ENTRESTO
NOVARTIS
valsartana
Tratamento da insuficiência cardíaca crônica.
Apresentações.
Entresto® 24 mg/26 mg (sacubitril/valsartana) - embalagens contendo 28 comprimidos revestidos.
Entresto® 49 mg/51 mg (sacubitril/valsartana) - embalagens contendo 28 ou 60 comprimidos revestidos.
Entresto® 97 mg/103 mg (sacubitril/valsartana) - embalagens contendo 28 ou 60 comprimidos revestidos.
VIA ORAL
USO ADULTO
Composição.
Cada comprimido revestido de Entresto® 24 mg/26 mg (sacubitril/valsartana) contém 24,3 mg de sacubitril e 25,7 mg de valsartana como 56,551 mg de sacubitril valsartana sódica hidratada (equivalente a 50 mg de ácido anidro livre).
Cada comprimido revestido de Entresto® 49 mg/51 mg (sacubitril/valsartana) contém 48,6 mg de sacubitril e 51,4 mg de valsartana como 113,103 mg de sacubitril valsartana sódica hidratada (equivalente a 100 mg de ácido anidro livre).
Cada comprimido revestido de Entresto® 97 mg/103 mg (sacubitril/valsartana) contém 97,2 mg de sacubitril e 102,8 mg de valsartana como 226,206 mg de sacubitril valsartana sódica hidratada (equivalente a 200 mg de ácido anidro livre).
Entresto® contém sacubitril valsartana sódica hidratada que é um complexo de sal das formas aniônicas de sacubitril e valsartana, cátions de sódio e moléculas de água na razão molar de 1:1:3:2:5, respectivamente. Após a administração oral, Entresto® se dissocia em sacubitril (que é metabolizado adicionalmente ao LBQ657 [sacubitrilato]) e valsartana.
Excipientes: celulose microcristalina, hiprolose, crospovidona, estearato de magnésio, talco e dióxido de silício. Excipientes do revestimento: hipromelose, macrogol, talco, dióxido de titânio, óxido de ferro vermelho, óxido de ferro preto (para 24 mg/26 mg e 97 mg/103 mg) e óxido de ferro amarelo (para 49 mg/51 mg).
Informações técnicas.
1. INDICAÇÕES
Entresto® é indicado para o tratamento de pacientes adultos com insuficiência cardíaca crônica sintomática (NYHA classe II-IV) com fração de ejeção reduzida.
2. RESULTADOS DE EFICÁCIA
As concentrações 24 mg/26 mg, 49 mg/51 mg e 97 mg/103 mg são referenciadas em algumas publicações como 50 mg, 100 mg e 200 mg, respectivamente.
PARADIGM-HF
PARADIGM-HF foi um estudo multinacional, randomizado, duplo-cego, com 8.442 pacientes1. Comparando Entresto® com enalapril, ambos administrados a pacientes adultos com insuficiência cardíaca crônica, classe II - IV da NYHA e fração de ejeção reduzida (fração de ejeção ventricular esquerda [FEVE] ≤ 40%, posteriormente alterada para ≤ 35%), além de outra terapia para insuficiência cardíaca2. O desfecho primário foi o composto de morte cardiovascular (CV) ou hospitalização por insuficiência cardíaca (HF)3.
Antes da participação no estudo, os pacientes foram bem tratados com a terapia padrão, que incluiu inibidores no estudo, os pacientes foram bem tratados com a terapia padrão, que incluiu inibidores de ECA/BRAs ( > 99%), betabloqueadores (94%), antagonistas mineralocorticoides (58%) e diuréticos (83%)4. A duração mediana do acompanhamento foi de 27 meses e os pacientes foram tratados por até 4,3 anos5.
Os pacientes precisavam descontinuar sua terapia atual com inibidor da ECA ou BRA e entrar em um período de introdução (run-in) simples-cego sequencial durante o qual receberam tratamento com enalapril 10 mg duas vezes ao dia, seguido pelo tratamento com Entresto® 100 mg duas vezes ao dia, aumentando para 200 mg duas vezes ao dia2. Os pacientes foram então randomizados para o período duplo-cego do estudo para receber Entresto® 200 mg ou enalapril 10 mg duas vezes ao dia [Entresto® (n=4.209); enalapril (n=4.233) ] 1.
A idade média da população estudada foi de 64 anos e 19% tinham 75 anos ou mais. Na randomização, 70% dos pacientes eram classe II e 25% eram Classe III/IV da NYHA6. A FEVE média foi de 29% e houve 963 (11,4%) pacientes com FEVE basal de > 35% e ≤ 40%. No grupo Entresto®, 76% dos pacientes permaneceram na dose alvo de 200 mg duas vezes ao dia no final do estudo (dose média diária de 375 mg). No grupo enalapril, 75% dos pacientes permaneceram na dose alvo de 10 mg duas vezes ao dia no final do estudo (dose média diária de 18,9 mg)7.
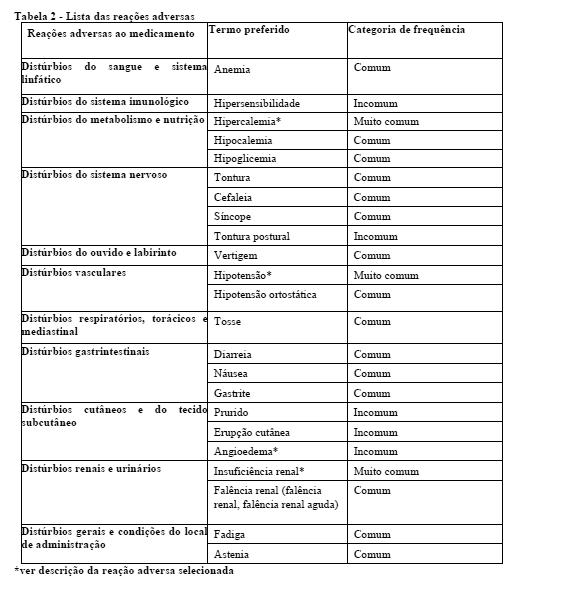
Entresto® demonstrou superioridade clinicamente relevante e estatisticamente significativa em relação ao enalapril, reduzindo o risco de morte cardiovascular ou hospitalizações por insuficiência cardíaca em 20% (razão de risco (RR): 0,80, IC 95% [0,73; 0,87], p unicaudal =0,0000002) versus enalapril 8. Este efeito foi observado inicialmente e foi sustentado ao longo da duração do estudo 9. A redução absoluta do risco foi de 4,69%10. Uma redução estatisticamente significativa para morte CV e primeira hospitalização HF foi observada (morte CV, RRR 20%, RR 0,80; IC 95% [0,71, 0,89], p unicaudal= 0,00004; e hospitalização por insuficiência cardíaca RRR 21%; RR 0,79; IC 95% [0,71, 0,89], p unicaudal= 0,00004)8; vide Tabela 1 e figura 1. Morte súbita foi responsável por 45% das mortes cardiovasculares e foi reduzida em 20% nos pacientes tratados com Entresto® em comparação com pacientes tratados com enalapril (RR 0,80, p= 0,0082)11. Falência cardiaca foi responsável por 26% das mortes cardiovasculares e foi reduzida em 21% nos pacientes tratados com Entresto® em comparação com pacientes tratados com enalapril (RR 0,79, p= 0,0338)12.
Esta redução de risco foi observada consistentemente entre subgrupos incluindo: idade, gênero, raça, geografia, classe NYHA, fração de ejeção, função renal, história de diabetes ou hipertensão, terapia anterior para insuficiência cardíaca e fibrilação atrial13.
Entresto® também reduziu significativamente a mortalidade por todas as causas em 16% em comparação com enalapril (RRR 16%, RR 0,84; IC 95% [0,76 a 0,93], p unicaudal=0,0005) (Tabela 1). A redução absoluta de risco foi de 2,84%14.
O Kaplan-Meier apresentado na figura abaixo (esquerda) mostra o tempo até a primeira ocorrência do desfecho composto primário de morte CV ou hospitalização por insuficiência cardíaca. O efeito do tratamento com Entresto® foi evidente inicialmente e sustentado pela duração do estudo. A figura Kaplan-Meier apresentada abaixo (direita) mostra o tempo até o desfecho morte CV.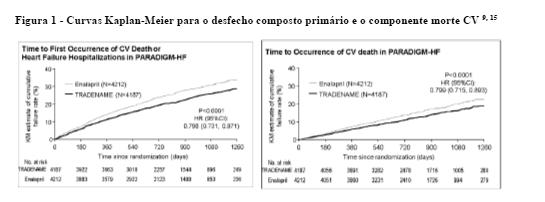
Em geral, houve menos hospitalizações por todas as causas em pacientes tratados com Entresto® em comparação com enalapril, incluindo uma redução no risco relativo de 12% para a primeira hospitalização (RR 0,88 [IC 95%: 0,82, 0,94], P < 0,001), e uma redução da taxa relativa de 16% para o número total de hospitalizações (RR 0,84 [IC 95%: 0,78, 0,91], P < 0,001)16, 17.
Entresto® demonstrou uma pontuação de dados clínicos significativamente melhor para os domínios relacionados a sintomas de HF e limitações físicas avaliadas pelo Kansas City Cardiomyopathy Questionnaire (KCCQ) [Questionário de Cardiomiopatia de Kansas City], um questionário autoadministrado18. Mais pacientes melhoraram a classe funcional do NYHA do baseline até o Mês 8 com Entresto® (16%) em comparação com enalapril (14%) e menos pacientes apresentaram piora na classe funcional do NYHA (10% vs 13%, respectivamente)19.
TITRATION
TITRATION foi um estudo de 12 semanas de segurança e tolerabilidade em 538 pacientes com insuficiência cardíaca crônica (NYHA classe II - IV) e disfunção sistólica (fração de ejeção ventricular esquerda ≤ 35%) virgens de terapia com inibidor da ECA ou BRA ou estavam recebendo doses variáveis de inibidores da ECA ou BRAs antes da entrada no estudo20,21. Os pacientes começaram com Entresto® 50 mg duas vezes ao dia, fizeram titulação crescente para 100 mg duas vezes ao dia e então para a dose alvo de 200 mg duas vezes ao dia com um regime de 3 semanas ou 6 semanas21.
Em geral, 76% dos pacientes atingiram e mantiveram a dose alvo de Entresto® 200 mg duas vezes ao dia sem nenhuma interrupção da dose ou titulação decrescente ao longo de 12 semanas22. Mais pacientes que eram virgens de terapia anterior com inibidor da ECA ou BRA ou estavam recebendo terapia de baixa dose (equivalente a < 10 mg de enalapril/dia) conseguiram atingir e manter Entresto® 200 mg quando a titulação ascendente foi feita em 6 semanas versus 3 semanas23.
PARAMOUNT
PARAMOUNT, um estudo randomizado, duplo-cego, em pacientes com fração de ejeção ventricular esquerda ≥ 45%, comparando 200 mg de Entresto® (n=149) com 160 mg de valsartana (n=152) duas vezes ao dia, demonstrou redução estatisticamente maior (p= 0,0050) em NT pro-BNP desde o período basal até a Semana 1224,25,26. A redução desde o período basal em NT-proBNP foi semelhante nas Semanas 12 e 36 em pacientes tratados com Entresto®, enquanto o NT-proBNP diminuiu desde a Semana 12 a 36 em pacientes tratados com valsartana27. Reduções significativas no tamanho atrial esquerdo, tanto do volume atrial esquerdo indexado (p=0,0069) como na dimensão atrial esquerda (p=0,0337) foram observadas na Semana 3628. Uma melhora estatisticamente significativa na classe NYHA foi observada na Semana 36 (p=0,0488)29.
Dados de segurança pré-clínicos
Estudos de segurança pré-clínicos conduzidos com Entresto® incluíram a avaliação de farmacologia de segurança, toxicidade de doses repetidas, genotoxicidade, carcinogenicidade e toxicidade reprodutiva e do desenvolvimento; Entresto® não apresentou efeitos adversos nos sistemas orgânicos vitais. A maioria dos achados observados nos estudos de toxicidade repetida foi reversível e atribuível à farmacologia do bloqueio do receptor AT1.
Carcinogenicidade, mutagênese e toxicidade genética
Estudos de carcinogenicidade conduzidos em camundongos e ratos com sacubitril e valsartana não identificaram nenhum potencial carcinogênico para Entresto®. As doses de sacubitril estudadas (alta dose de 1.200 e 400 mg/kg/dia em camundongos e ratos, respectivamente) foram cerca de 29 e 19 vezes, respectivamente, a dose humana máxima recomendada (DHMR) em uma base mg/m2. As doses de valsartana estudadas (alta dose de 160 e 200 mg/kg/dia em camundongos e ratos, respectivamente) foram cerca de 4 e 10 vezes, respectivamente, a dose humana máxima recomendada em uma base mg/m2.
Estudos de mutagenicidade e clastogenicidade conduzidos com Entresto®, sacubitril e valsartana não revelaram nenhum efeito em nível genético ou cromossômico.
Fertilidade, reprodução e desenvolvimento
Entresto® não apresentou nenhum efeito na fertilidade ou desenvolvimento embrionário inicial em ratos em uma dose de até 150 mg/kg/dia (≤1,0 vez e ≤0,18 vez a DHMR com base na AUC de valsartana e sacubitrilato, respectivamente). O tratamento com Entresto® durante a organogênese resultou em letalidade embriofetal elevada em ratos a doses ≥100 mg/kg/dia [≤0,72 vez a DHMR com base na AUC] e coelhos a doses ≥10 mg/kg/dia [2 vezes e 0,03 vez a DHMR com base na AUC de valsartana e sacubitrilato, respectivamente]. Entresto® é teratogênico com base em uma baixa incidência de hidrocefalia fetal, associada a doses maternalmente tóxicas, que foi observada em coelhos a uma dose de Entresto® ≥10 mg/kg/dia. Os efeitos embriofetais adversos de Entresto® são atribuídos à atividade do antagonista do receptor de angiotensina (vide "Advertências e precauções - Mulheres com potencial para engravidar (e medidas contraceptivas, se aplicáveis), Gravidez, Amamentação e Fertilidade").
Estudos de desenvolvimento pré e pós-natal em ratos, conduzidos com sacubitril a doses de até 750 mg/kg/dia [2,2 vezes a DHMR com base na AUC] e valsartana a doses de até 600 mg/kg/dia [0,86 vez a DHMR com base na AUC] indicam que o tratamento com Entresto® durante a organogênese, gestação e lactação podem afetar o desenvolvimento e sobrevida dos filhotes.
Outros achados pré-clínicos
Os efeitos de Entresto® nas concentrações de beta amiloide no líquido cefalorraquidiano (LCR) e tecido cerebral foram avaliados em macacos cynomolgus jovens (2 a 4 anos) tratados com Entresto® (50 mg/kg/dia) por 2 semanas. Neste estudo, Entresto® apresentou um efeito farmacodinâmico no clearance (depuração) de A no LCR em macacos cynomolgus, elevando os níveis de A-beta 1-40, 1-42 e 1-38 no LCR; não houve uma elevação correspondente nos níveis de A-beta no cérebro. Elevações no A-beta 1-40 e 1-42 no LCR não foram observados em um estudo com voluntários saudáveis de 2 semanas em humanos (vide "Características farmacológicas").
Referências bibliográficas
1. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.1- 1.1 [64].
2. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Section 9.1 [19].
3. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Section 8.1 [27].
4. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.1- 3.2.a [92].
5. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.3- 1.1 [36].
6. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.1- 3.1 [93].
7. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.3- 1.9 [66].
8. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.2- 1.1.post.14 [94].
9. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Figure14.2- 1.2 [98].
10. [Clinical Overview] 2.5- EU 2.5 Table 4-1 [62].
11. [Summary of Clinical Efficacy] 2.7.3- EU 2.7.3 Appendix 1 Table 14.2- 1.5 [78].
12. [Summary of Clinical Efficacy] 2.7.3- EU 2.7.3 Appendix 1 Table 14.2- 1.4 [77].
13. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Figure 11-6 [96].
14. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.2- 2.1 [97].
15. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Figure 14.2- 1.2.1 [99].
16. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.2- 3.2 [100].
17. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.2-3. 5 [101].
18. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.2- 2.4 [102].
19. [Study LCZ696B2314 report (2014)]: A multicenter, randomized, double-blind, parallel group, active-controlled study to evaluate the efficacy and safety of LCZ696 compared to enalapril on morbidity and mortality in patients with chronic heart failure (HF) and reduced ejection fraction. Table 14.2- 3.19.post.01 [103].
20. [Study LCZ696B2228 report (2014)] A multicenter, randomized, double-blind, parallel group study to assess the safety and tolerability of initiating LCZ696 in heart failure patients comparing two titration regimens. Section 10.1 [104].
21. [Study LCZ696B2228 report (2014)] A multicenter, randomized, double-blind, parallel group study to assess the safety and tolerability of initiating LCZ696 in heart failure patients comparing two titration regimens. Section 9 [105].
22. [Study LCZ696B2228 report (2014)] A multicenter, randomized, double-blind, parallel group study to assess the safety and tolerability of initiating LCZ696 in heart failure patients comparing two titration regimens. Table 14.2- 2.8 [106].
23. [Study LCZ696B2228 report (2014)] A multicenter, randomized, double-blind, parallel group study to assess the safety and tolerability of initiating LCZ696 in heart failure patients comparing two titration regimens. Table 14.2- 2.1 [107].
24. [Study LCZ696B2214 report (2013)] A 36-Week, randomized, double-blind, multi-center, parallel group, active controlled study to evaluate the efficacy, safety and tolerability of LCZ696 compared to valsartan in patients with CHF and preserved leftventricular ejection fraction Table10-1 [30].
25. [Study LCZ696B2214 report (2013)]: A 36-Week, randomized, double-blind, multi-center, parallel group, active controlled study to evaluate the efficacy, safety and tolerability of LCZ696 compared to valsartan in patients with CHF and preserved leftventricular ejection fraction. Table 14.2- 1.1 [73].
26. [Study LCZ696B2214 report (2013)]: A 36-Week, randomized, double-blind, multi-center, parallel group, active controlled study to evaluate the efficacy, safety and tolerability of LCZ696 compared to valsartan in patients with CHF and preserved leftventricular ejection fraction. Section 9.1 [76].
27. [Study LCZ696B2214 report (2013)] A 36-Week, randomized, double-blind, multi-center, parallel group, active controlled study to evaluate the efficacy, safety and tolerability of LCZ696 compared to valsartan in patients with CHF and preserved leftventricular ejection fraction Figure 11.1 [109].
28. [Study LCZ696B2214 report (2013)] A 36-Week, randomized, double-blind, multi-center, parallel group, active controlled study to evaluate the efficacy, safety and tolerability of LCZ696 compared to valsartan in patients with CHF and preserved leftventricular ejection fraction Table 14.2- 4.1c ext. [110].
29. [Study LCZ696B2214 report (2013)] A 36-Week, randomized, double-blind, multi-center, parallel group, active controlled study to evaluate the efficacy, safety and tolerability of LCZ696 compared to valsartan in patients with CHF and preserved leftventricular ejection fraction Table 14.2- 5.1a ext. [111]
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Entresto® exibe um mecanismo de ação inovador de um inibidor de neprilisina e do receptor da angiotensina (ARNI) inibindo simultaneamente a neprilisina (endopeptidase neutra; NEP) através do sacubitrilato, o metabólito ativo do pró- fármaco sacubitril, e bloqueando o receptor da angiotensina II tipo-1 (AT1) através da valsartana. Os benefícios cardiovasculares complementares e efeitos renais de Entresto® em pacientes com insuficiência cardíaca são atribuídos ao aumento dos peptídeos que são degradados pela neprilisina, como peptídeos natriuréticos (PN), pelo sacubitrilato e a inibição simultânea dos efeitos deletérios da angiotensina II pela valsartana. Os PNs exercem seus efeitos ativando receptores de guanilil ciclase acoplados à membrana, resultando em concentrações elevadas do segundo mensageiro guanosina monofosfato cíclica (cGMP), promovendo assim vasodilatação, natriurese e diurese, aumento da taxa de filtração glomerular e fluxo sanguíneo renal, inibição da liberação de renina e aldosterona, redução da atividade simpática e efeitos anti-hipertróficos e antifibróticos. A ativação contínua do sistema renina-angiotensina-aldosterona resulta em vasoconstrição, retenção de sódio renal e de fluidos, ativação de crescimento e proliferação celular e subsequente remodelagem cardiovascular de má adaptação. A valsartana inibe os efeitos cardiovasculares e renais prejudiciais da angiotensina II bloqueando seletivamente o receptor AT1 e também inibe a liberação de aldosterona dependente da angiotensina II.
Farmacodinâmica
Os efeitos farmacodinâmicos de Entresto® foram avaliados após administrações de doses únicas e múltiplas a indivíduos saudáveis e a pacientes com insuficiência cardíaca, e são consistentes com inibição da neprilisina e bloqueio do SRAA simultâneos. Em um estudo controlado de valsartana de 7 dias em pacientes com fração de ejeção (HFrEF) reduzida, a administração de Entresto® resultou em uma elevação não sustentada significativa na natriurese, aumento do cGMP na urina e MR-proANP e NT-proBNP plasmáticos diminuídos em comparação com valsartana. Em um estudo de 21 dias em pacientes HFrEF, Entresto® aumentou significativamente a ANP e cGMP na urina e cGMP no plasma, e diminuiu NT- proBNP, aldosterona e endotelina-1 no plasma em comparação com o período basal. Entresto® também bloqueou o receptor AT1 como evidenciado pela atividade aumentada da renina plasmática e concentrações de renina plasmática. No PARADIGM-HF, Entresto® diminuiu NT-proBNP no plasma e aumentou BNP no plasma e cGMP na urina em comparação com enalapril. Ao passo que BNP é um substrato da neprilisina, NT-proBNP não é, portanto, NT- proBNP (mas não BNP) é um biomarcador adequado para o monitoramento de pacientes com insuficiência cardíaca tratados com Entresto®.
Em um estudo clínico de QTc detalhado em indivíduos saudáveis do sexo masculino, doses únicas de 194 mg/206 mg e 583 mg/617 mg de Entresto® não apresentaram efeito na repolarização cardíaca.
A neprilisina é uma das múltiplas enzimas envolvidas no clearance (depuração) de beta-amiloide (A-beta) do cérebro e líquido cefalorraquidiano (LCR). A administração de Entresto® 400 mg uma vez ao dia por 2 semanas a voluntários saudáveis foi associada a uma elevação no LCR A-beta 1-38 em comparação com placebo; não houve alterações nas concentrações de LCR A-beta 1-40 e 1-42. A relevância clinica deste achado é desconhecida (vide "Dados de segurança pré-clínicos").
Farmacocinética
- Absorção
Após a administração oral, Entresto® se dissocia em sacubitril e valsartana. Sacubitril é posteriormente metabolizado em LBQ657 (sacubitrilato). Sacubitril, sacubitrilato e valsartana atingem as concentrações plasmáticas máximas em 0,5 hora, 2 horas e 1,5 hora, respectivamente. Estima-se que a biodisponibilidade oral absoluta de sacubitril e valsartana seja ≥ 60% e 23%, respectivamente.
A valsartana de Entresto® é mais biodisponível que a valsartana em outras formulações de comprimidos comercializadas; 26 mg, 51 mg, e 103 mg de valsartana no Entresto® é equivalente a 40 mg, 80 mg, e 160 mg de valsartana nas formulações de comprimidos comercializadas, respectivamente. Considere a biodisponibilidade aumentada da valsartana em Entresto® quando da substituição de outras formulações comercializadas de valsartana.
Após administrações duas vezes ao dia de Entresto®, os níveis no estado de equilíbrio de sacubitril, sacubitrilato e valsartana são atingidos em 3 dias. No estado de equilíbrio, sacubitril e valsartana não se acumulam significativamente, enquanto sacubitrilato se acumula em 1,6 vezes. A administração de Entresto® com alimentos não tem impacto clinicamente significativo nas exposições sistêmicas de sacubitril, sacubitrilato e valsartana. Embora haja uma diminuição na exposição de valsartana quando Entresto® é administrado com alimentos, essa diminuição não é acompanhada por uma redução clinicamente significativa no efeito terapêutico. Entresto® pode, portanto, ser administrado com ou sem alimentos.
- Distribuição
Entresto® é altamente ligado às proteínas plasmáticas (94% - 97%). Com base na comparação do plasma e das exposições de LCR, sacubitrilato cruza a barreira hematoencefálica em um grau limitado (0,28%). Entresto® tem um volume de distribuição aparente variando de 75 L a 103 L.
- Biotransformação/metabolismo
O sacubitril é prontamente convertido a sacubitrilato por esterases; sacubitrilato não é metabolizado adicionalmente em um grau significativo. A valsartana é minimamente metabolizada, já que apenas 20% da dose é recuperada como metabólitos. Um metabólito hidroxil foi identificado noplasma em baixas concentrações ( < 10%). Já que o metabolismo mediado pela enzima CYP450 de sacubitril e valsartana é mínimo, não se espera que a coadministração com medicamentos que impactam as enzimas CYP450 impacte a farmacocinética.
- Eliminação
Após a administração oral, 52 a 68% de sacubitril (principalmente como sacubitrilato) e ~13% da valsartana e seus metabólitos são excretados na urina; 37-48% do sacubitril (principalmente como sacubitrilato) e 86% da valsartana e seus metabólitos são excretados nas fezes.
O sacubitril, sacubitrilato e a valsartana são eliminados do plasma com uma meia-vida (T1/2) de eliminação média de aproximadamente 1,43 horas, 11,48 horas e 9,90 horas, respectivamente.
- Linearidade/não linearidade
A farmacocinética de sacubitril, sacubitrilato e valsartana são lineares no intervalo de dose testado (24 mg/26 mg - 194 mg/206 mg de Entresto®).
Populações especiais
- Pacientes idosos (com idade acima de 65 anos)
As exposições ao sacubitrilato e valsartana são elevadas em indivíduos idosos em 42% e 30%, respectivamente, em comparação com indivíduos mais jovens. Entretanto, isto não está associado a efeitos clinicamente relevantes e, portanto, não é necessário nenhum ajuste de dose.
- Pacientes pediátricos (com idade abaixo de 18 anos)
Entresto® não foi estudado em pacientes pediátricos.
- Função renal comprometida- Insuficiência renal
Foi observada uma correlação entre a função renal e exposição sistêmica ao sacubitrilato, mas não à valsartana. Em pacientes com insuficiência renal leve (TFGe 60-90 mL/min/1,73 m2) a moderada (TFGe 30-60 mL/min/1,73 m2), a AUC para sacubitrilato foi até 2 vezes mais alta. Uma AUC 2,7 vezes mais alta para sacubitrilato foi observada em pacientes com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2). Não é necessário ajuste de dose em pacientes com insuficiência renal leve (Taxa de Filtração Glomerular estimada [TFGe] 60-90mL/min/1,73m2). Uma dose inicial de 24 mg/26 mg duas vezes ao dia deve ser considerada em pacientes com insuficiência renal moderada (TFGe 30-60 mL/min/1,73m2).
Como existem dados clínicos limitados em pacientes com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2); Entresto® deve ser administrado com cuidado, e a dose inicial recomendada é de 24 mg/26 mg, duas vezes ao dia. Não há estudos em pacientes com doença renal em estágio terminal, portanto o uso não é recomendado.
Não foi realizado nenhum estudo em pacientes sendo submetidos a diálise. Entretanto, sacubitrilato e valsartana são altamente ligados a proteínas plasmáticas e, portanto, é improvável que sejam removidos com eficácia por diálise.
- Função hepática comprometida
Em pacientes com insuficiência hepática leve a moderada, as exposições do sacubitril aumentaram em 1,5 e 3,4 vezes, o sacubitrilato aumentou em 1,5 e 1,9 vezes e a valsartana aumentou em 1,2 vezes e 2,1 vezes, respectivamente, em comparação com indivíduos saudáveis correspondentes. Nenhum ajuste de dose é necessário ao administrar Entresto® a pacientes com insuficiência hepática leve (Child-Pugh classificação A). Os estudos clínicos são limitados em pacientes com insuficiência hepática moderada (Child-Pugh classificação B) ou com valores de TGO/TGP superiores a duas vezes o limite superior do intervalo normal. Entresto® deve ser utilizado com cautela nestes pacientes e a dose inicial recomendada é de 24 mg/26 mg, duas vezes ao dia.
Entresto® é contraindicado em pacientes com insuficiência hepática grave, cirrose biliar e colestase (Child-Pugh classificação C).
- Grupos étnicos
A farmacocinética de Entresto® (sacubitril, sacubitrilato e valsartana) é comparável entre diferentes grupos raciais e étnicos (Caucasianos, Negros, Asiáticos, Japoneses e outros).
- Efeito do gênero
A farmacocinética de Entresto® (sacubitril, sacubitrilato e valsartana) é semelhante entre indivíduos do sexo masculino e feminino.
4. CONTRAINDICAÇÕES
• Hipersensibilidade ao princípio ativo, a sacubitril, a valsartana ou a qualquer um dos excipientes;
• Uso concomitante com inibidores da ECA (vide "Advertências e precauções", "Posologia e modo de usar" e "Interações medicamentosas"). Entresto® não deve ser administrado em até 36 horas após a descontinuação da terapia com inibidor da ECA;
• História conhecida de angioedema relacionado a terapia anterior com inibidor da ECA ou BRA;
• Angioedema hereditário ou idiopático (vide "Advertências e precauções");
• Uso concomitante com alisquireno em pacientes com diabetes Tipo 2 (vide "Advertências e precauções", e "Interações medicamentosas");
• Insuficiência hepática grave, cirrose biliar e colestase (vide "Advertências e precauções");
• Gravidez (vide "Advertências e precauções - gravidez - amamentação - fertilidade").
Este medicamento é contraindicado para uso por mulheres grávidas.
Categoria de risco na gravidez: D
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
5. ADVERTÊNCIAS E PRECAUÇÕES
Bloqueio duplo do Sistema Renina-Angiotensina-Aldosterona (SRAA)
• Entresto® não deve ser administrado com um inibidor da ECA devido ao risco de angioedema. Entresto® não deve ser iniciado em até 36 horas após tomar a última dose da terapia com inibidor da ECA. Se o tratamento com Entresto® for interrompido, a terapia com inibidor da ECA não deve ser iniciada em até 36 horas após a última dose de Entresto® (vide "Contraindicações", "Posologia e modo de usar" e "Interações medicamentosas");
• É necessário cautela ao coadministrar Entresto® com inibidores diretos da renina, como alisquireno (vide "Contraindicações" e "Interações medicamentosas"). Entresto® não deve ser administrado com alisquireno em pacientes com diabetes Tipo 2 (vide "Contraindicações");
• Entresto® não deve ser coadministrado com um BRA devido à atividade de bloqueio do receptor de angiotensina II de Entresto® (vide "Posologia e modo de usar" e "Interações medicamentosas").
Hipotensão
Casos de hipotensão sintomática foram relatados em pacientes tratados com Entresto® durante estudos clínicos, especialmente em pacientes ≥ 65 anos, pacientes com doença renal e pacientes com PAS baixa ( < 112 mmHg). Quando iniciada a terapia ou durante a dosagem com Entresto, deve-se monitorar rotineiramente a pressão arterial. Se ocorrer hipotensão, deve-se considerar o ajuste da dose de diuréticos, o uso de medicamentos anti-hipertensivos concomitantes e o tratamento de outras causas de hipotensão (p.ex.: hipovolemia). Se a hipotensão persistir apesar de tais medidas, a dose de Entresto® deve ser reduzida ou o medicamento deve ser temporariamente descontinuado (vide "Posologia e modo de usar"). Normalmente não é necessária a descontinuação permanente da terapia. A hipotensão sintomática tem uma probabilidade maior de ocorrer se o paciente sofreu depleção de volume, p.ex.: por terapia com diurético, restrição dietética de sal, diarreia ou vômitos. A depleção de sódio e/ou volume devem ser corrigidos antes do início do tratamento com Entresto®.
Função renal comprometida
A avaliação de pacientes com insuficiência cardíaca deve sempre incluir a avaliação da função renal. Pacientes com insuficiência renal leve e moderada têm mais riscos de desenvolver hipotensão (vide "Posologia e Modo de usar"). Os estudos clínicos em pacientes com insuficiência renal grave (TFG estimado < 30mL/min/1,73m2) são limitados e estes pacientes podem apresentar maior risco de hipotensão (vide "Posologia e Modo de usar"). Não há estudos em pacientes com doença renal em estágio terminal, portanto o uso de Entresto® não é recomendado.
Piora da função renal
O uso de Entresto® pode estar associado com função renal diminuída. O risco pode ser ainda maior quando há desidratação ou uso concomitante de anti-inflamatório não esteroidal (AINES) (vide "Interações medicamentosas"). A redução de dose deve ser considerada em pacientes que desenvolvem diminuição clinicamente significativa da função renal.
Hipercalemia
Como para qualquer medicamento que age no sistema renina-angiotensina-aldosterona, o uso de Entresto® pode estar associado a um risco elevado de hipercalemia. No estudo PARADIGM- HF, a incidência de hipercalemia clinicamente relevante foi baixa, resultando em descontinuação do tratamento em 0,26% de pacientes tratados com Entresto® em comparação com 0,35% de pacientes tratados com enalapril. Medicações que são conhecidas por aumentar os níveis de potássio (p.ex.: diuréticos poupadores de potássio, suplementos de potássio) devem ser usadas com cuidado na coadministração com Entresto®. Se ocorrer hipercalemia clinicamente significativa, medidas como a redução do potássio da dieta ou ajuste da dose de medicações concomitantes devem ser consideradas. O monitoramento de potássio sérico é recomendado especialmente em pacientes com fatores de risco como insuficiência renal grave, diabetes mellitus, hipoaldosteronismo ou que estejam recebendo uma dieta rica em potássio e em pacientes idosos (vide "Posologia e modo de usar").
Angioedema
Angioedema foi relatado em pacientes tratados com Entresto®. Se ocorrer angioedema, Entresto® deve ser descontinuado imediatamente e devem ser fornecidos terapia e monitoramento adequados até que ocorra a resolução completa e sustentada dos sinais e sintomas. Entresto® não deve ser administrado novamente. Em casos de angioedema confirmado onde o inchaço estava confinado à face e lábios, a condição geralmente se resolveu sem tratamento, embora anti-histamínicos tenham sido úteis no alívio de sintomas.
Angioedema associado a edema de laringe pode ser fatal. Onde há envolvimento da língua, glote ou laringe, com probabilidade de causar obstrução das vias aéreas, devem ser prontamente administradas terapia adequada, p.ex.: solução subcutânea de epinefrina/adrenalina 1:1000 (0,3 mL a 0,5 mL) e/ou medidas necessárias para garantir vias aéreas patentes.
Pacientes com uma história anterior de angioedema não foram estudados. Como eles podem estar em risco mais alto de angioedema, recomenda-se cautela se Entresto® for usado nestes pacientes. Entresto® não deve ser usado em pacientes com uma história conhecida de angioedema relacionado à terapia anterior com inibidor da ECA ou BRA, ou em pacientes com angioedema hereditário (vide "Contraindicações").
Pacientes negros podem ter maior suscetibilidade para desenvolver angioedema.
Pacientes com estenose da artéria renal
Semelhante a outros medicamentos que afetam o sistema renina-angiotensina-aldosterona, Entresto® pode elevar os níveis de ureia sanguínea e creatinina sérica em pacientes com estenose da artéria renal bilateral ou unilateral. É necessário cuidado no uso em pacientes com estenose da artéria renal, e é recomendado o monitoramento da função renal.
Mulheres com potencial para engravidar (e medidas contraceptivas, se aplicáveis)
Mulheres com potencial para engravidar devem - ser avisadas sobre as consequências da exposição ao Entresto® durante a gravidez. Recomenda-se a utilização de contracepção durante o tratamento com Entresto® e por 1 semana após sua última dose.
Gravidez
Assim como para outros medicamentos que também agem diretamente no SRAA, Entresto® não deve ser usado durante a gravidez (vide "Contraindicações"). Entresto® exerce seus efeitos através do antagonismo à angiotensina II. Como resultado, um risco ao feto não pode ser excluído. Houve relatos de dano ao feto em desenvolvimento (p.ex.: aborto espontâneo, oligohidrâmnio e disfunção renal do recém-nascido), quando mulheres grávidas tomaram valsartana. Pacientes devem ser orientados a procurar seu médico e descontinuar o uso de Entresto® assim que a gravidez for detectada.
Amamentação
Não se sabe se Entresto® é excretado no leite humano. Os componentes de Entresto®, sacubitril e valsartana, foram excretados no leite de ratas lactantes (vide