("downstream") em ensaios in vitro e in vivo. O brigatinibe inibiu a proliferação in vitro de linhagens celulares expressando proteínas de fusão EML4-ALK e NPM-ALK e demonstrou inibição dose-dependente do crescimento de xenoenxerto para CPNPC positivo para EML4-ALK em camundongos.
Em concentrações ≤ 500 nM, que são clinicamente alcançadas, o brigatinibe inibiu a viabilidade in vitro de células expressando EML4-ALK e 17 mutações associadas à resistência aos inibidores de ALK, incluindo crizotinibe. Nenhuma mutação de ALK associada à resistência ao brigatinibe foi observada (o painel de mutações investigadas não contempla todas as mutações observadas na clínica). O brigatinibe demonstrou atividade não clínica in vivo contra múltiplas mutações de EML4-ALK, incluindo as mutações G1202R e L1196M identificadas nos tumores de CPNPC em pacientes que progrediram com crizotinibe. A investigação clínica continua em andamento.
A administração de brigatinibe resultou em atividade antitumoral e prolongou a sobrevida em camundongos com linhagem de célula tumoral orientada por ALK implantada via intracraniana.
Propriedades farmacocinéticas
Absorção
Após a administração de doses orais únicas de 30 a 240 mg de brigatinibe, o tempo mediano até o pico de concentração (Tmax) variou de 1 a 4 horas após a dose. A média geométrica (CV%) da concentração máxima (Cmax) de brigatinibe no estado de equilíbrio nas doses de 90 mg e 180 mg uma vez ao dia foi de 552 (65%) e 1452 (60%) ng/mL, respectivamente, e a ASC0-tau correspondente foi de 8165 (57%) e 20276 (56%) h?ng/mL, respectivamente. Após a administração de dose única e também de doses repetidas de brigatinibe, a exposição sistêmica foi proporcional à dose ao longo da faixa de 60 mg a 240 mg, uma vez ao dia. A razão de acumulação média após a administração repetida foi de 1,9 a 2,4.
A Cmax de brigatinibe foi reduzida em 13% sem efeito na área sob a curva (ASC) de sujeitos de pesquisa sadios que receberam brigatinibe após uma refeição com alto teor de gordura em comparação à Cmax e ASC após jejum durante toda a noite.
Distribuição
A ligação de brigatinibe às proteínas plasmáticas humanas foi de 91% e não foi dependente da concentração. A razão da concentração do sangue para o plasma é de 0,69. Após a administração oral de 180 mg de brigatinibe, uma vez ao dia, a média geométrica do volume de distribuição médio aparente (Vz/F) no estado de equilíbrio foi de 153 L.
Metabolismo
Estudos in vitro demonstraram que o brigatinibe é metabolizado principalmente pela CYP2C8 e CYP3A4. Após a administração oral de uma dose única de 180 mg de [14C]-brigatinibe em indivíduos sadios, a conjugação com cisteína e a N-desmetilação foram as duas principais vias metabolização. O brigatinibe inalterado (92%) e seu metabólito primário, AP26123 (3,5%), foram os principais componentes radioativos circulantes. Em pacientes, a ASC no estado de equilíbrio de AP26123 foi inferior a 10% da exposição ao brigatinibe. O metabólito, AP26123, inibiu ALK com potência aproximadamente 3 vezes menor do que o brigatinibe in vitro.
Excreção e Eliminação
Após a administração oral de 180 mg de brigatinibe, uma vez ao dia, a média geométrica do clearance oral aparente (CL/F) de brigatinibe no estado de equilíbrio foi de 13 L/h e a meiavida de eliminação plasmática média foi de 25h.
Após a administração de uma dose oral única de 180 mg de [14C]-brigatinibe em seis indivíduos sadios do sexo masculino, houve recuperação de 65% da dose administrada nas fezes e de 25% na urina. O brigatinibe inalterado representou 41% e 86% da radioatividade total nas fezes e na urina, respectivamente.
- Populações Especiais
Função Renal Comprometida
A farmacocinética de brigatinibe é similar em indivíduos com função renal normal e em pacientes com comprometimento renal leve ou moderado (TFGe ≥ 30 mL/min/1,73 m2) com base nos resultados das análises da farmacocinética da população. Em um estudo farmacocinético, a ASC0-INF não ligada foi 92% maior em pacientes com comprometimento renal grave (TFGe < 30 mL/min/1,73 m2, n=8) em comparação a indivíduos com função renal normal (TFGe ≥ 90 mL/min/1,73 m2, n=8) (ver item 8.POSOLOGIA E MODO DE USAR).
Função Hepática Comprometida
A farmacocinética de brigatinibe foi avaliada em indivíduos com função hepática normal (n=9), com comprometimento hepático leve (Child-Pugh classe A, n=6), com comprometimento hepático moderado (Child-Pugh classe B, n=6) e com comprometimento hepático grave (Child-Pugh classe C, n=6). A farmacocinética de brigatinibe foi similar entre os indivíduos com função hepática normal e pacientes com comprometimento hepático leve (Child-Pugh classe A) ou moderado (Child-Pugh classe B). A ASC0-INF não ligada foi 37% maior em pacientes com comprometimento hepático grave (Child-Pugh classe C) em comparação aos indivíduos com função hepática normal (ver item 8.POSOLOGIA E MODO DE USAR).
Idade, Sexo, Etnia
As análises farmacocinéticas da população mostraram que idade, sexo ou etnia não apresentam efeito clinicamente significativo na farmacocinética de brigatinibe.
Interações Medicamentosas
- Agentes que podem aumentar as concentrações plasmáticas de Brigatinibe
Inibidores do CYP3A
Estudos in vitro demonstraram que o brigatinibe é um substrato do CYP3A4/5. A coadministração de doses múltiplas de 200 mg duas vezes ao dia de itraconazol, um forte inibidor do CYP3A, em conjunto com uma dose única de 90 mg de brigatinibe aumentou a Cmax de brigatinibe em 21%, ASC0-INF em 101% (2 vezes) e ASC0-120 em 82% ( < 2 vezes) em relação à dose de 90 mg de brigatinibe administrada isoladamente. O uso concomitante de inibidores fortes do CYP3A com brigatinibe, incluindo, dentre outros, determinados antivirais (por exemplo, indinavir, nelfinavir, ritonavir, saquinavir), antibióticos macrolídeos (por exemplo, claritromicina, telitromicina, troleandomicina), antifúngicos (por exemplo, cetoconazol, voriconazol) e nefazodona deve ser evitado. Se o uso concomitante de inibidores fortes do CYP3A não puder ser evitado, a dose de brigatinibe deve ser reduzida em aproximadamente 50% (isto é, de 180 mg a 90 mg, ou de 90 mg a 60 mg). Após a descontinuação de um inibidor forte do CYP3A, o brigatinibe deve ser retomado na dose que foi tolerada antes do início da administração do inibidor forte do CYP3A.
Os inibidores moderados do CYP3A (por exemplo, diltiazem e verapamil) podem aumentar a ASC de brigatinibe em 40% com base nas simulações de um modelo farmacocinético baseado em fisiologia. Nenhum ajuste de dose é necessário para brigatinibe em associação com inibidores moderados do CYP3A. Os pacientes devem ser monitorados rigorosamente quando o brigatinibe é coadministrado com inibidores moderados do CYP3A.
Toranja ("grapefruit") ou suco de toranja ("grapefruit") também pode aumentar as concentrações plasmáticas de brigatinibe e devem ser evitados.
Inibidores do CYP2C8
Estudos in vitro demonstraram que o brigatinibe é um substrato do CYP2C8. A coadministração de doses múltiplas de 600 mg, duas vezes ao dia, de genfibrozila, um inibidor forte do CYP2C8, com uma dose única de 90 mg de brigatinibe reduziu a Cmax de brigatinibe em 41%, ASC0-INF em 12% e ASC0-120 em 15% em relação à dose de 90 mg de brigatinibe administrada isoladamente. Nenhum ajuste de dose é necessário para brigatinibe durante a coadministração com inibidores fortes do CYP2C8.
Inibidores de P-gp e BCRP
O brigatinibe é um substrato de glicoproteína P (P-gp) e proteína de resistência ao câncer de mama (BCRP) in vitro. O brigatinibe exibe alta solubilidade e alta permeabilidade in vitro. Não se espera que seja necessário ajuste de dose para brigatinibe durante a coadministração com inibidores de P-gp e BCRP.
- Agentes que podem reduzir as concentrações plasmáticas de Brigatinibe
Indutores do CYP3A
A coadministração de doses múltiplas de 600 mg, uma vez ao dia, de rifampicina, um indutor forte do CYP3A, com uma dose única de 180 mg de brigatinibe reduziu a Cmax de brigatinibe em 60%, ASC0-INF em 80% (5 vezes) e ASC0-120 em 80% (5 vezes) em relação à dose de 180 mg de brigatinibe administrada isoladamente. O uso concomitante de indutores fortes do CYP3A com brigatinibe, incluindo, dentre outros, rifampicina, carbamazepina, fenitoína, rifabutina, fenobarbital e Erva de São João, deve ser evitado.
Indutores moderados do CYP3A podem reduzir a ASC de brigatinibe em aproximadamente 50% com base em simulações de um modelo farmacocinético baseado em fisiologia. O uso concomitante de indutores moderados do CYP3A com brigatinibe, incluindo, dentre outros, efavirenz, modafinila, bosentana, etravirina e nafcilina, deve ser evitado.
-Agentes que podem sofrer alteração em suas concentrações plasmáticas pelo Brigatinibe
Substratos do CYP3A
Estudos in vitro em hepatócitos mostraram que o brigatinibe é um indutor do CYP3A. Estudos clínicos de interação medicamentosa com substratos do CYP3A sensíveis não foram conduzidos. O brigatinibe pode reduzir as concentrações plasmáticas de medicações coadministradas que são predominantemente metabolizadas pelo CYP3A.
O brigatinibe também pode induzir outras enzimas (por exemplo, CYP2C) por meio dos mesmos mecanismos responsáveis pela indução do CYP3A (por exemplo, ativação do receptor pregnano X).
Substratos de Transportadores
O brigatinibe é um inibidor de P-gp, BCRP, OCT1, MATE1 e MATE2K in vitro. A coadministração de brigatinibe com substratos de P-gp, (por exemplo digoxina, dabigatrana, colchicina, pravastatina), BCRP (por exemplo, metotrexato, rosuvastatina, sulfasalazina), OCT1, MATE1 e MATE2K pode aumentar suas concentrações plasmáticas.
• Dados de Segurança Não Clínicos
Carcinogênese, Mutagênese, Comprometimento de Fertilidade
Carcinogenicidade
Estudos de carcinogenicidade não foram realizados com brigatinibe.
Mutagenicidade
O brigatinibe não foi mutagênico in vitro nos ensaios de mutação reversa bacteriana (Ames) ou de aberração cromossômica de células de mamíferos, mas aumentou ligeiramente o número de micronúcleos no teste de micronúcleos de medula óssea de ratos. O mecanismo de indução de micronúcleos foi segregação cromossômica anormal (aneugenicidade) e não um efeito clastogênico nos cromossomos. Esse efeito foi observado no que corresponde a aproximadamente cinco vezes a exposição humana, na dose de 180 mg, uma vez ao dia.
Comprometimento da Fertilidade
O brigatinibe pode comprometer a fertilidade masculina. A toxicidade testicular foi observada em estudos de doses repetidas em animais. Em ratos, os achados incluíram menor peso dos testículos, das vesículas seminais e da próstata, e degeneração tubular testicular; esses efeitos não foram reversíveis durante o período de recuperação. Em macacos, os achados incluíram redução do tamanho dos testículos com evidência microscópica de hipoespermatogênese; esses efeitos foram reversíveis durante o período de recuperação. Em geral, esses efeitos nos órgãos reprodutores de machos em ratos e macacos ocorreram em exposições tão baixas quanto 0,2 vezes a ASC em pacientes tratados com a dose de 180 mg uma vez ao dia. Nenhum efeito adverso aparente nos órgãos reprodutores de fêmeas foi observado nos estudos de toxicologia geral em macacos. No estudo de 28 dias de toxicidade de dose repetida em ratos, observaramse diminuições do peso uterino e/ou atrofia uterina mínima a moderada com doses ≥ 30 mg/kg. Estes achados ocorreram com exposições tão baixas quanto 2,4 vezes a ASC em pacientes com a dose de 180 mg uma vez ao dia. Suspeita-se que a atrofia uterina esteja relacionada à má condição clínica dos animais e/ou diminuição do peso corporal final e não diretamente relacionada ao brigatinibe. A atrofia uterina foi geralmente observada apenas em animais que morreram ou foram removidos precocemente do estudo; e foi observada em apenas 1 animal (dose administrada de 30 mg/kg) que sobreviveu até o final do estudo. A atrofia uterina não foi observada após um período de recuperação de 28 dias. Não foram observadas alterações uterinas relacionados com o brigatinibe no estudo de 6 meses em ratos, em exposições que foram até 1,9 vezes a ASC em pacientes com a dose de 180 mg uma vez ao dia.
Farmacologia e/ou Toxicologia em Animais
Uma avaliação de segurança não clínica em ratos e macacos identificou um risco potencial de toxicidade em vários órgãos, como sistema gastrointestinal, medula óssea, olhos, testículos, fígado, rins, ossos, coração, timo, baço e pâncreas. Esses efeitos foram geralmente reversíveis durante o período de recuperação sem tratamento; entretanto, os efeitos nos olhos e testículos foram exceções notáveis devido à ausência de recuperação.
4. CONTRAINDICAÇÕES
O uso de EVOBRIG é contraindicado em pacientes com hipersensibilidade ao brigatinibe ou a qualquer um dos excipientes.
5. ADVERTÊNCIAS E PRECAUÇÕES
Reações Adversas Pulmonares
Reações adversas pulmonares graves, de risco à vida e fatais, incluindo aquelas com características consistentes de DPI/pneumonite, podem ocorrer em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS).
A maioria das reações adversas pulmonares foi observada nos primeiros 7 dias de tratamento. As reações adversas pulmonares Grau 1-2 foram resolvidas com a interrupção do tratamento ou a modificação da dose. O aumento da idade e um menor intervalo (inferior a 7 dias) entre a última dose de crizotinibe e a primeira dose de brigatinibe foram independentemente associados com um aumento na taxa dessas reações adversas pulmonares. Esses fatores devem ser considerados ao iniciar o tratamento com brigatinibe.
Alguns pacientes apresentaram pneumonite tardiamente no tratamento com brigatinibe. Os pacientes devem ser monitorados quanto a sintomas respiratórios novos ou agravados (por exemplo, dispneia, tosse, etc), principalmente na primeira semana de tratamento. Caso haja evidência de pneumonite em qualquer paciente com sintomas respiratórios agravados, deve ser realizada investigação imediatamente. Se houver suspeita de pneumonite, brigatinibe deverá ser suspenso, o paciente deve ser avaliado quanto a outras causas dos sintomas (por exemplo, embolia pulmonar, progressão tumoral e pneumonite infecciosa) e a dose deve ser modificada de acordo com a necessidade (ver item 8. POSOLOGIA E MODO DE USAR).
Hipertensão
A hipertensão ocorreu em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS).
A pressão arterial deve ser monitorada regularmente durante o tratamento com brigatinibe. A hipertensão deve ser tratada de acordo com as diretrizes padrões para controle da pressão arterial. Para hipertensão grave (≥ Grau 3), o brigatinibe deverá ser suspenso até que a hipertensão tenha sido recuperada para Grau 1 ou basal. A dose deve ser modificada de acordo com a necessidade (ver item 8. POSOLOGIA E MODO DE USAR).
Bradicardia
Bradicardia ocorreu em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). A frequência cardíaca e a pressão arterial devem ser monitoradas regularmente. A frequência cardíaca deve ser monitorada mais frequentemente em pacientes se o uso concomitante de um medicamento conhecido por causar bradicardia não puder ser evitado. Deve-se ter cautela ao administrar brigatinibe em associação com outros agentes conhecidos por causar bradicardia.
Se a bradicardia sintomática ocorrer, o tratamento com brigatinibe deverá ser suspenso e as medicações concomitantes conhecidas por causar bradicardia deverão ser avaliadas. Após a recuperação, a dose deverá ser modificada de acordo com a necessidade (ver item 8. POSOLOGIA E MODO DE USAR). Em caso de bradicardia com risco à vida, se nenhuma medicação concomitante que contribui para a condição for identificada ou em caso de recidiva, o tratamento com brigatinibe deverá ser descontinuado (ver item 8. POSOLOGIA E MODO DE USAR).
Distúrbio Visual
Reações adversas de distúrbio visual ocorreram em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). Os pacientes devem ser aconselhados a relatarem todos os sintomas visuais. Caso ocorram sintomas visuais novos ou em caso de piora dos sintomas visuais, uma avaliação oftalmológica e uma redução da dose devem ser consideradas (ver item 8. POSOLOGIA E MODO DE USAR).
Elevação de Creatinofosfoquinase
Elevações da creatinofosfoquinase (CPK) ocorreram em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). Os pacientes devem ser aconselhados a relatarem qualquer dor muscular sem causa aparente, sensibilidade ou fraqueza. Os níveis de CPK devem ser monitorados regularmente durante o tratamento com brigatinibe. Com base na gravidade da elevação de CPK, o tratamento com brigatinibe deve ser suspenso e a dose modificada de acordo com a necessidade (ver item 8. POSOLOGIA E MODO DE USAR).
Elevação de Enzimas Pancreáticas
Elevações de amilase e lipase ocorreram em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). Lipase e amilase devem ser monitoradas regularmente durante o tratamento com brigatinibe. Com base na gravidade das anormalidades laboratoriais, o tratamento com brigatinibe deve ser suspenso e a dose modificada de acordo com a necessidade (ver item 8. POSOLOGIA E MODO DE USAR).
Hepatotoxicidade
Elevações das enzimas hepáticas (aspartato aminotransferase, alanina aminotransferase) e bilirrubina ocorreram em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). A função hepática, incluindo AST, ALT e bilirrubina total, deve ser avaliada antes do início do tratamento com brigatinibe e, depois a cada 2 semanas, durante os primeiros 3 meses de tratamento. Posteriormente, o monitoramento deve ser realizado periodicamente. Com base na gravidade das alterações laboratoriais, o tratamento deve ser interrompido e a dose modificada conforme item 8. POSOLOGIA E MODO DE USAR.
Hiperglicemia
Elevações da glicose sérica ocorreram em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). A glicose sérica de jejum deve ser avaliada antes do início de brigatinibe e, em seguida, monitorada periodicamente. Medicações anti-hiperglicêmicas devem ser iniciadas ou otimizadas, conforme necessário. Se o controle hiperglicêmico adequado não puder ser obtido com o tratamento médico ideal, brigatinibe deverá ser suspenso até que o controle hiperglicêmico adequado seja obtido; após a recuperação, a redução da dose de brigatinibe pode ser considerada ou brigatinibe pode ser descontinuado permanentemente (ver item 8. POSOLOGIA E MODO DE USAR).
Fotossensibilidade
Fotossensibilidade à luz solar ocorreu em pacientes tratados com brigatinibe (ver item 9. REAÇÕES ADVERSAS). Os pacientes devem ser aconselhados a evitar a exposição prolongada ao sol durante o tratamento com brigatinibe e por pelo menos 5 dias após a interrupção do tratamento. Quando ao ar livre, os pacientes devem ser aconselhados a usar chapéu e roupas de proteção, e a usar protetor solar e protetor labial Ultravioleta A (UVA) / Ultravioleta B (UVB) de amplo espectro (FPS = 30) para ajudar na proteção contra possíveis queimaduras solares. Para reações de fotossensibilidade graves (≥ Grau 3), brigatinibe deve ser suspenso até a recuperação ao basal. A dose deve ser modificada conforme item 8. POSOLOGIA E MODO DE USAR.
Toxicidade Embriofetal
Com base em seu mecanismo de ação e achados em animais, brigatinibe pode causar dano fetal ao ser administrado em gestantes.
Não há dados clínicos sobre o uso de brigatinibe em mulheres gestantes. Mulheres gestantes devem ser aconselhadas sobre o potencial risco fetal. As mulheres com potencial reprodutivo devem ser orientadas a utilizarem um método contraceptivo não hormonal eficaz durante o tratamento com brigatinibe e por pelo menos 4 meses após a dose final. Os homens com parceiras com potencial reprodutivo devem ser orientados a utilizarem um método contraceptivo eficaz durante o tratamento e por pelo menos 3 meses após a última dose de brigatinibe (ver Uso durante a gravidez e a lactação).
Uso durante a gravidez e a lactação
Categoria D de Risco na Gravidez - Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Gravidez
Brigatinibe pode causar dano fetal quando administrado em gestantes. Estudos em animais demonstraram toxicidade reprodutiva (ver Dados de Segurança Não Clínicos). Não há dados clínicos sobre o uso de brigatinibe em gestantes. O brigatinibe não deve ser utilizado durante a gravidez, a menos que a condição clínica da mãe exija o tratamento. Se o brigatinibe for utilizado durante a gravidez ou se a paciente engravidar durante o tratamento com esse medicamento, a paciente deve ser informada sobre o risco potencial ao feto.
Mulheres em idade fértil que são tratadas com brigatinibe devem ser orientadas a não engravidarem e homens sendo tratados com brigatinibe devem ser orientados a não terem filhos durante o tratamento.
Mulheres com potencial reprodutivo devem ser orientadas a utilizarem contracepção não hormonal eficaz durante o tratamento com brigatinibe e por pelo menos 4 meses após a dose final. Os pacientes do sexo masculino com parceiras com potencial reprodutivo devem ser orientados a utilizar contracepção eficaz durante o tratamento e por pelo menos 3 meses após a última dose de brigatinibe.
Em um estudo de desenvolvimento embrio-fetal em que ratas grávidas foram tratadas com doses diárias de brigatinibe durante a organogênese, anomalias viscerais e esqueléticas (ossificação incompleta, incisivos pequenos) relacionadas à dose foram observadas em baixa dosagem, 12,5 mg/kg/dia (aproximadamente 0,7 vezes a exposição humana por ASC, com 180 mg uma vez ao dia). Foram observadas más-formações com 25 mg/kg/dia (aproximadamente 1,26 vezes a ASC humana, com 180 mg uma vez ao dia), incluindo anasarca (edema subcutâneo generalizado), anoftalmia (ausência de olhos), hiperflexão de membro anterior, membros pequenos, curtos e/ou curvados, múltiplas costelas fundidas, escápula curvada, onfalocele (intestino saliente no umbigo) e gastrosquise (intestinos salientes por meio de um defeito na parede abdominal), juntamente com achados viscerais de dilatação bilateral moderada dos ventrículos laterais.
Lactação
Não se sabe se o brigatinibe é excretado no leite humano. Os dados disponíveis não podem excluir o potencial de excreção no leite humano. A amamentação deve ser interrompida durante o tratamento com brigatinibe.
Fertilidade
Não estão disponíveis dados sobre o efeito de brigatinibe na fertilidade de humanos. Com base nos estudos de reprodução em animais machos, o brigatinibe pode reduzir a fertilidade masculina (ver Dados de Segurança Não Clínicos). A relevância clínica desses achados na fertilidade em humanos é desconhecida.
Efeitos na capacidade de dirigir veículos e operar máquinas
Não há dados sobre o efeito de EVOBRIG na capacidade de dirigir e operar máquinas. Distúrbio visual, tontura e fadiga haviam sido observados nos estudos clínicos. Os pacientes devem ser orientados a não dirigir ou operar máquinas se apresentarem qualquer um desses sintomas ao utilizar brigatinibe.
Abuso de Drogas e Dependência
EVOBRIG não tem potencial conhecido para abuso ou dependência.
Lactose
EVOBRIG contém lactose monoidratada. Os pacientes com problemas hereditários raros de intolerância à galactose, deficiência total de lactase ou má absorção de glucose-galactose não devem utilizar este medicamento.
6. INTERAÇÕES MEDICAMENTOSAS
• Agentes que Podem Aumentar as Concentrações Plasmáticas de Brigatinibe
Inibidores do CYP3A
Deve-se evitar o uso concomitante de fortes inibidores do CYP3A com brigatinibe, incluindo, dentre outros, certos antivirais (por exemplo, indinavir, nelfinavir, ritonavir, saquinavir), antibióticos macrolídeos (por exemplo, claritromicina, telitromicina, troleandomicina), antifúngicos (por exemplo, cetoconazol, voriconazol) e nefazodona. Se o uso concomitante de inibidores fortes do CYP3A não puder ser evitado, a dose de brigatinibe deverá ser reduzida em aproximadamente 50% (isto é, de 180 mg a 90 mg, ou de 90 mg para 60 mg). Após a descontinuação de um inibidor forte do CYP3A, o brigatinibe deverá ser retomado na dose que era tolerada antes do início da administração do inibidor forte do CYP3A.
Nenhum ajuste de dose é necessário para brigatinibe em associação com inibidores moderados do CYP3A (por exemplo, diltiazem e verapamil). Os pacientes devem ser monitorados rigorosamente quando o brigatinibe é coadministrado com inibidores moderados do CYP3A. Toranja ("grapefruit") ou suco de toranja ("grapefruit") também pode aumentar as concentrações plasmáticas de brigatinibe e deve ser evitado.
Inibidores do CYP2C8
Nenhum ajuste de dose é necessário para brigatinibe durante a coadministração como inibidores fortes do CYP2C8.
Inibidores de P-gp e BCRP
Não se espera que seja necessário ajuste de dose para brigatinibe durante a coadministração com inibidores de P-gp e BCRP.
• Agentes que Podem Reduzir as Concentrações Plasmáticas de Brigatinibe Indutores do CYP3A
O uso concomitante de indutores fortes do CYP3A com brigatinibe, incluindo, dentre outros, rifampicina, carbamazepina, fenitoína, rifabutina, fenobarbital e Erva de São João, deve ser evitado.
O uso concomitante de indutores moderados do CYP3A com brigatinibe, incluindo, dentre outros, efavirenz, modafinila, bosentana, etravirina e nafcilina, deve ser evitado.
• Agentes que Podem Sofrer Alterações em suas Concentrações Plasmáticas pelo Brigatinibe
Substratos do CYP3A
O brigatinibe pode reduzir as concentrações plasmáticas de medicações coadministradas que são metabolizadas predominantemente por CYP3A.
Portanto, a co-administração de brigatinibe com substratos da CYP3A que possuem índice terapêutico estreito (por exemplo, alfentanil, fentanil, quinidina, ciclosporina, sirolimus, tacrolimo) deve ser evitada, uma vez que a eficácia destes fármacos pode ser reduzida.
O brigatinibe também pode induzir outras enzimas (por exemplo, CYP2C) por meio dos mesmos mecanismos responsáveis pela indução do CYP3A (por exemplo, ativação do receptor pregnano X).
Substratos de Transportador
Brigatinibe é um inibidor de P-gp, BCRP, OCT1, MATE1 e MATE2K in vitro. A coadministração de brigatinibe com substratos de P-gp, (por exemplo, digoxina, dabigatrana, colchicina, pravastatina), BCRP (por exemplo, metotrexato, rosuvastatina, sulfasalazina), OCT1, MATE1 e MATE2K, pode aumentar suas concentrações plasmáticas.
Os pacientes devem ser cuidadosamente monitorados quando brigatinibe é co-administrado com substratos destes transportadores que possuem índice terapêutico estreito (por exemplo, digoxina, dabigatrana, metotrexato).
7. CONDIÇÕES DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente entre 15°C e 30°C.
EVOBRIG comprimidos revestidos tem validade de 36 meses a partir da data de sua fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Os comprimidos de EVOBRIG são brancos a quase brancos, a concentração de 30 mg possui "U3" gravado em um dos lados, e o outro lado liso, 90 mg possui "U7" gravado em um dos lados e o outro lado liso e 180 mg possui "U13 gravado em um dos lados e o outro lado liso.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Teste ALK
Um teste de ALK validado é necessário para a seleção de pacientes com CPNPC positivos para ALK. O status de CPNPC positivo para ALK deve ser estabelecido antes do início da terapia com EVOBRIG.
Posologia
A dose inicial recomendada de EVOBRIG é 90 mg uma vez ao dia para os primeiros 7 dias, e então 180 mg uma vez ao dia.
O tratamento deve continuar enquanto o benefício clínico for observado. Se uma dose de EVOBRIG for esquecida, esta não deve ser administrada e a dose seguinte de brigatinibe deverá ser administrada na hora programada para a próxima dose.
Se ocorrer vômito após tomar a dose de EVOBRIG, uma dose adicional não deve ser administrada e a dose seguinte de brigatinibe deverá ser administrada na hora programada para a próxima dose
Ajuste de Dose
A interrupção da administração e/ou a redução da dose pode ser necessária com base na segurança individual e tolerabilidade. Os níveis de redução da dose de EVOBRIG são resumidos na Tabela 4.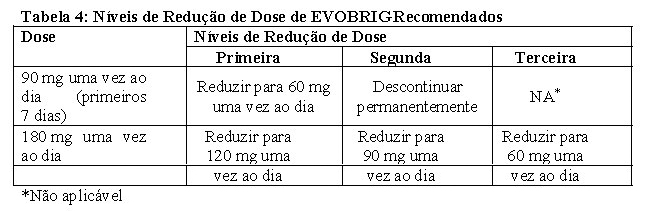
EVOBRIG deve ser permanentemente descontinuado se o paciente não conseguir tolerar a dose de 60 mg uma vez ao dia. Se EVOBRIG for interrompido por 14 dias ou mais, por motivos que não sejam reações adversas, o tratamento deverá ser retomado com a dose de 90 mg uma vez ao dia por 7 dias antes de aumentar para a dose tolerada anteriormente. As recomendações para modificações da dose de EVOBRIG para o manejo de reações adversas são resumidas na Tabela 5.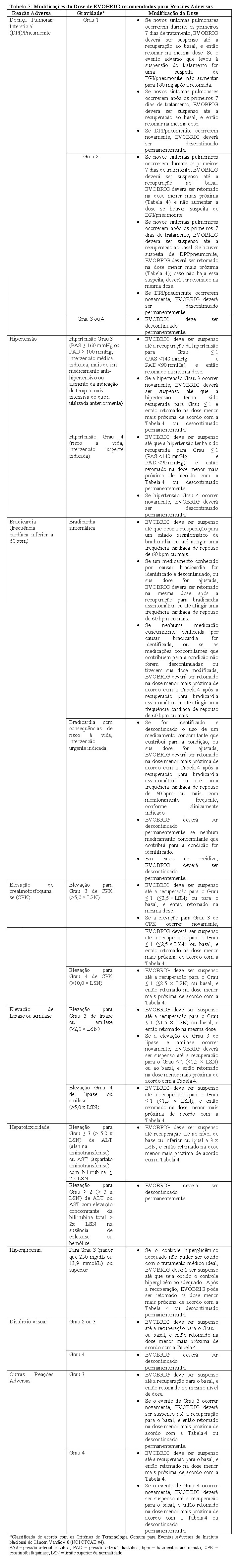
Populações Especiais
- Pacientes Idosos
Os dados limitados sobre a segurança e eficácia de brigatinibe em pacientes com 65 anos de idade ou mais sugerem que o ajuste de dose não é necessário em pacientes idosos. Existem dados limitados sobre pacientes com mais de 85 anos de idade. Os pacientes idosos devem ser cuidadosamente monitorados durante o tratamento com brigatinibe.
- Pacientes Pediátricos
Não foram estabelecidas segurança e eficácia de brigatinibe em pacientes com menos de 18 anos de idade. Não há dados disponíveis.
- Função Renal Comprometida
Nenhum ajuste de dose de brigatinibe é necessário para pacientes com comprometimento renal leve ou moderado (taxa de filtração glomerular estimada [TFGe] ≥ 30 mL/min/1,73 m2). Recomenda-se uma dose inicial reduzida de 60 mg uma vez ao dia para os primeiros 7 dias, e então uma dose de 90 mg uma vez ao dia para pacientes com comprometimento renal grave (TFGe < 30 mL/min/1,73 m2) (ver Propriedades Farmacocinéticas).
Os pacientes com comprometimento renal grave devem ser cuidadosamente monitorados quanto a eventos adversos, incluindo sintomas respiratórios novos ou agravados que possam indicar DPI / pneumonite (por exemplo, dispneia, tosse, etc), particularmente na primeira semana de tratamento (ver item 5. ADVERTÊNCIAS E PRECAUÇÕES).
- Função Hepática Comprometida
Nenhum ajuste de dose de brigatinibe é necessário para pacientes com comprometimento hepático leve (Child-Pugh classe A) ou comprometimento hepático moderado (Child-Pugh classe B). Recomenda-se uma dose inicial reduzida de 60 mg uma vez ao dia para os primeiros 7 dias, e então uma dose de 120 mg uma vez ao dia para pacientes com comprometimento hepático grave (Child-Pugh classe C) (ver Propriedades Farmacocinéticas).
Método de Administração
EVOBRIG é destinado ao uso oral. Os comprimidos devem ser engolidos inteiros e com água. Não esmague ou mastigue os comprimidos. EVOBRIG pode ser administrado com ou sem alimento.
Este medicamento não deve ser partido, aberto ou mastigado.
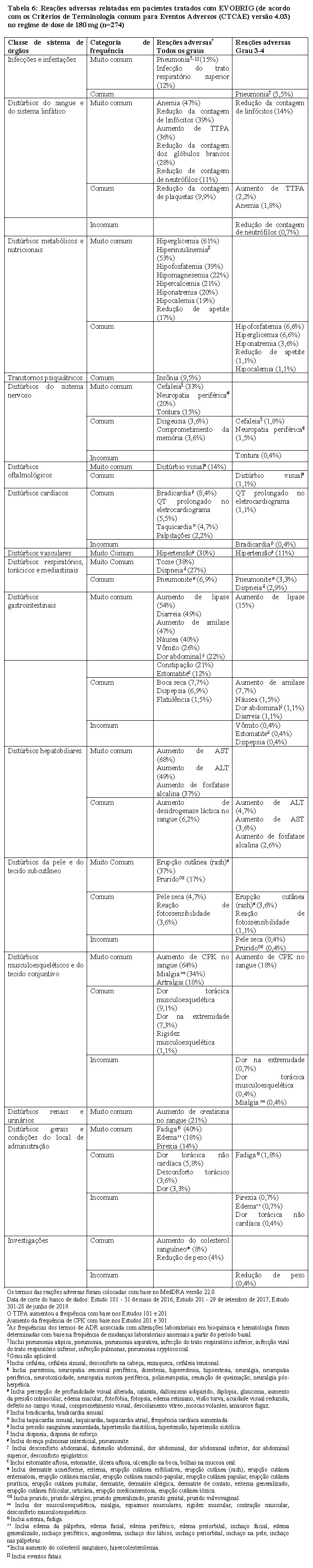
9. REAÇÕES ADVERSAS
Estudos Clínicos
As reações adversas descritas nessa seção foram identificadas a partir de três estudos clínicos: ·
• Estudo 301 (ALTA 1L): Um estudo randomizado, aberto, multicêntrico em pacientes com CPNPC avançado ALK positivo que não haviam recebido anteriormente uma terapia direcionada para ALK. Os pacientes foram randomizados em uma proporção de 1: 1 para receber 180 mg de brigatinibe uma vez ao dia com introdução de 7 dias na dose de 90 mg uma vez ao dia (n = 137) ou crizotinibe 250 mg duas vezes ao dia (n = 138). Embora conclusões quanto à eficácia se darão após a segunda análise interina do Estudo 301, dados de segurança obtidos quando da segunda análise interina compuseram os resultados de segurança ora reportados. ·
• Estudo 201 (ALTA): Um estudo randomizado, aberto, multicêntrico em pacientes tratados com brigatinibe com CPNPC ALK positivo que progrediram anteriormente com crizotinibe. Os pacientes foram randomizados em uma razão 1:1 para receber 90 mg de brigatinibe uma vez ao dia continuamente (regime de 90 mg, n=112) ou 180 mg uma vez ao dia com dose de 90 mg uma vez ao dia nos primeiros 7 dias (regime de 180 mg, n=110). ·
• Estudo 101: Um estudo de Fase 1/2 aberto, multicêntrico, de expansão/aumento de dose em pacientes com malignidades avançadas.
As reações adversas mais comuns relatadas em pacientes (≥25%) tratados com brigatinibe no regime de dose de 180 mg foram aumento de AST (68%), aumento de