ESBRIET
ROCHE
pirfenidona
Imunossupressor.
Apresentações.
Cápsulas gelatinosas duras de 267 mg, em frasco com 270 cápsulas.
VIA ORAL
USO ADULTO
Composição.
Princípio ativo: 267 mg de pirfenidona
Excipientes: croscarmelose sódica, celulose microcristalina, povidona, estearato de magnésio e água purificada.
Cápsula: dióxido de titânio e gelatina.
Informações técnicas.
1. INDICAÇÕES
Esbriet® está indicado para tratamento de fibrose pulmonar idiopática (FPI).
2-RESULTADOS DE EFICÁCIA
A eficácia clínica de Esbriet® foi estudada em três estudos multinacionais, Fase 3, multicêntricos, randomizados, duplo-cegos, controlados por placebo em pacientes com FPI.
O PIPF-004 e o PIPF-006 compararam o tratamento com Esbriet® 2403 mg/dia com placebo. Os critérios de inclusão do estudo foram: capacidade predita de CVF ≥ 50%; capacidade predita de difusão de DLCO ≥ 35%; porcentagem da capacidade predita de CVF ou porcentagem da capacidade predita de difusão DLCO ≤ 90%; tempo desde o diagnóstico ≤ 48 meses; VEF1 /CVF ≥ 0,7 e teste de caminhada de 6 minutos (TC6) ≥ 150 m (e saturação do oxigênio ≥83% com suplementação de oxigênio ≤6 L/minuto durante o teste de caminhada de 6 minutos no processo de titulação do oxigênio na triagem dos pacientes. Os estudos foram praticamente idênticos no desenho, com poucas exceções, incluindo um grupo com dose intermediária (1197 mg/dia) no PIPF-004. Nos dois estudos, o tratamento foi administrado três vezes por dia durante 72 semanas, no mínimo. O desfecho primário nos dois estudos foi a alteração em percentual de Capacidade Vital Forçada (CVF) prevista entre o período basal e a Semana 72.
No estudo PIPF-004, o declínio percentual previsto de CVF entre o período basal e a Semana 72 de tratamento foi significativamente reduzido em pacientes que receberam Esbriet® (N = 174) em comparação com pacientes recebendo placebo (N = 174; p = 0,001, classificação ANCOVA). O tratamento com Esbriet® também reduziu significativamente o declínio percentual previsto de CVF entre o período basal e as Semanas 24 (p = 0,014), 36 (p < 0,001), 48 (p < 0,001) e 60 (p < 0,001). Na Semana 72, um declínio percentual previsto de CVF ≥ 10% a partir do valor basal (um limiar indicativo do risco de mortalidade em FPI) foi observado em 20% dos pacientes que receberam Esbriet® em comparação com 35% dos que receberam placebo (Tabela 1).
Embora não exista nenhuma diferença na alteração da distância caminhada durante um teste de caminhada de seis minutos (TC6) entre pacientes recebendo Esbriet® em comparação com placebo entre o valor basal e a Semana 72 pela classificação predeterminada de ANCOVA, em uma análise ad hoc 37% dos pacientes recebendo Esbriet® apresentaram um declínio de ≥ 50 m em distância TC6 em comparação com 47% dos pacientes que receberam placebo no PIPF-004.
No estudo PIPF-006, o tratamento com Esbriet® (N = 171) não reduziu o declínio percentual previsto de CVF entre o período basal e a Semana 72, em comparação com o placebo (N = 173; p = 0,501). No entanto, o tratamento com Esbriet® reduziu o declínio percentual previsto de CVF entre o valor basal e as Semanas 24 (p < 0,001), 36 (p = 0,011) e 48 (p = 0,005). Na Semana 72, um declínio ≥ 10% de CVF foi observado em 23% dos pacientes que receberam Esbriet® e 27% dos que receberam placebo (Tabela 2).
O declínio na distância TC6 entre o valor basal e a Semana 72 foi significativamente reduzido em comparação com o placebo no estudo PIPF-006 (p < 0,001, classificação ANCOVA). Além disso, em uma análise ad hoc, 33% dos pacientes que receberam Esbriet® apresentaram um declínio ≥ 50 m em distância TC6 em comparação com 47% dos pacientes recebendo placebo no PIPF-016.
Em uma análise agrupada de sobrevida no PIPF-004 e no PIPF-006, a taxa de mortalidade no grupo com Esbriet® 2403 mg/dia foi de 7,8% em comparação com 9,8% com placebo (HR 0,77 [IC 95%, 0,47-1,28]).
O estudo PIPF-016 comparou o tratamento com Esbriet® 2403 mg/dia com placebo. Os critérios de inclusão do estudo foram: porcentagem da capacidade predita de CVF ≥ 50% e ≤ 90%; porcentagem da capacidade predita de difusão DLCO ≥ 30% e ≤ 90%; tempo desde o diagnóstico ≥ 6 e ≤ 48 meses; VEF1 /CVF ≥ 0,8 e teste de caminhada de 6 minutos (TC6) ≥ 150 m. O tratamento foi administrado três vezes por dia diariamente durante 52 semanas. O desfecho primário foi a alteração em percentual previsto de CVF entre o valor basal e a Semana 52.
No estudo PIPF-016, o declínio em percentual previsto de CVF entre o valor basal e a Semana 52 de tratamento foi significativamente reduzido em pacientes que receberam Esbriet® (N = 278) em comparação com pacientes que receberam placebo (N = 277; p < 0,000001, classificação ANCOVA). O tratamento com Esbriet® também reduziu significativamente o declínio em percentual previsto de CVF entre o valor basal e as Semanas 13 (p < 0,000001), 26 (p < 0,000001) e 39 (p = 0,000002). Na Semana 52, foi observado declínio em relação ao valor basal em CVF percentual previsto ≥ 10% ou óbito em 17% dos pacientes que receberam Esbriet® em comparação com 32% dos que receberam placebo (Tabela 3).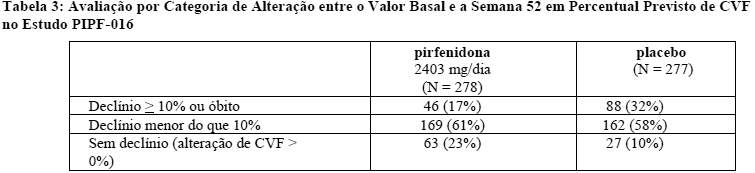
O declínio em distância caminhada durante um TC6 entre o valor basal e a Semana 52 foi significativamente reduzido em pacientes que receberam Esbriet® em comparação com pacientes que receberam placebo no estudo PIPF-016 (p = 0,036, classificação ANCOVA); 26% dos pacientes que receberam Esbriet® apresentaram uma redução ≥ 50 m na distância TC6 em comparação com 36% dos pacientes recebendo placebo.
Em uma análise agrupada predeterminada dos estudos PIPF-016, PIPF-004 e PIPF-006 no Mês 12, a mortalidade por todas as causas foi significativamente menor no grupo Esbriet® 2403 mg/dia (3,5%, 22 de 623 pacientes) comparado ao placebo (6,7%, 42 de 624 pacientes), resultando em uma redução de 48% no risco de mortalidade por todas as causas dentro dos primeiros 12 meses (HR 0,52 [IC 95%, 0,31-0,87], p = 0,0107, teste log-rank)1.
Referências Bibliográficas
1. Clinical Study Report PIPF-016, A Randomized, Double-blind, Placebo-controlled, Phase 3 Study of the Efficacy and Safety of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. 22 May 2014.
2. Clinical Study Report PIPF-004: A randomized, double-blind, placebo-controlled, phase 3, three-arm study of the safety and efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis. Report No. 1065293, June 2009.
3. Clinical Study Report PIPF-006: A randomized, double-blind, placebo-controlled, phase 3 study of the safety and efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis. Report No. 1065296, June 2009.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de Ação
O mecanismo de ação da pirfenidona não foi completamente estabelecido. No entanto, os dados existentes indicam que a pirfenidona exerce tanto propriedades antifibróticas quanto anti-inflamatórias em diversos sistemas in vitro e modelos animais de fibrose pulmonar (fibrose induzida por bleomicina e transplante).
A FPI é uma doença pulmonar fibrótica e inflamatória crônica afetada pela síntese e liberação de citocinas pró-inflamatórias incluindo fator de necrose tumoral alfa (TNF-a) e interleucina-1-beta (IL-1b) e Esbriet® demonstrou reduzir o acúmulo de células inflamatórias em resposta a diversos estímulos.
Esbriet® atenua a proliferação de fibroblastos, produção de proteínas associadas à fibrose e citocinas e o aumento de biossíntese e acúmulo de matriz extracelular em reposta aos fatores de crescimento (citocinas), como fator de transformação de crescimento beta (TGF-b) e fator de crescimento derivado de plaquetas (PDGF).
Propriedades Farmacocinéticas
Absorção
Após a administração de uma única dose oral de 801 mg de Esbriet®, a máxima concentração plasmática observada (Cmáx) foi alcançada entre 30 minutos a 4 horas (tempo mediano de 0,5 hora). Alimentos reduzem a taxa e extensão de absorção. O Tmáx mediano aumentou de 0,5 hora a 3 horas com alimento.
A administração de Esbriet® com alimentos resulta em grande redução na Cmáx (em 50%) e um menor efeito sobre ASC em comparação com a administração em jejum. Depois de administração oral de dose única de 801 mg a voluntários adultos saudáveis (50 - 66 anos de idade) no estado alimentado, a velocidade de absorção da pirfenidona é reduzida, enquanto que ASC no estado alimentado foi aproximadamente 80-85% da ASC observada no jejum. Uma incidência reduzida de eventos adversos (náuseas e tontura) foi observada em indivíduos que receberam o medicamento no estado alimentado em comparação com o grupo que tomou em jejum. Portanto, recomenda-se que Esbriet® seja administrado com alimentos para reduzir a incidência de náuseas e tontura.
A biodisponibilidade absoluta da pirfenidona não foi determinada em humanos.
Distribuição
A pirfenidona se liga às proteínas plasmáticas humanas, principalmente à albumina sérica. A ligação média geral variou de 50% a 58% em concentrações observadas em estudos clínicos (1 a 100 mg/mL). O volume médio aparente de distribuição oral no estado de equilíbrio é de aproximadamente 70 litros, indicando que a distribuição de pirfenidona para os tecidos é pequena.
Metabolismo
Estudos de metabolismo in vitro com microssomos hepáticos indicam que pirfenidona é metabolizada principalmente através de CYP1A2 com menor contribuição de outras isoenzimas CYP, incluindo CYP2C9, 2C19, 2D6 e 2E1. Estudos in vitro e in vivo até hoje não detectaram nenhuma atividade do principal metabólito (5-carboxi-pirfenidona), mesmo em concentrações ou doses muito acima das associadas com a atividade da pirfenidona.
Eliminação
A depuração oral da pirfenidona parece ser discretamente saturável. Em um estudo de múltiplas doses, com pesquisa de dose em adultos idosos saudáveis que receberam doses variando de 267 mg a 1335 mg três vezes por dia, a média de depuração diminuiu em aproximadamente 25% acima da dose de 801 mg três vezes por dia. Depois de uma administração de dose única de pirfenidona em adultos idosos saudáveis, a meia-vida média de eliminação terminal aparente foi de aproximadamente 2,4 horas.
Aproximadamente 80% de uma dose administrada por via oral de pirfenidona é eliminada na urina em 24 horas após a administração. A maior parte da pirfenidona é excretada na forma do metabólito 5-carboxi-pirfenidona ( > 95% do que foi recuperado), com menos de 1% de pirfenidona excretada inalterada na urina.
Farmacocinética em Populações Especiais
Insuficiência Hepática
A farmacocinética da pirfenidona e do metabólito 5-carboxi-pirfenidona foi comparável em indivíduos com insuficiência hepática moderada (Classe B de Child-Pugh) e indivíduos com função hepática normal. Os resultados mostraram que houve um aumento médio de 60% na exposição à pirfenidona após administração de dose única de 801 mg de pirfenidona (3 x cápsulas de 267 mg) em pacientes com insuficiência hepática moderada. A pirfenidona deve ser utilizada com cautela em pacientes com insuficiência hepática leve a moderada e os pacientes devem ser acompanhados de perto em relação a sinais de toxicidade, especialmente se estiverem recebendo concomitantemente um inibidor conhecido de CYP1A2 (vide itens "Posologia e Modo de Usar" e "Advertências e Precauções").
Insuficiência Renal
Não foram observadas diferenças clinicamente relevantes na farmacocinética da pirfenidona em indivíduos com insuficiência renal leve a grave em comparação com indivíduos com função renal normal. A droga original é metabolizada predominantemente para 5-carboxi-pirfenidona, cuja farmacodinâmica e margens de segurança não foram estabelecidas. A ASC0-∞ da 5-carboxi-pirfenidona foi significativamente maior nos grupos com insuficiência renal moderada (p = 0,009) e grave (p < 0,0001) comparado ao grupo com função renal normal. A quantidade prevista de acúmulo de metabólitos no estado de equilíbrio não é importante farmacodinamicamente, porque a meia-vida de eliminação terminal é de apenas 1 a 2 horas nesses indivíduos e há atividade farmacológica inexistente ou mínima do metabólito, conforme mensurado por efeitos inibitório da TNF. Análises de farmacocinética populacional de 4 estudos incluindo indivíduos saudáveis ou indivíduos com insuficiência renal e um estudo incluindo pacientes com FPI não mostraram nenhum efeito clinicamente relevante de idade, sexo ou tamanho corporal sobre a farmacocinética da pirfenidona.
Segurança Pré-Clínica
Dados não clínicos não revelam nenhum risco especial para o ser humano com base em estudos convencionais de segurança farmacológica, toxicidade com doses repetidas, genotoxicidade e potencial carcinogênico.
Carcinogenicidade
Em estudos de toxicidade com doses repetidas, foram observados aumentos no peso do fígado em camundongos, ratos e cães; isto foi frequentemente acompanhado de hipertrofia centrilobular hepática. Foi observada reversibilidade depois da descontinuação do tratamento. Observou-se aumento da incidência de tumores hepáticos em estudos de carcinogenicidade conduzidos em ratos e camundongos. Esses achados hepáticos são compatíveis com uma indução de enzimas microssomais hepáticas, um efeito que não foi observado em pacientes que estavam recebendo Esbriet®. Esses achados não são considerados relevantes para o ser humano.
Um aumento estatisticamente significativo de tumores uterinos foi observado em ratos fêmeas que receberam 1500 mg/kg/dia, 37 vezes a dose humana, de 2403 mg/dia. Os resultados de estudos mecanísticos indicam que a ocorrência de tumores uterinos está provavelmente relacionada com um desequilíbrio crônico de hormônios sexuais mediado por dopamina envolvendo um mecanismo endócrino espécie-específico no rato, que não está presente no ser humano.
Mutagenicidade
Não foi demonstrada nenhuma indicação de atividade mutagênica nem genotóxica da pirfenidona em uma bateria padronizada de testes e quando testada sob exposição UV não foi mutagênica. A pirfenidona foi positiva em um ensaio fotoclastogênico em células de pulmão de hâmster chinês, quando testada sob exposição UV.
Comprometimento da fertilidade
Em animais, a transferência placentária de pirfenidona e/ou dos seus metabólitos ocorre com o potencial para acúmulo de pirfenidona e/ou seus metabólitos no líquido amniótico. Em doses elevadas (≥450 mg/kg/dia), os ratos apresentaram um prolongamento do ciclo estral e uma incidência elevada de ciclos irregulares. Em doses elevadas (≥1000 mg/kg/dia), os ratos apresentaram um prolongamento de gestação e redução da viabilidade fetal. Estudos em ratas em lactação indicam que a pirfenidona e/ou seus metabólitos são excretados no leite, com o potencial para acúmulo de pirfenidona e/ou seus metabólitos no leite.
Teratogenicidade
Estudos de toxicologia reprodutiva não demonstraram efeitos adversos em fertilidade masculina e feminina nem sobre o desenvolvimento pós-natal de crias em ratos e não houve nenhuma evidência de teratogenicidade em ratos (1000 mg/kg/dia) ou coelhos (300 mg/kg/dia).
Outros
Fototoxicidade e irritação foram observadas em cobaias depois da administração oral de pirfenidona e com a exposição à luz UVA/UVB. A gravidade das lesões fototóxicas foi reduzida pela aplicação de filtro solar.
4. CONTRAINDICAÇÕES
Esbriet® está contraindicado nos casos de:
-hipersensibilidade a substância ativa ou qualquer um de seus componentes;
-histórico de angioedema devido ao uso de pirfenidona (vide item "Advertências e precauções");
-insuficiência hepática grave ou doença hepática terminal (vide itens "Posologia e modo de usar" e "Advertências e precauções")
-insuficiência renal grave (CrCl < 30mL/min) ou doença renal terminal com necessidade de diálise (vide itens "Posologia e modo de usar" e "Advertências e precauções").
O uso concomitante de fluvoxamina e Esbriet® está contraindicado (vide item "Interações Medicamentosas").
5. ADVERTÊNCIAS E PRECAUÇÕES
Gerais
Função Hepática
Elevações de alanina aminotransferase (ALT) e aspartato aminotransferase (AST) > 3 x o limite superior da normalidade (LSN) foram reportadas em pacientes recebendo terapia com Esbriet® . Raramente, essas estavam associadas com elevações concomitantes da bilirrubina. Provas de função hepática (ALT, AST e bilirrubinas) devem ser realizadas antes do início do tratamento com Esbriet® e subsequentemente em intervalos mensais nos 6 primeiros meses e depois a cada 3 meses a partir de então. No caso de elevação significativa de aminotransferases hepáticas, a dose de Esbriet® deve ser ajustada ou o tratamento descontinuado de acordo com as orientações apresentadas no item "Posologia e Modo de Usar". Para pacientes com elevações confirmadas de ALT, AST ou bilirrubinas durante o tratamento, podem ser necessários ajustes da dose (vide item "Posologia e Modo de Usar").
Reação de hipersensibilidade e erupção cutânea
A exposição direta à luz solar (incluindo bronzeamento artificial) deve ser evitada ou reduzida durante o tratamento com Esbriet® . Os pacientes devem ser orientados a usar bloqueador solar eficaz diariamente, usar roupas que protejam contra a exposição solar e evitar outros medicamentos que reconhecidamente provoquem fotossensibilidade. Os pacientes devem ser orientados a reportar sintomas de fotossensibilidade ou erupção cutânea ao seu médico. Ajustes de dose ou descontinuação temporária de tratamento podem ser necessários no caso de reação de fotossensibilidade ou erupção (vide item "Posologia e Modo de Usar").
Angioedema
Há relatos de angioedema (alguns sérios) tais como inchaço da face, lábios e / ou língua, que podem ser associados com dificuldade em respirar ou respiração ofegante em associação com o uso de Esbriet® no período de pós-comercialização. Assim, os pacientes que desenvolvam sinais ou sintomas de angioedema após a administração de Esbriet® devem interromper imediatamente o tratamento. Os pacientes com angioedema devem ser manejados de acordo com padrão de tratamento. Esbriet® não deve ser utilizado em pacientes com história de angioedema devido ao Esbriet® (vide item "Contraindicações").
Tontura
Tonturas têm sido relatadas em pacientes tomando Esbriet® . Portanto, os pacientes devem saber como eles reagem a este medicamento antes de se envolver em atividades que exigem prontidão ou coordenação mentais (vide item "Advertências e precauções -Capacidade para dirigir e operar máquinas"). Em estudos clínicos, a maioria dos pacientes que apresentaram tontura tinham um único evento, e a maioria dos eventos resolvidos, com uma duração média de 22 dias. Se a tontura não melhorar ou se agravar, pode ser necessário um ajuste da dose ou até mesmo a interrupção de Esbriet® .
Fadiga
Fadiga tem sido relatada em pacientes tomando Esbriet® . Portanto, os pacientes devem saber como eles reagem a este medicamento antes de se envolver em atividades que exigem prontidão ou coordenação mental (vide item "Advertências e precauções -Capacidade para dirigir e operar máquinas").
Perda de peso
A perda de peso tem sido relatada em pacientes tratados com Esbriet® (vide item "Reações adversas"). Os médicos devem monitorar o peso dos pacientes, e, quando necessário, incentivar o desenvolvimento do consumo de calorias se a perda de peso é considerada de importância clínica.
Distúrbios gastrintestinais
Nos estudos clínicos, os eventos gastrintestinais de náuseas, diarreia, dispepsia, vômitos, doença do refluxo gastresofágico e dor abdominal foram mais frequentemente relatados pelos pacientes nos grupos de tratamento com Esbriet® do que naqueles que receberam o placebo. Foi necessária a redução de dose ou interrupção por eventos gastrointestinais em 18,5% dos pacientes no grupo de 2.403 mg/dia, em comparação com 5,8% dos pacientes no grupo com placebo; 2,2% dos pacientes do grupo com Esbriet® 2.403 mg/dia descontinuaram o tratamento, devido a um evento gastrointestinal, em comparação com 1,0% no grupo do placebo. Os eventos gastrointestinais mais comuns ( > 2%) que levaram à redução da dose ou interrupção foram náuseas, diarreia, vômitos e dispepsia.
A incidência de eventos gastrointestinais foi mais elevada no início do curso do tratamento (com maior incidência ocorrendo durante os primeiros 3 meses) e diminuiu ao longo do tempo. Modificações de dosagem podem ser necessárias em alguns casos de reacções adversas do trato gastrointestinal.
Capacidade para dirigir e operar máquinas
Não foram realizados estudos para avaliar os efeitos na capacidade de dirigir e operar máquinas. Esbriet® pode provocar tontura e fadiga, o que poderia influenciar a capacidade de dirigir ou operar máquinas.
Uso em Populações Especiais
Gravidez
Categoria de risco na gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Efeitos Teratogênicos
Não existem dados sobre uso de Esbriet® em gestantes.
Em animais, a transferência placentária de pirfenidona e/ou seus metabólitos ocorre com o potencial para acúmulo de pirfenidona e/ou seus metabólitos no líquido amniótico.
Em doses elevadas (≥1000 mg/kg/dia), ratas apresentaram prolongamento de gestação e redução de viabilidade fetal. Como medida de cautela, é preferível evitar o uso de Esbriet® durante a gravidez.
Fertilidade
Não foram observados efeitos adversos sobre a fertilidade em estudos pré-clínicos (vide "Segurança Pré-Clínica" do item "Características Farmacológicas".
Lactação
Não se sabe se a pirfenidona ou seus metabólitos são excretados no leite humano. Dados farmacocinéticos em animais mostraram a excreção de pirfenidona e/ou seus metabólitos no leite, com potencial para acúmulo de pirfenidona e/ou seus metabólitos no leite (vide "Segurança Pré-Clínica" do item "Características Farmacológicas"). Não se pode afastar um risco para a criança em aleitamento.
Deve-se optar entre descontinuar a amamentação ou descontinuar a terapia com Esbriet® , considerando o benefício da amamentação para a criança e o benefício da terapia com Esbriet® para a mãe.
Uso em pediatria
A segurança e eficácia de Esbriet® não foram estabelecidas em pacientes pediátricos.
Uso em idosos
Não é necessário ajuste da dose de acordo com a idade.
Insuficiência Renal
Esbriet® deve ser utilizado com cautela em pacientes com insuficiência renal leve a moderada.
A segurança, eficácia e farmacocinética de Esbriet® não foram estudadas em pacientes com insuficiência renal grave (CrCl < 30 mL/min) ou doença renal terminal com necessidade de diálise. Dessa forma, Esbriet® não deve ser usado nesses pacientes.
Insuficiência Hepática
Esbriet® deve ser utilizado com cautela em pacientes com insuficiência hepática leve a moderada. A segurança, eficácia e farmacocinética de Esbriet® não foram estudadas em pacientes com insuficiência hepática grave ou doença hepática terminal, Dessa forma, Esbriet® não deve ser administrado nesses pacientes.
Até o momento, não há informações de que a pirfenidona possa causar doping.
6. INTERAÇÕES MEDICAMENTOSAS
Interações com outros medicamentos e outras formas de interação
A pirfenidona é metabolizada principalmente através de CYP1A2, com mínimas contribuições de outras isoenzimas CYP, incluindo CYP2C9, 2C19, 2D6 e 2E1.
Fluvoxamina e inibidores da CYP1A2
Em um estudo Fase 1, a coadministração de Esbriet® e fluvoxamina (um potente inibidor de CYP1A2 com efeitos inibitórios sobre outras isoenzimas CYP [CYP2C9, 2C19 e 2D6]) resultou em um aumento de 4 vezes na exposição a pirfenidona em não fumantes.
Esbriet® é contraindicado em pacientes em uso concomitante de fluvoxamina (vide item "Contraindicações"). A fluvoxamina deve ser descontinuada antes do início da terapia com Esbriet® e evitada durante o tratamento, devido à depuração reduzida da pirfenidona.
Extrapolações in vitro e in vivo indicam que inibidores potentes e seletivos de CYP1A2 têm o potencial para aumentar a exposição a pirfenidona em aproximadamente 2 a 4 vezes. Se o uso concomitante de Esbriet® com um inibidor potente e seletivo de CYP1A2 não puder ser evitado, a dose de Esbriet® deve ser reduzida para 801 mg por dia (uma cápsula, três vezes por dia). Os pacientes devem ser monitorados com cuidado em relação à emergência de reações adversas associadas com a terapia de Esbriet® . Descontinue Esbriet®, se necessário (vide itens "Posologia e Modo de Usar" e "Advertências e Precauções").
A coadministração de Esbriet® e ciprofloxacino 750 mg (inibidor moderado e seletivo de CYP1A2) aumentou a exposição a pirfenidona em 81%. Se o uso de ciprofloxacino 750 mg duas vezes por dia não puder ser evitado, a dose de Esbriet® deve ser reduzida para 1602 mg por dia (duas cápsulas, três vezes por dia). Esbriet® deve ser usado com cautela quando o ciprofloxacino for utilizado em dose de 250 mg ou 500 mg, uma ou duas vezes por dia.
Esbriet® deve ser utilizado com cautela em pacientes tratados com outros inibidores moderados de CYP1A2.
Agentes ou combinações de agentes que sejam inibidores moderados ou potentes de CYP1A2 e também uma ou mais das outras isoenzimas CYP envolvidas no metabolismo da pirfenidona (CYP2C9, 2C19, 2D6 e 2E1) devem ser evitadas durante o tratamento com Esbriet® .
Uso de cigarro e indutores de CYP1A2
Um estudo Fase 1 de interação avaliou o efeito do cigarro (indutor CYP1A2) sobre a farmacocinética de Esbriet®. A exposição a pirfenidona em tabagistas foi 50% da observada em não fumantes. O tabagismo tem o potencial para induzir a produção de enzimas hepáticas e por isso aumenta a depuração e reduz a exposição ao Esbriet®. O uso concomitante de indutores potentes de CYP1A2, incluindo o fumo, deve ser evitado durante a terapia com Esbriet® com base na relação observada entre o uso de cigarro e seu potencial para induzir CYP1A2. Os pacientes devem ser estimulados a descontinuar o uso de indutores potentes de CYP1A2 e interromper o tabagismo antes e durante o tratamento com pirfenidona.
No caso de indutores moderados de CYP1A2 (p.ex., omeprazol), o uso concomitante pode teoricamente resultar em redução dos níveis plasmáticos de pirfenidona.
A coadministração de medicamentos que atuem como indutores potenciais tanto de CYP1A2 quanto de outras isoenzimas CYP envolvidas no metabolismo da pirfenidona (p.ex., rifampicina) pode resultar em redução significativa dos níveis plasmáticos de pirfenidona. Esses medicamentos devem ser evitados sempre que possível.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Esbriet® cápsulas duras deve ser conservado em temperatura ambiente (entre 15 e 30°C).
Prazo de validade
Este medicamento possui prazo de validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Descarte de medicamentos não utilizados e/ou com data de validade vencida
O descarte de medicamentos no meio ambiente deve ser minimizado. Os medicamentos não devem ser descartados no esgoto, e o descarte em lixo doméstico deve ser evitado. Utilize o sistema de coleta local estabelecido, se disponível.
Antes de usar, observe o aspecto do medicamento.
As cápsulas de Esbriet® apresentam coloração branca, com a inscrição "PFD 267 mg".
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Modo de Administração
Esbriet® deve ser administrado inteiro com água e com alimento para reduzir a possibilidade de náuseas e tontura (vide item
"Reações Adversas" e "Propriedades Farmacocinéticas" do item "Características Farmacológicas").
Posologia
Adultos
Ao iniciar o tratamento, a dose deve ser escalonada em um período de 14 dias até a dose diária recomendada de nove cápsulas por dia, como se segue:
Dias 1 a 7: uma cápsula, três vezes por dia (801 mg/dia)
Dias 8 a 14: duas cápsulas, três vezes por dia (1602 mg/dia)
Dias 15 em diante: três cápsulas, três vezes por dia (2403 mg/dia)
A dose diária recomendada de Esbriet® para pacientes com FPI é de três cápsulas de 267 mg três vezes por dia com alimentos até um total de 2403 mg/dia.
Doses acima de 2403 mg/dia não são recomendadas para nenhum paciente (vide item "Superdose").
Este medicamento não deve ser aberto, partido ou mastigado.
Conduta em caso de esquecimento
Pacientes que perderem 14 dias consecutivos ou mais de tratamento com Esbriet® devem reiniciar a terapia se submetendo ao esquema de escalonamento inicial de 2 semanas até a dose diária recomendada.
Para interrupção de tratamento de menos de 14 dias consecutivos, a dose pode ser reiniciada na dose diária recomendada previamente sem escalonamento.
Ajustes de dose e outras considerações
Eventos gastrintestinais: Em pacientes que apresentarem intolerância à terapia por efeitos colaterais gastrintestinais, eles devem ser orientados a ingerir o medicamento com alimentos. Se os sintomas persistirem, o tratamento com Esbriet® pode ser reduzido para 1-2 cápsulas (267 mg - 534 mg), 2-3 vezes/dia com alimentos com reescalonamento até a dose diára recomendada conforme a tolerância. Se os sintomas persistirem, os pacientes podem ser orientados para interromper o tratamento durante 1 ou 2 semanas para permitir que os sintomas sejam resolvidos.
Reação de Fotossensibilidade ou erupção cutânea: Os pacientes que apresentarem reação de fotossensibilidade ou erupção cutânea leve a moderada devem ser orientados sobre a necessidade de usar bloqueador solar diariamente e evitar a exposição ao sol (vide item "Advertências e Precauções"). A dose de Esbriet® pode ser reduzida para 3 cápsulas/dia (1 cápsula três vezes por dia). Se a erupção persistir depois de 7 dias, Esbriet® deve ser descontinuado durante 15 dias, com reescalonamento até a dose diária recomendada da mesma forma que o período de escalonamento de dose.
Os pacientes que apresentarem reação de fotossensibilidade ou erupção graves devem ser orientados a interromper o tratamento e buscar atendimento médico (vide item "Advertências e Precauções"). Depois de resolvida a erupção, Esbriet® pode ser reintroduzido e reescalonado até a dose diária recomendada, a critério do médico.
Função hepática: No caso de elevação significativa de alanina e/ou aspartato aminotransferases (ALT/AST) com ou sem elevação de bilirrubinas, a dose de Esbriet® deve ser ajustada ou o tratamento descontinuado.
Recomendações em caso de elevações em ALT, AST e bilirrubina sérica: Se um paciente apresentar uma elevação de aminotransferase > 3 a ≤5 x LSN depois de iniciar a terapia Esbriet, medicamentos que possam ser fatores de confusão devem ser descontinuados, outras causas excluídas e o paciente monitorado com cuidado. Se for clinicamente adequado, a dose de Esbriet® deve ser reduzida ou o tratamento interrompido. Depois que as provas de função hepática estiverem dentro dos limites normais, Esbriet® pode ser re-escalonado até a dose diária recomendada, se tolerada.
Se um paciente apresentar uma elevação de aminotransferase até ≤5 x LSN acompanhada de sintomas ou hiperbilirrubinemia, o tratamento com Esbriet® deve ser descontinuado e o paciente não deve receber o medicamento novamente.
Se um paciente apresentar uma elevação de aminotransferase > 5 x LSN, Esbriet® deve ser descontinuado e o paciente não deve receber o medicamento novamente.
Modificações de dose devido a interação com medicamentos:
-Forte inibidor da CYP1A2 (por exemplo, fluvoxamina): reduzir a dose de Esbriet® para 3 cápsulas/dia (1 cápsula três vezes a dia).
-Moderado inibidor da CYP1A2 (por exemplo, ciprofloxacina): com o uso de ciprofloxacina na dose de 750 mg duas vezes ao dia, reduzir a dose de Esbriet® para 6 cápsulas/dia (2 cápsulas três vezes por dia).
Populações Especiais
Idosos
Não é necessário ajuste em pacientes com 65 anos de idade ou mais (vide item "Características Farmacológicas")
Insuficiência Hepática
Não é necessário ajuste de dose em pacientes com insuficiência hepática leve a moderada (Classe Ae B de Child-Pugh). No entanto, como os níveis plasmáticos de pirfenidona podem aumentar em alguns indivíduos com insuficiência hepática leve a moderada, deve-se ter cautela no tratamento com Esbriet® nesta população. Os pacientes devem ser monitorados rigorosamente em relação a sinais de toxicidade, especialmente se estiverem recebendo concomitantemente um inibidor conhecido de CYP1A2 (vide item "Interações Medicamentosas" e "Características Farmacológicas"). Esbriet® não foi estudado em pacientes com insuficiência hepática grave ou com doença hepática terminal e não deve ser utilizado em pacientes com essas condições. Recomenda-se monitorar a função hepática durante o tratamento, podendo ser necessários ajustes de dose em caso de elevações (vide item "Advertências e Precauções" e "Farmacocinética em Populações Especiais" do item "Características Farmacológicas").
Insuficiência Renal
Não é necessário nenhum ajuste de dose em pacientes com insuficiência renal leve. Esbriet® deve ser utilizado com cautela em pacientes com insuficiência renal moderada (CrCl 30 - 50 mL/min) por causa da falta de informações referentes ao metabólito (vide item "Farmacocinética em Populações Especiais). A terapia com Esbriet® não deve ser usada em pacientes com insuficiência renal grave (CrCl < 30 ml/min) nem doença renal terminal com necessidade de diálise (vide "Farmacocinética em Populações Especiais" do item "Características Farmacológicas").
9. REAÇÕES ADVERSAS
Estudos Clínicos
A segurança de Esbriet® foi avaliada em 623 pacientes, a partir de três estudos clínicos Fase 3.
A Tabela 4 resume as reações adversas ao medicamento (RAM) que foram reportadas em associação com o uso de Esbriet® em estudos clínicos. As reações adversas ao medicamento estão listadas de acordo com a classe de sistema de órgãos, as categorias de frequências são: Muito comum ≥1/10 e Comum ≥1/100 e < 1/10.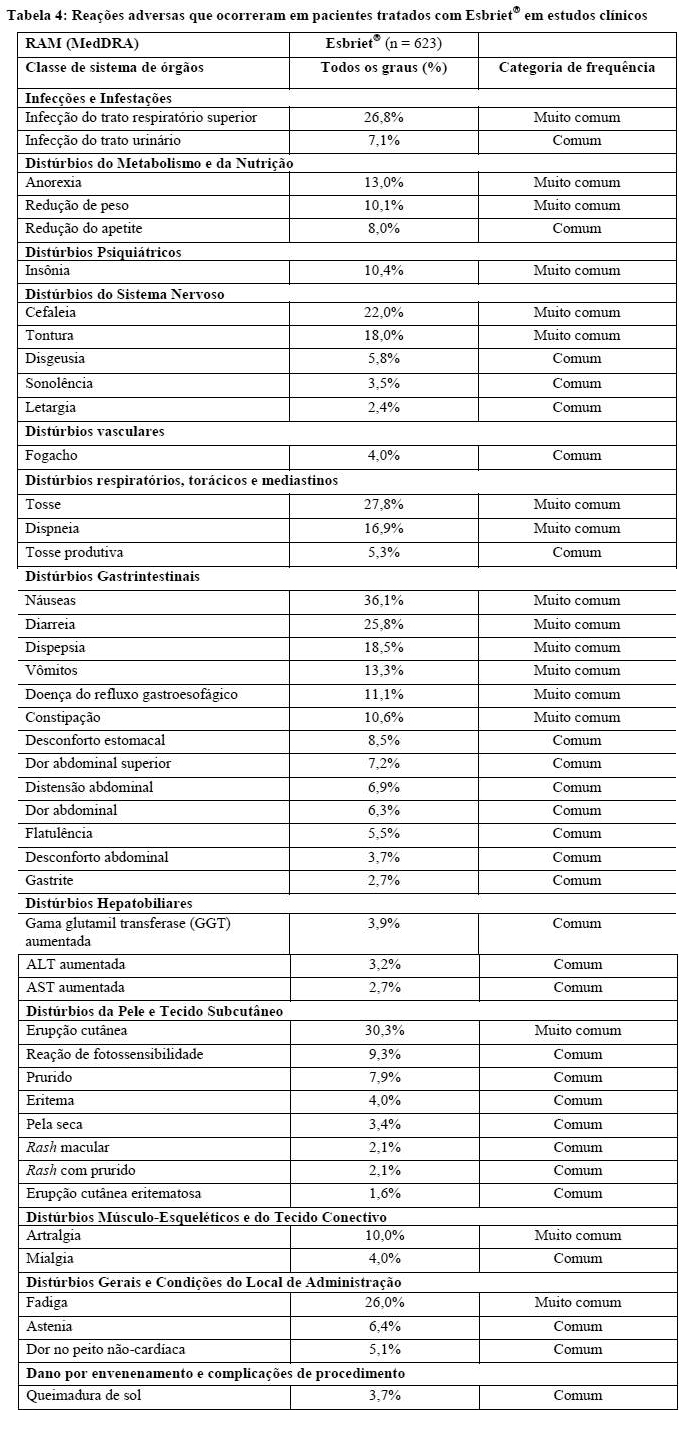
Pós-Comercialização
Além das reações adversas identificadas a partir de estudos clínicos, as seguintes reações adversas foram identificadas durante a utilização pós-aprovação de pirfenidona. Como essas reações podem ser reportadas voluntariamente a partir de uma população de tamanho incerto, nem sempre é possível estimar confiavelmente sua frequência.
Distúrbios do Sangue e Sistema Linfático
Agranulocitose
Distúrbios de Sistema Imunológico
Angioedema
Distúrbios Hepatobiliares:
Bilirrubina aumentada em combinação com aumentos de ALT e AST
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
A experiência clínica com superdosagem é muito limitada. Múltiplas doses de Esbriet® até uma dose total de 4806 mg/dia foram administradas na forma de seis cápsulas de 267 mg três vezes por dia a voluntários adultos saudáveis durante um período de escalonamento de dose de 12 dias. As reações adversas foram leves, transitórias e compatíveis com as reações adversas mais frequentemente reportadas para Esbriet® .
No caso de uma suspeita de superdosagem, devem ser fornecidos cuidados médicos de suporte de sinais vitais e observação cuidadosa da situação clínica do paciente.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
DIZERES LEGAIS
MS-1.0100.0663
VENDA SOB PRESCRIÇÃO MÉDICA
Esta bula foi aprovada pela ANVISA em 12/07/2016.