ERFANDEL
JANSSEN-CILAG
erdafitinibe
Antineoplásico.
Apresentações.
Comprimidos revestidos com 3 mg de erdafitinibe em 2 blisters com 28 comprimidos.
Comprimidos revestidos com 3 mg de erdafitinibe em 2 blisters com 42 comprimidos.
Comprimidos revestidos com 4 mg de erdafitinibe em 1 blister com 28 comprimidos.
Comprimidos revestidos com 4 mg de erdafitinibe em 2 blister com 28 comprimidos.
Comprimidos revestidos com 5 mg de erdafitinibe em 1 blister com 28 comprimidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém 3, 4 ou 5 mg de erdafitinibe.
Excipientes: manitol, celulose microcristalina, meglumina, croscarmelose sódica, estearato de magnésio, álcool polivinílico, dióxido de titânio, talco, monocaprilocaprato de glicerila, laurilsulfato de sódio, óxido de ferro amarelo, óxido de ferro vermelho (apenas para comprimidos laranja e marrom), óxido de ferro preto (apenas para comprimido marrom).
Informações técnicas.
1. INDICAÇÕES
ERFANDEL® é indicado para o tratamento de pacientes adultos com carcinoma urotelial (UC) localmente avançado ou metastático, cujos tumores apresentam determinadas alterações genéticas de receptores de fator de crescimento de fibroblastos (FGFR), que apresentam progressão da doença durante ou após pelo menos uma linha de quimioterapia anterior, ou até 12 meses após quimioterapia neoadjuvante ou adjuvante.
2. RESULTADOS DE EFICÁCIA
Tumores de carcinoma urotelial com alterações genéticas selecionadas de FGFR
O estudo BLC2001 foi um estudo multicêntrico, aberto, de Fase 2 para avaliar a eficácia e segurança de ERFANDEL® em 99 pacientes com carcinoma urotelial localmente avançado ou metastático, incluindo 12 pacientes nunca tratados com quimioterapia com base na inelegibilidade para cisplatina. Todos os pacientes foram incluídos com base na avaliação do investigador da mensuração da doença e foi requerido que apresentassem tecidos tumorais com pelo menos 1 das seguintes mutações genéticas de FGFR3: R248C, S249C, G370C, Y373C ou uma das seguintes fusões genéticas FGFR: FGFR3TACC3, FGFR3-BAIAP2L1, FGFR2-BICC1, FGFR2-CASP7, conforme determinado pelo ensaio de estudo clínico realizado em um laboratório central. A análise de eficácia foi baseada em 87 pacientes cuja doença progrediu durante ou após pelo menos uma quimioterapia anterior. Os pacientes receberam uma dose inicial de ERFANDEL® de 8 mg uma vez ao dia com titulação crescente guiada de forma farmacodinâmica de 9 mg uma vez ao dia em pacientes em que os níveis de fosfato sérico entre os dias 14 e 17 estavam abaixo da meta de 5,5 mg/dL; a titulação crescente ocorreu em 41% dos pacientes. ERFANDEL® foi administrado até a progressão da doença ou toxicidade inaceitável.
A idade mediana foi 67 anos (variação: 36 a 87 anos), 79% foram homens e 74% foram caucasianos. A maioria dos pacientes (92%) apresentou status de desempenho do Eastern Cooperative Oncology Group (ECOG) basal de 0 ou 1. A metade dos pacientes (51%) recebeu uma linha anterior de terapia, 49% receberam duas ou mais e 79% apresentaram metástases viscerais. Os resultados de eficácia foram baseados na taxa de resposta objetiva (TRO) usando os Critérios de Avaliação de Resposta em Tumores Sólidos (RECIST) v1.1 (veja Tabela 1).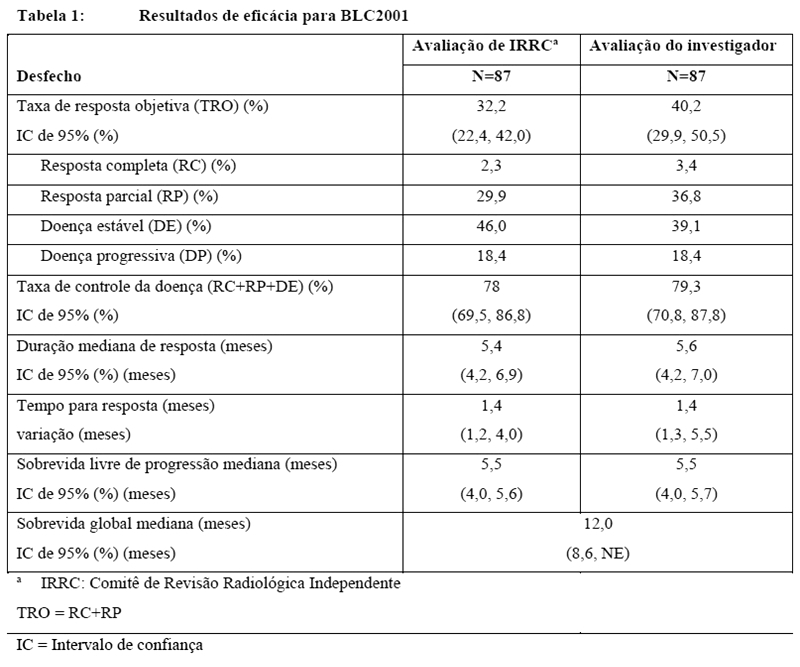
A avaliação de IRRC demonstrou que as TROs para pacientes que recebem ERFANDEL® foram compatíveis independentemente da quantidade de linhas de terapia sistêmica anterior e variou de 36% a 60% e a Taxa de Controle da Doença (TCDs) variou de 75% a 90%.
A TRO foi maior em pacientes com fosfato sérico ≥ 5,5 mg/dL (43,5% com fosfato sérico ≥ 5.5 mg/dL versus 33,3% com fosfato sérico < 5,5 mg/dL conforme obtido nos primeiros 3 meses de tratamento). A sobrevida global foi mais longa em pacientes com fosfato sérico ≥ 5,5 mg/dL (sobrevida global média 13,8 meses com fosfato sérico ≥ 5,5 mg/dL versus 7,23 meses com fosfato sérico < 5,5 mg/dL).
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Erdafitinibe é um inibidor oral de tirosina quinase pan-FGFR altamente seletivo e potente com alta afinidade e atividade inibitória em níveis nanomolares baixos para todos os membros da família FGFR, 1, 2, 3 e 4. Nas linhagens celulares cancerígenas ativadas pela via FGFR, a concentração exigida para 50% de inibição do crescimento tumoral (IC50) está na variação nanomolar baixa de 0,1 a 129,2 nM.
Erdafitinibe demonstrou atividade antitumoral nas linhagens celulares dirigidas pelo FGFR e modelos de xenoenxerto, derivados de múltiplos tipos de tumores, incluindo câncer de bexiga.
Propriedades farmacodinâmicas
Eletrofisiologia cardíaca
Erdafitinibe não apresentou efeitos na repolarização cardíaca ou outros parâmetros eletrocardiográficos em humanos. As análises da relação exposição-QT foram realizadas sobre uma variação de dose de 0,5 a 12 mg de 187 indivíduos com câncer em um estudo de Fase 1, aberto, de aumento de dose. As análises de resposta à exposição não indicaram nenhuma uma relação significativa entre concentração plasmática de erdafitinibe e alteração nos intervalos de QTc. Os two-sided ICs de 90% superiores na Cmáx a partir da maior dose clínica (9 mg) foram 2,5 ms ou menos.
Fosfato sérico
Erdafitinibe aumentou o nível de fosfato sérico, um biomarcador farmacodinâmico da inibição de FGFR. Alcançar os níveis de fosfato sérico alvo ≥ 5,5 mg/dL em ciclos iniciais com administração diária contínua está associado à resposta clínica melhorada (vide "Posologia e Modo De Usar").
Propriedades farmacocinéticas
Após administração única e repetida uma vez ao dia, a exposição ao erdafitinibe [concentração plasmática máxima observada (Cmáx) e área sob a curva (ASC)] aumentou de forma proporcional à dose em relação à variação de dose de 0,5 a 12 mg. O estado de equilíbrio foi alcançado após 2 semanas com administração uma vez ao dia e a proporção de acumulação mediana foi de 4 vezes. Após a administração de 8 mg uma vez ao dia, a dose inicial proposta, Cmáx de estado de equilíbrio mediano [coeficiente de variação (CV%)] de erdafitinibe, ASCt, e concentração plasmática mínima observada (Cmín) foram 1399 ng/mL (50,8%), 29268 ng.h/mL (59,9%) e 936 ng/mL (64,9%). As flutuações diárias nas concentrações plasmáticas de erdafitinibe foram baixas, com uma proporção média (CV%) de máximo-mínimo de 1,47 (23%) no estado de equilíbrio na administração diária.
Absorção
Após administração oral de dose única, o tempo médio para alcançar a concentração plasmática máxima (tmáx) foi de 2,5 horas (variação: 2 a 6 horas) e a absorção oral é quase completa.
Efeito de alimentos
A administração de erdafitinibe em indivíduos saudáveis em condições de jejum e com uma refeição de alto teor de gordura não resultou em alterações clinicamente relevantes na Cmáx e ASC. O tempo médio para alcançar tmáx foi postergado em aproximadamente 1,5 hora com alimento (vide "Posologia e Modo De Usar").
Distribuição
O volume médio aparente de distribuição de erdafitinibe em indivíduos com câncer foi 28,8 L.
Em pacientes com câncer, erdafitinibe estava 99,76% ligado às proteínas plasmáticas humanas, preferivelmente a a?-glicoproteína ácida AGP.
Eliminação
A depuração aparente total média (CL/F) de erdafitinibe foi 0,362 L/h nos pacientes.
A meia vida efetiva média de erdafitinibe nos pacientes foi de 58,9 horas.
Metabolismo
O metabolismo é a via principal de eliminação para erdafitinibe. Erdafitinibe é metabolizado principalmente em humanos por CYP2C9 e CYP3A4 para formar o metabólito principal O-desmetilado. A contribuição de CYP2C9 e CYP3A4 na depuração total de erdafitinibe é estimada como sendo 39% e 20%, respectivamente. Erdafitinibe inalterado foi a maior parte relacionada à droga no plasma, não houve metabólitos circulantes.
Excreção
Até 16 dias após uma administração oral única de [14C]-erdafitinibe radiomarcado, 69% da dose foi recuperada nas fezes (14-21% como erdafitinibe inalterado) e 19% na urina (13% como erdafitinibe inalterado).
Populações especiais
Nenhuma diferença clinicamente significativa na farmacocinética de erdafitinibe foi observada com base na idade (21-88 anos), sexo, raça (hispânico ou asiático), peso corporal (36-132 kg), comprometimento renal leve ou moderado ou comprometimento hepático leve.
Pacientes Pediátricos
A farmacocinética de erdafitinibe não foi estudada em pacientes pediátricos.
Comprometimento Renal
Nenhuma diferença clinicamente significativa na farmacocinética de erdafitinibe foi observada entre indivíduos com função renal normal [TFGe-MDRD (taxa de filtração glomerular estimada -modificação dietética na doença renal) ≥ 90 mL/min/1,73 m²], e indivíduos com comprometimento renal leve (TFGe-MDRD 60 a 89 mL/min/1,73 m²) e moderado (TFGe-MDRD 30-59 mL/min/1,73 m²).
Comprometimento hepático
Nenhuma diferença clinicamente significativa no perfil farmacocinético de erdafitinibe foi observada em indivíduos com comprometimento hepático leve (conforme definido pelos critérios do Instituto Nacional do Câncer) e indivíduos com função hepática normal com base na análise de farmacocinética da população.
Metabolizador fraco de CYP2C9
A exposição ao erdafitinibe foi comparável em indivíduos com genótipos CYP2C9 *1/*2 e *1/*3 em relação a indivíduos com tipo selvagem. Não há dados disponíveis em indivíduos caracterizados por outros genótipos (por exemplo, *2/*2, *2/*3 e *3/*3). A simulação sugeriu nenhuma alteração clinicamente significativa da exposição ao erdafitinibe em indivíduos CYP2C9 *2/*2 e *2/*3. A exposição ao erdafitinibe é prevista para aumentar aproximadamente 50% em indivíduos de genótipo CYP2C9 *3/*3, estimado como sendo 0,4% a 3% da população entre diversos grupos étnicos e representando o pior cenário entre as diversas populações heterogêneas de pobres metabolizadores de 2C9.
Informação não clínica
Em estudos de toxicidade com doses repetidas em ratos e cães, distúrbios da homeostase do fosfato, caracterizados pela elevação sérica do fosfato, FGF-23 e 1,25 dihidroxivitamina D3 foram observados com exposições menores que as exposições humanas em todas as doses estudadas (vide "Posologia e Modo De Usar"). A displasia de cartilagem e a mineralização dos tecidos moles, associada à hiperfosfatemia, foram observadas como toxicidades primárias relacionadas a medicamentos em animais. Quando os ratos receberam uma dieta suplementada com o sequestrador de fosfato, sevelamer, as mineralizações dos tecidos moles foram reduzidas. Foram observadas atrofia das glândulas e estruturas epiteliais (alterações dentárias, afinamento da glândula lacrimal do epitélio da córnea, alterações no couro cabeludo e unhas).
As mineralizações de tecido mole (exceto a mineralização da aorta em cães) e a displasia condroide em ratos e cães e a atrofia da glândula mamária em ratos foram parcialmente a completamente recuperadas ao final de um período de recuperação de 4 semanas sem medicação.
Toxicologia Reprodutiva
O erdafitinibe foi teratogênico e embriotóxico em ratos com ≥ 4 mg/kg/dia e exposições inferiores â exposição humana em todas as doses estudadas (vide "Posologia e Modo de Usar"). A toxicidade fetal foi caracterizada por defeitos de mão/pé e malformações de alguns vasos sanguíneos principais, como a aorta.
4. CONTRAINDICAÇÕES
ERFANDEL® é contraindicado em pacientes com hipersensibilidade conhecida ao produto ou qualquer um dos componentes de sua fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Distúrbios oculares
Assim como outros inibidores de tirosina quinase, podem ocorrer distúrbios oculares com a administração de ERFANDEL® . Os eventos mais comumente relatados de retinopatia serosa central (CSR) foram coriorretinopatia (8%), descolamento da retina (5%) e descolamento do epitélio pigmentado da retina (5%). A CSR foi observada em 23 pacientes (23%) tratados com ERFANDEL® no estudo BLC2001 na dose diária de 8 mg. Um resultado de teste de tela de Amsler anormal foi identificado na maioria (70%) dos pacientes que desenvolveram CSR, a maioria grau 1 e 2. No estudo BLC2001, a CSR se resolveu em 12 pacientes e 11 pacientes apresentaram eventos em andamento, dos quais muitos melhoraram em gravidade e a maioria foi Grau 1. A CSR levou a interrupções e reduções de dose em 8,1% e 13,1% dos pacientes, respectivamente, e três pacientes (3%) descontinuaram ERFANDEL®. Outros distúrbios oculares além de CSR ocorreram em 55% dos pacientes, incluindo olho seco (19%) e visão embaçada (17%).
Examine os pacientes quanto a distúrbios oculares antes do início do tratamento com ERFANDEL® usando um teste de tela de Amsler, fundoscopia, acuidade visual e, se disponível, uma tomografia de coerência ótica (OCT). Para prevenir e tratar olhos secos, frequentemente use lágrimas artificiais substitutas, géis hidratantes ou lubrificantes ou pomadas oculares, pelo menos a cada 2 horas durante as horas despertas. Encaminhar casos graves de olho seco relacionados ao tratamento a um oftalmologista para avaliação. Examine os pacientes mensalmente e consequentemente, se qualquer anormalidade for observada, ou em qualquer momento um paciente relatar eventos relacionados ao olho ou distúrbio visual, siga as diretrizes de tratamento na Tabela 4 (vide "Posologia e Modo De Usar").
Toxicidade embrionário/fetal
Com base nos achados em estudos de reprodução animal, erdafitinibe pode causar dano fetal quando administrado a uma gestante. Em um estudo de toxicidade embrionário/fetal em ratas, erdafitinibe foi embriotóxico e teratogênico nas exposições menores que as exposições humanas em todas as doses estudadas (vide "Posologia e Modo De Usar"). Orientar as gestantes sobre o risco potencial ao feto. Orientar as pacientes com potencial reprodutivo sobre o uso de contracepção altamente eficaz antes e durante o tratamento e por 3 meses após a última dose.
Gravidez
Não existem dados em humanos disponíveis que informem o risco associado ao erdafitinibe. Com base nos resultados de estudos de reprodução animal, o erdafitinibe pode causar danos fetais quando administrado a uma mulher grávida. Em um estudo de toxicidade embrionário/fetal em ratos, o erdafitinibe se mostrou embriotóxico e teratogênico, com exposições inferiores às exposições humanas, em todas as doses estudadas (vide "Posologia e Modo de Uso"). A toxicidade fetal foi caracterizada por defeitos de mão/pé e malformações de alguns vasos sanguíneos principais, como a aorta.
Se ERFANDEL® for usado durante a gravidez, ou se a paciente engravidar durante o tratamento com ERFANDEL®, orientar a paciente do risco potencial ao feto e aconselhe a paciente sobre suas opções clínicas e terapêuticas. Orientar as pacientes a contatar seu médico caso engravidem ou haja suspeita de gravidez durante o tratamento com ERFANDEL® e em até 3 meses depois.
Categoria B de gravidez
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Lactação
Não há dados sobre a presença de erdafitinibe em leite humano, ou os efeitos de ERFANDEL® no bebê amamentado, ou na produção de leite. Devido ao potencial para reações adversas sérias de ERFANDEL® em bebês amamentados, orientar as mulheres a não amamentarem durante o tratamento com ERFANDEL® e por 3 meses após a última dose de ERFANDEL®.
Teste de gravidez
Teste de gravidez altamente sensível é recomendado para mulheres com potencial reprodutivo antes de iniciar ERFANDEL®.
Contracepção
ERFANDEL® pode causar danos fetais quando administrado a mulheres grávidas. Orientar as pacientes mulheres com potencial reprodutivo a usar contracepção altamente eficaz antes e durante o tratamento, e por 3 meses após a última dose de ERFANDEL®. Orientar os pacientes homens a usar contracepção eficaz (por exemplo, preservativo) e a não doar ou armazenar sêmen durante o tratamento e por 3 meses após a última dose de ERFANDEL®.
Fertilidade
Nenhum dado está disponível para determinar efeitos potenciais de erdafitinibe na fertilidade de mulheres ou homens.
Carcinogênese, Mutagênese e Fertilidade
Estudos em animais de longo prazo não foram realizados para avaliar o potencial carcinogênico de erdafitinibe. Erdafitinibe não induziu mutações no teste de mutação reversa bacteriana (Ames) e não foi genotóxico no estudo de micronúcleos em medula óssea de ratos in vitro ou estudo de micronúcleo in vivo. Estudos dedicados à fertilidade animal não foram realizados com erdafitinibe. Entretanto, em um estudo de toxicidade geral de 3 meses, erdafitinibe apresentou efeitos nos órgãos reprodutores femininos (necrose de corpora lutea) em ratos em uma exposição aproximando a ASC em pacientes na dose máxima recomendada de 9 mg, uma vez ao dia.
Efeitos na capacidade de dirigir e usar máquinas
Não foram realizados estudos para determinar os efeitos do erdafitinibe na capacidade de dirigir e usar máquinas. No entanto, distúrbios oculares, como retinopatia serosa central ou ceratite, foram observados com inibidores de FGFR (inibidores do receptor de fato de crescimento fibroblástico) e com o tratamento com ERFANDEL®. Se os pacientes apresentarem sintomas relacionados ao tratamento que afetam sua visão, recomenda-se que eles não conduzam ou usem máquinas até que o efeito desapareça (vide "Advertências e Precauções").
6. INTERAÇÕES MEDICAMENTOSAS
Efeito de outros medicamentos em ERFANDEL®
Inibidores potentes de CYP2C9 ou CYP3A4
A administração concomitante com um inibidor potente de CYP2C9 ou de CYP3A4 aumentou a exposição de erdafitinibe e pode levar ao aumento de toxicidade relacionada à medicação (vide "Propriedades Farmacocinéticas"). As proporções médias de erdafitinibe (IC de 90%) para Cmáx e ASC∞ foram 121% (99,9, 147) e 148% (120, 182), respectivamente, quando administrado concomitantemente com fluconazol, um inibidor potente de CYP2C9 e inibidor moderado de CYP3A4, em relação ao erdafitinibe isoladamente. A Cmáx de erdafitinibe foi 105% (IC de 90%: 86,7, 127) e ASC∞ foi 134% (IC de 90%: 109, 164) quando administrado concomitantemente com itraconazol, um inibidor potente de CYP3A4 e inibidor de gpP, em relação ao erdafitinibe isoladamente. Considere agentes alternativos sem potencial de inibição enzimática ou com potencial de inibição enzimática mínima. Se ERFANDEL® for administrado concomitantemente com um inibidor potente de CYP2C9 ou CYP3A4, reduza a dose de ERFANDEL® com base na tolerabilidade (vide "Posologia e Modo De Usar"). Se o inibidor potente for descontinuado, a dose de ERFANDEL® pode ser ajustada conforme tolerada.
Indutores potentes de CYP2C9 ou CYP3A4
A administração concomitante com indutores potentes de CYP2C9 ou CYP3A4 pode levar à toxicidade reduzida por exposição ao erdafitinibe (vide "Propriedades farmacocinéticas"). Os efeitos dos indutores de CYP3A4 ou CYP2C9 na farmacocinética de erdafitinibe não foram avaliados in vivo. As simulações sugeriram que rifampicina (um potente indutor de CYP3A4/2C9) pode levar a redução de aproximadamente 60% na exposição ao erdafitinibe (ASC e Cmáx). Considere agentes alternativos sem potencial de indução enzimática ou com potencial de indução enzimática mínima. Se ERFANDEL® for administrado concomitantemente com um indutor de CYP2C9 ou de CYP3A4, a dose pode ser cautelosamente elevada para aproximadamente 1 a 2 mg e ajustada gradualmente a cada duas ou três semanas com base no monitoramento clínico para reações adversas. Se o indutor potente for descontinuado, a dose de ERFANDEL® pode ser ajustada conforme tolerada.
Agentes redutores de ácido
Erdafitinibe é um composto Classe I da classificação BCS com solubilidade adequada entre a variação de pH de 1 a 7,4. Não se espera que agentes redutores de ácidos (por exemplo, antiácidos, antagonistas de H2 ou inibidores de bomba de próton) afetem a biodisponibilidade de erdafitinibe.
Transportadores que afetam medicamentos
Erdafitinibe é um substrato para P-glicoproteína (P-gp), mas não para BCRP, OATP1B1 e OATP1B3. Não se espera que os inibidores de P-gp afetem a farmacocinética de erdafitinibe de forma clinicamente relevante.
Sevelamer
Nenhuma diferença clinicamente significativa na farmacocinética de erdafitinibe foi observada em pacientes tratados com sevelamer.
Efeito de ERFANDEL® em outros medicamentos
Substratos de isoformas principais de CYP
Erdafitinibe não é um inibidor de isoenzimas principais de CYP em concentrações clinicamente relevantes; entretanto, ele tem demonstrado ser um inibidor dependente de tempo fraco em relação à atividade do CYP3A4, assim como um indutor de CYP3A4 fraco. A simulação demonstrou que não se espera que as interações medicamentosas com substratos de CYP3A4 sejam clinicamente relevantes.
Transportadores P-Glicoproteína (P-gp)
Erdafitinibe é um inibidor de P-gp in vitro e pode ser um inibidor clínico de P-gp intestinal. A simulação previu uma taxa de Cmáx de 1,45 e taxa de ASC de 1,18 para digoxina quando erdafitinibe foi administrado concomitantemente com digoxina ao mesmo tempo com uma taxa de Cmáx de 1,45 e uma taxa de ASC de 1,18, considerando que o intervalo entre as doses de 6 horas poderia evitar essa interação. A administração concomitante de ERFANDEL® com substratos de P-gp pode aumentar sua exposição sistêmica se administrado concomitantemente (vide "Propriedades farmacocinéticas"). Os substratos de P-gp orais com faixa terapêutica estreita, como digoxina, devem ser administrados pelo menos 6 horas antes ou depois de erdafitinibe para minimizar o potencial para interações.
Outros transportadores
Erdafitinibe não é um inibidor in vitro de OATP1B3, OAT1 e OAT3. Em concentrações clinicamente relevantes, erdafitinibe não é considerado um inibidor de transportadores BCRP, OATP1B, OCT1, MATE-1 e MATE-2K. Erdafitinibe é um inibidor de OCT2 in vitro. As simulações com metformina, um substrato de OCT2, previram uma ausência de interação clinicamente relevante com erdafitinibe.
7. CUIDADO DE ARMAZENAMENTO DO MEDICAMENTO
ERFANDEL® deve ser armazenado em temperatura ambiente (entre 15 °C a 30 °C).
A validade de ERFANDEL® é de 24 meses a partir da data de sua fabricação.
Aparência física
Comprimidos de 3 mg: amarelos, de formato redondo biconvexo, revestidos, gravados com "3" em um lado; e "EF" no outro lado.
Comprimidos de 4 mg: laranjas, de formato redondo biconvexo, revestidos, gravados com "4" em um lado; e "EF" no outro lado.
Comprimidos de 5 mg: marrons, de formato redondo biconvexo, revestidos, gravados com "5" em um lado; e "EF" no outro lado.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento antes do uso.
Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USAR
Dosagem recomendada
A dose inicial recomendada de ERFANDEL® é 8 mg via oral uma vez ao dia; com titulação crescente guiada pela farmacodinâmica, com base nos níveis de fosfato sérico, para 9 mg ao dia, se os critérios forem atendidos.
Administração
Antes de tomar ERFANDEL®, os pacientes devem ter confirmação de certas alterações do gene FGFR, como confirmado por um teste validado.
Os comprimidos devem ser engolidos inteiros, com ou sem alimento. Caso ocorra vômito a qualquer momento após a administração de ERFANDEL®, a próxima dose deve ser administrada no dia seguinte.
O tratamento deve continuar até que ocorra progressão da doença ou toxicidade inaceitável.
Dose perdida
Se uma dose de ERFANDEL® for perdida, ela pode ser administrada assim que possível. Reinicie o cronograma de dose diário regular para ERFANDEL® no dia seguinte. Comprimidos extras não devem ser administrados para compensar a dose perdida.
Modificação da dose
Titulação crescente guiada pela farmacodinâmica com base nos níveis de fosfato sérico
Os níveis de fosfato sérico (PO4) devem ser avaliados entre 14 e 21 dias após o início do tratamento. Aumente a dose para 9 mg ao dia assim que possível se o nível de fosfato sérico (PO4) for < 5,5 mg/dL, e se não houver toxicidade relacionada à medicação.
Redução de dose
Para possíveis reduções de dose e tratamento de reações adversas, veja as Tabelas 2 a 5.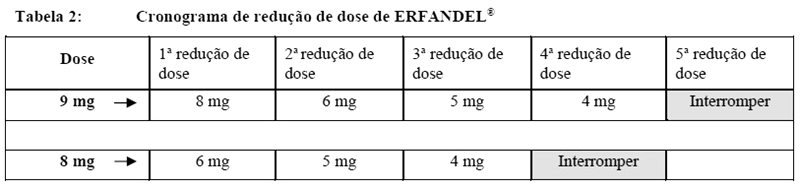
Hiperfosfatemia é uma anormalidade laboratorial temporária esperada de inibidores de FGFR (vide "Farmacodinâmica"). Os níveis de fosfato devem ser monitorados mensalmente. Para níveis de fosfato elevados em pacientes tratados com ERFANDEL® siga as diretrizes de modificação de dose da Tabela 3. Para níveis de fosfato persistentemente elevados, adicionar um ligante de fosfato não contendo cálcio (por exemplo, carbonato de sevelamer) pode ser considerado.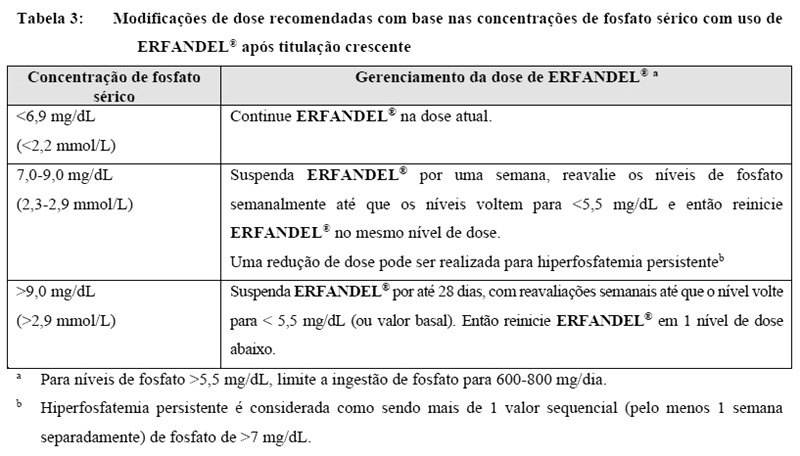
Tratamento de distúrbio ocular
Antes do início de ERFANDEL®, realize um exame oftalmológico basal incluindo um teste de tela de Amsler, fundoscopia, acuidade visual e, se disponível, uma tomografia de coerência ótica (OCT).
Para prevenir e tratar olhos secos, use lágrimas artificiais substitutas, géis hidratantes ou lubrificantes ou pomadas oculares frequentemente, pelo menos a cada 2 horas durante as horas despertas. Olho seco grave relacionado ao tratamento deve ser avaliado por um oftalmologista.
Subsequentemente, examine os pacientes mensalmente, incluindo um teste de tela de Amsler e, se qualquer anormalidade for observada, siga as diretrizes de tratamento na Tabela 4.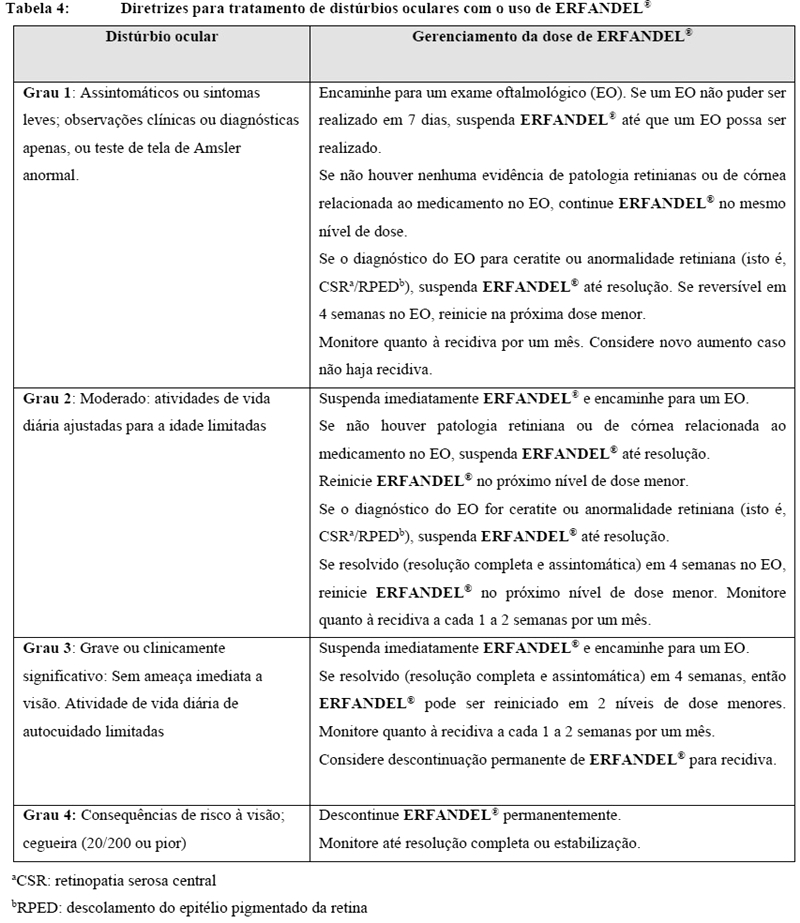
Modificações de dose para outras reações adversas
Alterações cutâneas, da mucosa e ungueais foram observadas com ERFANDEL®. Siga as diretrizes de modificação de dose na Tabela 5.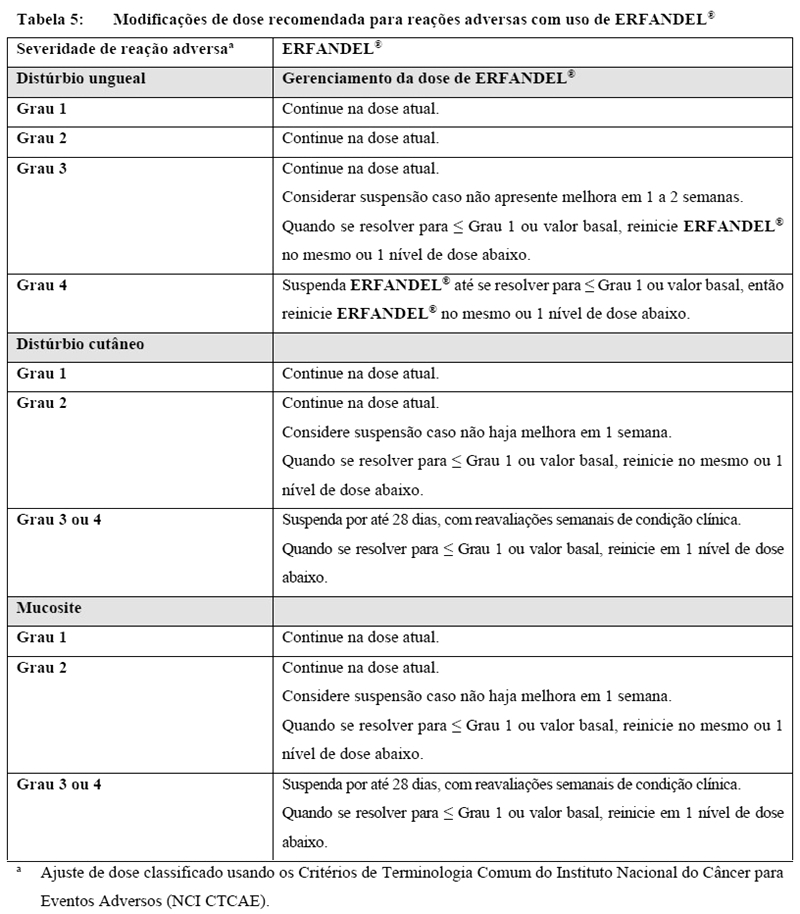
População especial
Uso pediátrico
A segurança e eficácia de ERFANDEL® não foram estabelecidas em pacientes pediátricos. Não há dados disponíveis.
Uso geriátrico
Nenhuma diferença geral na segurança ou eficácia foi observada entre esses pacientes e pacientes mais novos. Não são considerados necessários ajustes específicos de dose em pacientes idosos (vide "Propriedades Farmacocinéticas").
Insuficiência renal
Com base nas análises farmacocinéticas populacionais, não é necessário ajuste de dose para pacientes com comprometimento renal mínimo ou moderado (vide "Propriedades Farmacocinèticas"). Não há dados disponíveis em pacientes com insuficiência renal grave.
Insuficiência hepática
Com base na análise farmacocinética populacional, não é necessário ajuste de dose para pacientes com comprometimento hepático mínimo (vide "Propriedades Farmacocinèticas). Dados limitados ou inexistentes estão disponíveis em pacientes com insuficiência hepática moderada ou grave.
Este medicamento não deve ser partido, aberto ou mastigado.
9. REAÇÕES ADVERSAS
Ao longo desta seção, as reações adversas (RAs) são apresentadas. As reações adversas são eventos adversos (EAs) considerados como estando razoavelmente associados ao uso de erdafitinibe com base na avaliação abrangente da informação disponível sobre eventos adversos. Uma relação causal com o erdafitinibe não pode ser estabelecida de forma confiável em casos individuais. Além disso, como os estudos clínicos são conduzidos sob condições muito variadas, as taxas de reações adversas observadas nos estudos clínicos de um medicamento não podem ser comparadas diretamente às taxas nos estudos clínicos de outra droga e podem não refletir as taxas observadas na prática clínica. Os dados de segurança descritos abaixo refletem a exposição ao ERFANDEL® no estudo BLC2001, estudo de Fase 2 incluindo 99 pacientes com carcinoma urotelial localmente avançado ou metastático e cujos tumores apresentaram determinadas alterações genéticas de FGFR conforme detectadas por um ensaio de estudo clínico em um laboratório central, que apresentaram progressão da doença durante ou após pelo menos uma linha de quimioterapia anterior incluindo dentro de 12 meses de quimioterapia neoadjuvante ou adjuvante. Os pacientes foram tratados com ERFANDEL® na dose de 8 mg uma vez ao dia via oral; com titulação crescente guiada pela farmacodinâmica para 9 mg em pacientes com níveis de fosfato < 5,5 mg/dL. A duração média de tratamento foi 5,3 meses (variação: 0 a 17 meses).
Os eventos adversos mais comuns ≥15% foram hiperfosfatemia (77%), estomatite (58%), boca seca (45%), apetite reduzido (38%), pele seca (32%), alopecia (29%), síndrome de eritrodisestesia palmar-plantar (23%), olho seco (19%), onicólise (18%), paroníquia (17%) e distrofia ungueal (16%). Os eventos adversos Grau 3 mais comuns > 1% foram estomatite (10%), distrofia ungueal (6%), síndrome de eritrodisestesia palmar-plantar (5%), paroníquia (3%), distúrbio ungueal (3%), ceratite (3%), onicólise (2%) e hiperfosfatemia (2%). Reações adversas levando à redução de dose ocorreram em 52% de pacientes, incluindo vinte (20%) por distúrbios oculares. Apenas nove pacientes (9%) apresentaram eventos adversos levando à descontinuação do tratamento, incluindo três (3%) por distúrbios oculares. A Tabela 6 apresenta os eventos adversos relatados em ≥1% dos pacientes tratados com ERFANDEL® na dose de 8 mg uma vez ao dia no estudo BLC2001.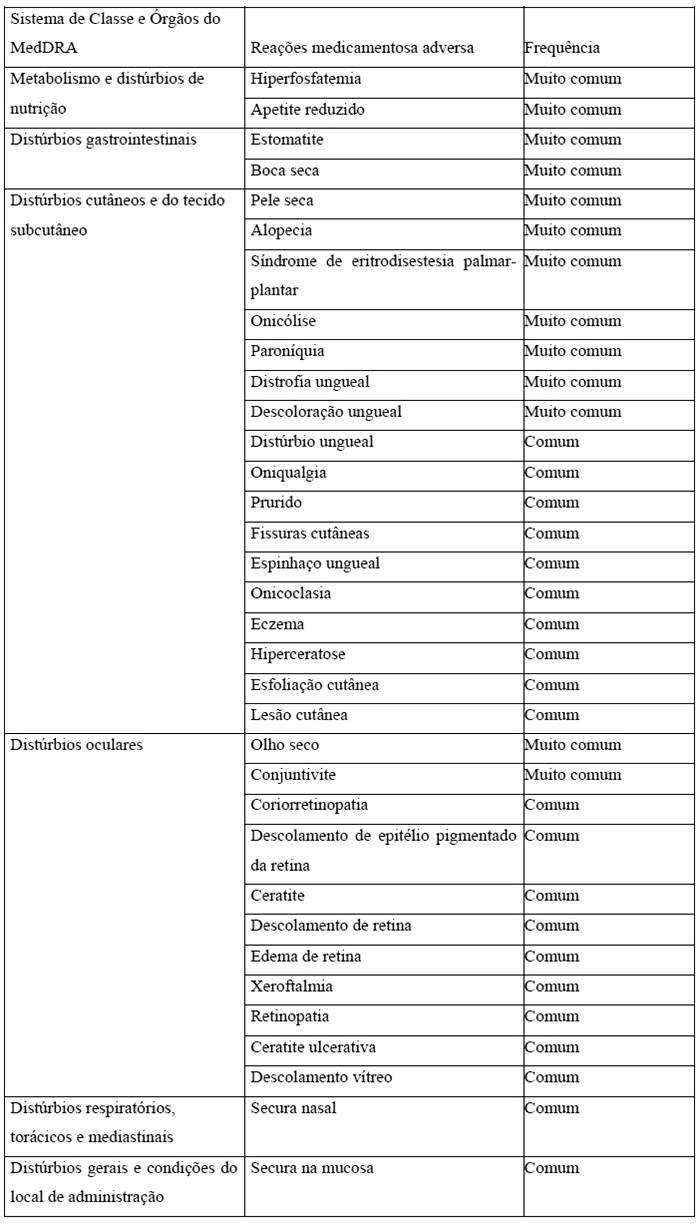
Os seguintes ADRs foram relatados com a administração de ERFANDEL® no estudo BLC2001 e outros estudos:
Retinopatia serosa central (CSR)
CSR foi relatado com o uso de ERFANDEL®, assim como com outros inibidores de FGFR. Reações adversas de CSR foram relatadas em 23% de pacientes; a CSR incluiu coriorretinopatia, descolamento de retina, descolamento de epitélio pigmentado macular da retina, edema de retina, retinopatia e descolamento vítreo (vide "Advertência e Precauções").
Distúrbios ungueais
Distúrbios ungueais foram relatados em 57% dos pacientes e incluíram onicólise, paroníquia, distrofia ungueal, descoloração ungueal, oniqualgia, paroníquia, espinhaço ungueal, onicoclasia, sangramento do leito ungueal e desconforto ungueal. A incidência de distúrbios ungueais aumentou com exposição elevada. O tempo médio para início para qualquer grau de distúrbio ungueal foi 68 dias.
Distúrbios cutâneos
Distúrbios cutâneos foram relatados em 51% dos pacientes e incluíram pele seca e síndrome de eritrodisestesia palmar-plantar, prurido, fissuras cutâneas, eczema, hiperceratose, esfoliação cutânea, lesão cutânea, xeroderma, atrofia cutânea, eczema numular e toxicidade cutânea. O tempo médio para início para qualquer grau de distúrbio cutâneo foi 40 dias.
Hiperfosfatemia
Aumentos nos níveis de fosfato é uma anormalidade laboratorial esperada e temporária (vide "Características Farmacológicas - Propriedades Farmacodinâmicas"). Hiperfosfatemia foi relatada como evento adverso em 77% dos pacientes tratados com ERFANDEL® . Nenhum evento de hiperfosfatemia foi relatado como sério. O tempo médio para início para qualquer grau de evento de hiperfosfatemia foi 20 dias. As elevações de fosfato médias atingiram seu pico aproximadamente 6 semanas após o início de ERFANDEL® e subsequentemente reduziram para abaixo de 4,5 mg/dL por volta aproximadamente do mês 5.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Neste caso, notifique os eventos adversos pelo ao Sistema de Notificação de Eventos Adversos a Medicamentos -VIGIMED, disponível em http://portal.anvisa.gov.br/vigimed, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Sinais e sintomas
Não há informação sobre superdose com ERFANDEL®.
Tratamento
Não há um antídoto específico conhecido para superdose de ERFANDEL®. Em caso de uma superdose, interrompa a administração de ERFANDEL®, tome medidas de suporte gerais até que a toxicidade clínica tenha diminuído ou se resolvido.
Em caso de intoxicação, ligue para 0800 722 6001, se você precisar de mais orientações.
Esse medicamento foi registrado por meio de um procedimento especial, conforme previsão da Resolução RDC n° 205, de 28 de dezembro de 2017, considerando a raridade da doença para qual está indicado e a condição séria debilitante que esta representa. Dados complementares e provas adicionais ainda serão submetidos à Anvisa, após a concessão do registro do medicamento. A revisão desses novos dados pela Anvisa poderá implicar a alteração das informações descritas nesta bula ou mesmo a alteração do status do registro do medicamento.
Dizeres legais.
MS-1.1236.3433
Esta bula foi aprovada pela ANVISA em 30/09/2019.
VENDA SOB PRESCRIÇÃO MÉDICA.