ENLUTOZA
DR. REDDY'S
enzalutamida
Antineoplásico.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
ENLUTOZA® é fornecido em cápsulas moles contendo 40 mg de enzalutamida e está disponível nas seguintes apresentações:
Embalagem com 120 cápsulas.
VIA ORAL
USO ADULTO
Composição.
Cada cápsula mole contém 40 mg de enzalutamida.
Excipientes: caprilocaprato de polioxilglicerídeos, butil-hidroxianisol, butil-hidroxitolueno, gelatina, Glycerinum, Sorbitol, dióxido de titânio, água purificada, álcool etílico e triglicerídeo.
Informações técnicas.
1. INDICAÇÕES
ENLUTOZA® está indicado para o tratamento de homens adultos com câncer de próstata metastático resistente à castração que são assintomáticos ou ligeiramente sintomáticos após falha de terapia de privação androgênica.
ENLUTOZA® está indicado para o tratamento de homens adultos com câncer de próstata metastático resistente à castração que tenham recebido terapia com docetaxel.
ENLUTOZA® está indicado para o tratamento de homens adultos com câncer de próstata não metastático resistente à castração.
ENLUTOZA® é indicado para o tratamento de homens adultos com câncer de próstata metastático sensível à castração (CPSCm), sem uso de docetaxel concomitante.
ENLUTOZA®, em monoterapia ou em combinação com leuprolida, é indicado para o tratamento de homens adultos com câncer de próstata não metastático sensível à castração (CPSCnm) com recidiva bioquímica (RB) de alto risco.
2. RESULTADOS DE EFICÁCIA
Eficácia clínica e segurança
A eficácia da enzalutamida foi estabelecida em três estudos clínicos de Fase 3, multicêntricos, randomizados e controlados por placebo [MDV3100-14 (PROSPER), CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] de pacientes com câncer de próstata progressivo que não apresentaram progressão da doença em uso de terapia de privação de andrógeno (análogo do hormônio liberador do hormônio luteinizante [LHRH] ou após orquiectomia bilateral). O estudo PREVAIL incluiu pacientes com câncer de próstata resistente à castração (CPRC) metastático não tratados previamente com quimioterapia, enquanto o estudo AFFIRM incluiu pacientes com CPRC metastático que já haviam recebido docetaxel; e o estudo PROSPER incluiu pacientes com CPRC não metastático. Além disso, a eficácia em pacientes com CPSCm também foi estabelecida em um estudo clínico de fase 3, multicêntrico, randomizado e controlado por placebo [9785-CL-0335 (ARCHES)]. Outro estudo clínico de Fase 3, multicêntrico, randomizado e controlado por placebo [MDV3100-13 (EMBARK)] estabeleceu a eficácia em pacientes com CPSCnm com recidiva bioquímica (RB) de alto risco. Todos os pacientes foram tratados com um análogo do LHRH ou foram submetidos previamente a orquiectomia bilateral, exceto quando indicado.
Enzalutamida foi administrada por via oral na dose diária de 160 mg no grupo de tratamento ativo. Nos cinco estudos clínicos (EMBARK, ARCHES, PROSPER, PREVAIL e AFFIRM), os pacientes do grupo controle receberam placebo e os pacientes não foram obrigados a tomar prednisona.
Alterações da concentração sanguínea de PSA nem sempre indicam benefício clínico de forma independente. Portanto, nos cinco estudos recomendou-se que os pacientes mantivessem seus tratamentos do estudo até que os critérios de suspensão ou descontinuação fossem atingidos, conforme especificado abaixo para cada um dos estudos.
Estudo MDV3100-13 (EMBARK) (pacientes com CPSC não metastático com recidiva bioquímica (RB) de alto risco)
O estudo EMBARK incluiu 1.068 pacientes com recidiva bioquímica (RB) de alto risco que foram randomizados 1:1:1 para receber tratamento com enzalutamida por via oral na dose de 160 mg uma vez ao dia concomitantemente com terapia de privação androgênica (TPA) (N = 355), enzalutamida por via oral na dose de 160 mg uma vez ao dia como monoterapia em caráter aberto (N = 355), ou placebo por via oral uma vez ao dia simultaneamente com TPA (N = 358) (TPA definida como leuprolida)1. Todos os pacientes tiveram terapia definitiva anterior com prostatectomia radical ou radioterapia (incluindo braquiterapia) ou ambas, com intenção curativa. Foi exigido dos pacientes que tivessem confirmação de doença não metastática por revisão central independente em caráter cego (BICR) e recidiva bioquímica de alto risco (definida por um tempo de duplicação do PSA ≤ 9 meses). Os pacientes também deveriam ter valores de PSA ≥ 1 ng/mL se tivessem sido submetidos a prostatectomia radical anterior (com ou sem radioterapia) como tratamento primário para câncer de próstata, ou valores de PSA pelo menos 2 ng/mL acima do nadir se tivessem sido submetidos a radioterapia anterior apenas.
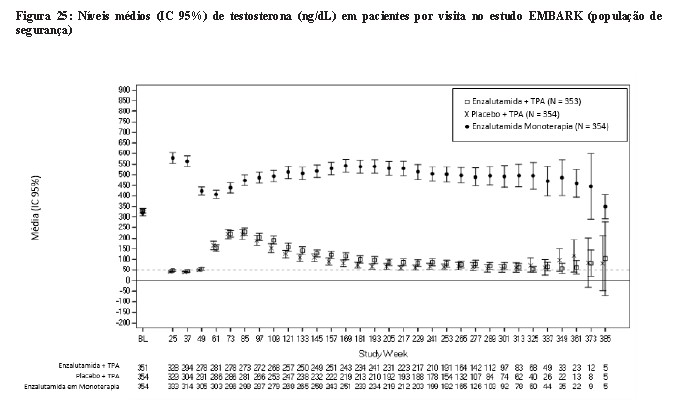
Os pacientes foram estratificados pelo PSA da triagem (≤ 10 ng/mL vs. > 10 ng/mL), tempo de duplicação do PSA (≤ 3 meses versus > 3 meses a ≤ 9 meses) e terapia hormonal anterior (terapia hormonal anterior vs. nenhuma terapia hormonal). Para pacientes cujos valores de PSA eram indetectáveis ( < 0,2 ng/mL) na semana 36, o tratamento foi suspenso na semana 37 e reiniciado quando os valores de PSA aumentaram para ≥ 2,0 ng/mL para pacientes com prostatectomia anterior ou ≥5,0 ng/mL para pacientes sem prostatectomia anterior. Para pacientes cujos valores de PSA foram detectáveis (≥ 0,2 ng/mL) na semana 36, o tratamento continuou sem suspensão até que os critérios de descontinuação permanente do tratamento fossem atendidos.
As características demográficas e basais estavam bem equilibradas entre os três grupos de tratamento. A idade mediana na randomização foi de 69 anos em ambos os grupos de tratamento de enzalutamida mais TPA e enzalutamida em monoterapia. A idade mediana na randomização foi de 70 anos no grupo de tratamento com placebo mais TPA. A maioria dos pacientes na população total eram caucasianos (83,2%), 7,3% eram asiáticos e 4,4% eram negros. O tempo mediano de duplicação do PSA foi de 4,9 meses. Setenta e quatro por cento (74%) dos pacientes tiveram terapia definitiva anterior com prostatectomia radical, 75% dos pacientes tiveram terapia anterior com radioterapia (incluindo braquiterapia) e 49% dos pacientes tiveram terapia anterior com ambas. Trinta e dois por cento (32%) dos pacientes tiveram uma pontuação de Gleason ≥ 8. A pontuação de status de desempenho do Eastern Cooperative Oncology Group (ECOG PS) foi 0 para 92,2% dos pacientes e 1 para 7,6% dos pacientes no entrada no estudo.
A sobrevida livre de metástases (SLM) em pacientes randomizados para receber enzalutamida mais TPA em comparação com pacientes randomizados para receber placebo mais TPA foi o desfecho primário. A sobrevida livre de metástases foi definida como o tempo desde a randomização até a progressão radiográfica ou morte no estudo, o que ocorrer primeiro.
Os principais desfechos secundários foram o tempo até a progressão do PSA, o tempo até o primeiro uso de terapia antineoplásica e a sobrevida global. Outro desfecho secundário importante foi SLM em pacientes randomizados para receber enzalutamida em monoterapia, em comparação com pacientes randomizados para receber placebo mais TPA.
Enzalutamida mais TPA demonstraram uma redução estatisticamente significativa de 58% no risco de desenvolver um evento de SLM em comparação com placebo mais TPA [HR = 0,42 (IC 95%: 0,30, 0,61) p < 0,0001]. O tempo mediano até um evento de SLM não foi alcançado em nenhum dos grupos de tratamento (Tabela 1, Figura 1).
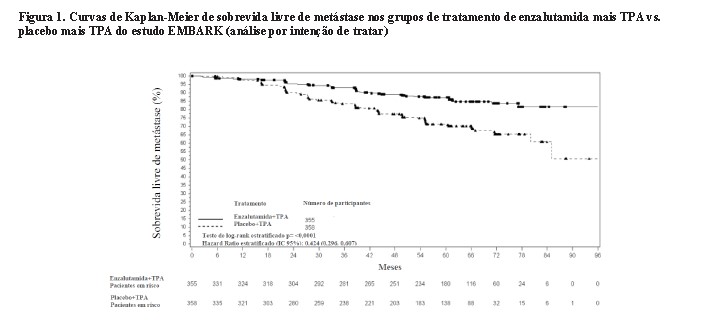
A enzalutamida em monoterapia também demonstrou uma redução estatisticamente significativa de 37% no risco de desenvolver um evento de SLM em comparação com placebo mais TPA [HR = 0,63 (IC 95%: 0,46; 0,87) p = 0,0049]. O tempo mediano até um evento de SLM não foi alcançado em nenhum dos grupos de tratamento (Tabela 1. Figura 2).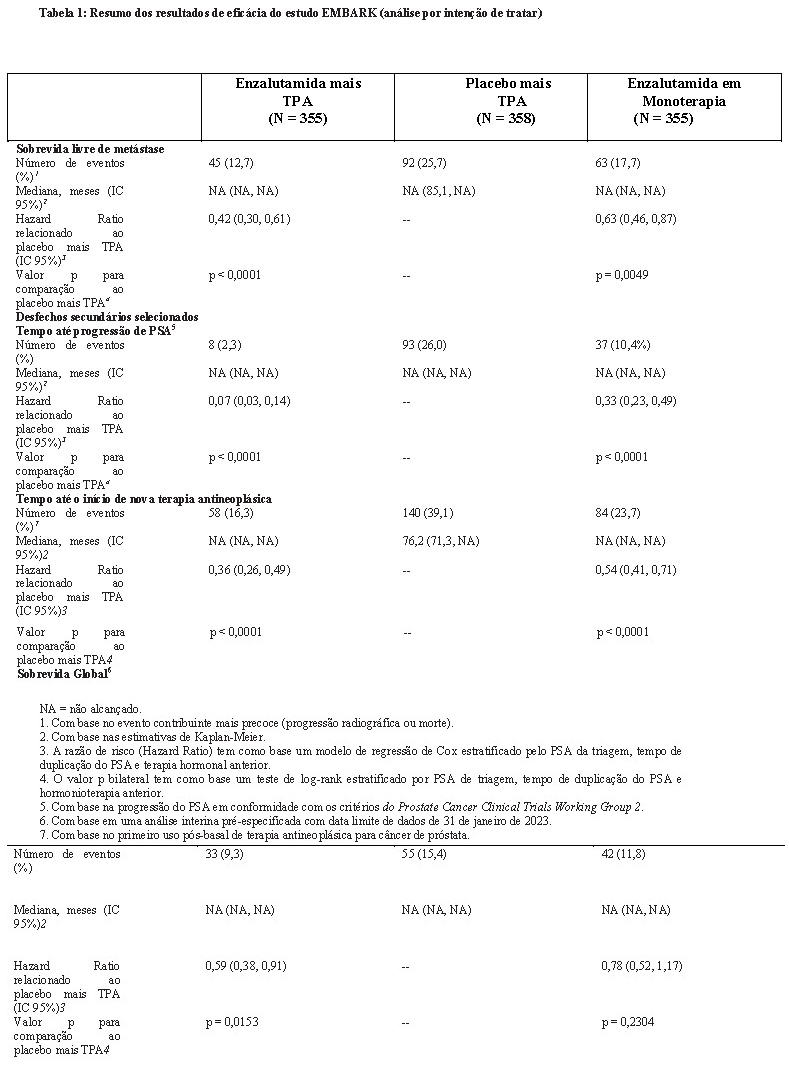
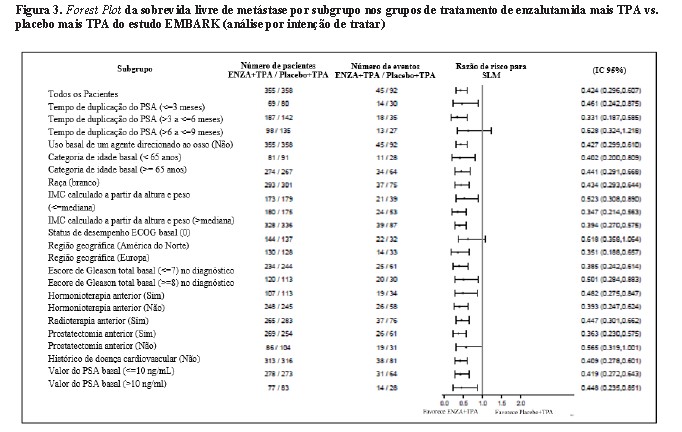
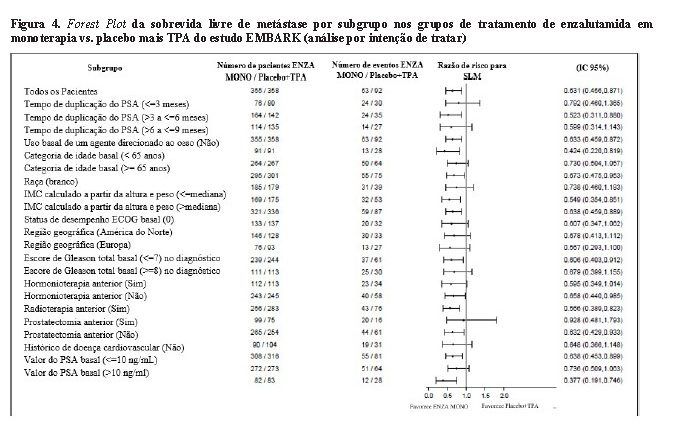
Enzalutamida mais TPA demonstraram uma redução estatisticamente significativa de 93% no risco de progressão do PSA em comparação com placebo mais TPA [HR = 0,07 (IC 95%: 0,03, 0,14) p < 0,0001]. A enzalutamida em monoterapia também demonstrou uma redução estatisticamente significativa de 67% no risco de progressão do PSA em comparação com placebo mais TPA [HR = 0,33 (IC 95%: 0,23, 0,49) p < 0,0001]. O tempo mediano para progressão do PSA não foi alcançado em nenhum dos grupos de tratamento.
Enzalutamida mais TPA demonstraram uma redução estatisticamente significativa de 64% no risco de nova terapia antineoplásica em comparação com placebo mais TPA [HR = 0,36 (IC 95%: 0,26, 0,49), p < 0,0001]. Enzalutamida em monoterapia também demonstrou uma redução estatisticamente significativa de 46% no risco de nova terapia antineoplásica em comparação com placebo mais TPA [HR = 0,54 (IC 95%: 0,41, 0,71), p < 0,0001]. O tempo mediano até o primeiro uso de nova terapia antineoplásica não foi alcançado no grupo de enzalutamida mais TPA ou no grupo de enzalutamida em monoterapia e foi de 76,2 meses (IC 95%: 71,3, NR) no grupo placebo mais TPA.
Uma análise interina pré-especificada para a sobrevida global foi planejada no momento da análise da SLM. No momento da análise interina, os dados de sobrevida global não estavam maduros e não mostraram uma diferença estatisticamente significativa em pacientes tratados com enzalutamida mais TPA em comparação com placebo mais TPA [HR = 0,59 (IC 95%: 0,38, 0,91) p = 0,0153] ou em pacientes tratados com enzalutamida em monoterapia em comparação ao placebo mais TPA [HR = 0,78 (IC 95%: 0,52, 1,17) p = 0,2304]. A sobrevida global mediana não foi alcançada em nenhum dos grupos de tratamento (Tabela 1).
ESTUDO 9785-CL-0335 (ARCHES) (PACIENTES COM CPHS METASTÁTICO)
O estudo ARCHES incluiu 1150 pacientes com CPSCm, que foram randomizados em proporção de 1:1 para receber tratamento com enzalutamida mais terapia de privação androgênica (TPA) ou placebo mais TPA (TPA definida como análogo do LHRH ou orquiectomia bilateral). Os pacientes receberam enzalutamida 160 mg uma vez ao dia (N=574) ou placebo (N=576).
Os dados demográficos e as características basais foram bem equilibrados entre os dois grupos de tratamento. A idade mediana no momento da randomização foi de 70 anos em ambos os grupos de tratamento. A maioria dos pacientes na população total eram caucasianos (80,5%); 13,5% eram asiáticos e 1,4% eram negros. O escore do estado de desempenho do Grupo Cooperativo Oriental de Oncologia [Eastern Cooperative Oncology Group Performance Status] (ECOG PS) foi 0 para 78% dos pacientes e 1 para 22% dos pacientes à entrada no estudo.
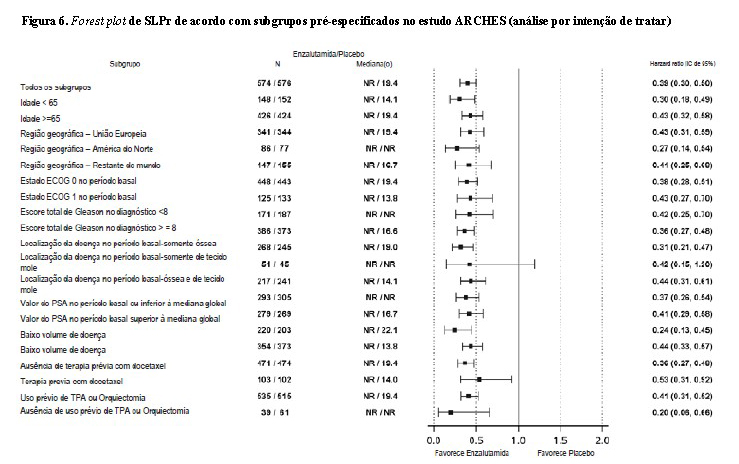
A sobrevida livre de progressão radiográfica (SLPr), com base em uma revisão central independente, foi o desfecho primário definido como o tempo desde a randomização até a primeira evidência objetiva da progressão radiográfica da doença ou o óbito (devido a qualquer causa desde o tempo da randomização até 24 semanas da descontinuação da droga em estudo), o que ocorresse primeiro. Os principais desfechos de eficácia secundários avaliados no estudo foram o tempo até a progressão do PSA, o tempo até o início de nova terapia antineoplásica, a taxa indetectável do PSA (diminuição para < 0,2 mg/L), a taxa de resposta objetiva (RECIST 1.1) com base na revisão independente e a sobrevida global. Consultar a Tabela 2 abaixo.
Enzalutamida demonstrou uma redução estatisticamente significativa de 61% no risco de um evento de SLPr em comparação ao placebo [HR = 0,39 (IC de 95%: 0,30, 0,50), p < 0,0001]. O tempo mediano para um evento de SLPr não foi alcançado no braço de enzalutamida, e foi de 19,4 meses (IC de 95%: 16,6, NA) no braço do placebo.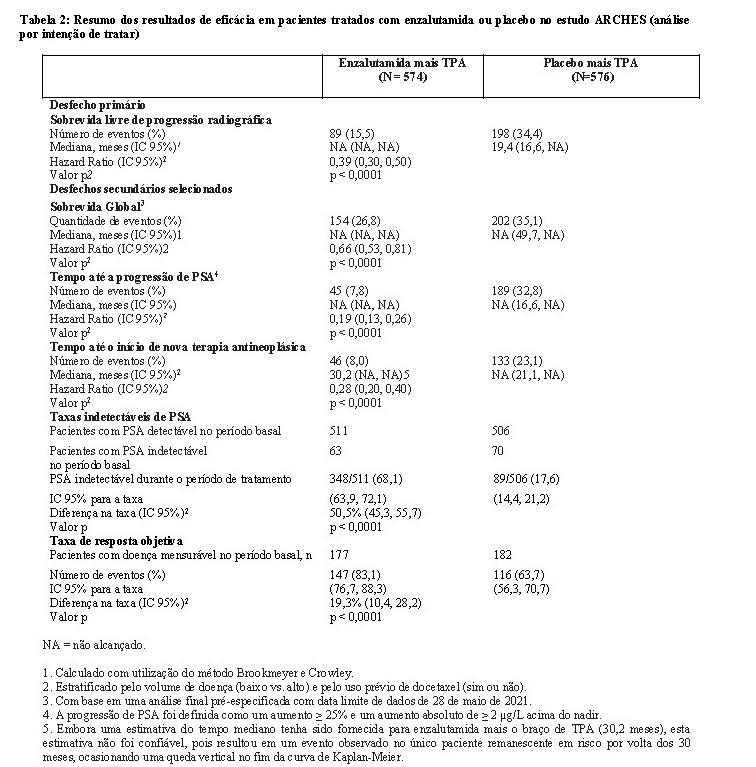
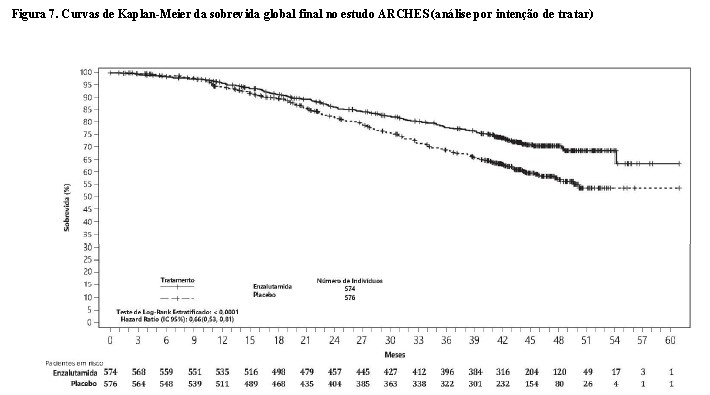
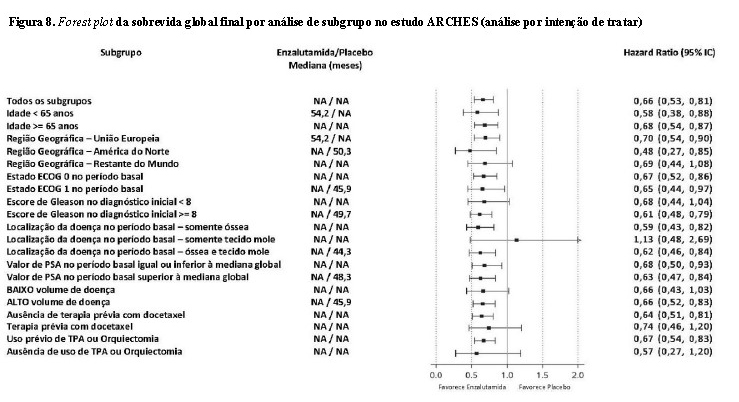
Na análise final pré-especificada de sobrevida global, conduzida quando 356 mortes foram observadas, uma redução de 34% estatisticamente significativa no risco de morte foi demonstrada no grupo randomizado para receber enzalutamida em comparação com o grupo randomizado para receber placebo [HR = 0,66, (IC 95%: 0,53; 0,81), p < 0,0001]. O tempo mediano para sobrevida global não foi atingido em nenhum grupo de tratamento (Tabela 2, Figura 7).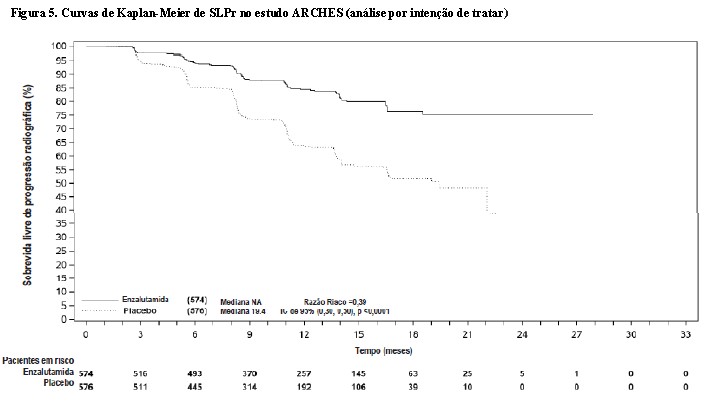
enzalutamida demonstrou uma redução estatisticamente significativa de 81% no risco de progressão de PSA em comparação ao placebo [HR = 0,19 (IC 95%: 0,13, 0,26), p < 0,0001]. O tempo mediano para a progressão de PSA (IC 95%) não foi alcançado para enzalutamida ou placebo.
enzalutamida demonstrou uma redução estatisticamente significativa de 72% no risco de início de uma nova terapia antineoplásica em comparação ao placebo [HR=0,28 (IC 95%: 0,20, 0,40); p < 0,0001].
enzalutamida aumentou significativamente a taxa de uma redução de PSA a um nível indetectável ( < 0,2 mg/L) em comparação ao tratamento com placebo. A taxa indetectável de PSA foi de 68,1% para enzalutamida e de 17,6% para placebo. A diferença da taxa é estatisticamente significativa [50,5% (IC 95%: 45,3, 55,7); p < 0,0001].
A taxa de resposta objetiva (calculada como a porcentagem de pacientes com doença mensurável no período basal que atingiram uma resposta completa ou parcial na sua doença nos tecidos moles) foi de 83,1% para pacientes no grupo de tratamento com enzalutamida e de 63,7% no grupo de placebo. Enzalutamida demonstrou uma melhora estatisticamente significativa de 19,3% na taxa de resposta objetiva em comparação ao placebo.
ESTUDO ANZUP 1304 (ENZAMET) (PACIENTES COM CPHS METASTÁTICO)
O estudo ENZAMET incluiu 1125 pacientes com CPSCm, que foram randomizados na proporção de 1:1 para receber tratamento por via oral, uma vez ao dia, com enzalutamida 160 mg (N=563) ou antiandrogênio não esteroidal (AANE, N=562). Todos os pacientes no estudo receberam um análogo do LHRH ou foram submetidos previamente a orquiectomia bilateral. Os pacientes foram estratificados de acordo com o volume de doença (baixo vs. alto), a terapia reabsortiva concomitante (sim vs. não), comorbidades (ACE-27: 0 a 1 vs. 2 a 3) e uso planejado de um total de 6 ciclos de docetaxel, sendo que foram autorizados 0-2 ciclos antes da randomização (sim vs. não). Foi necessário que os pacientes confirmassem o câncer de próstata metastático por cintilografia óssea positiva ou por lesões metastáticas no exame de tomografia computadorizada (TC) ou de ressonância magnética (RM). Os pacientes continuaram o tratamento até que fosse evidenciada progressão clínica por meio de TC, RM ou cintilografia óssea de corpo inteiro. 3
Os dados demográficos e as características basais do paciente a seguir foram equilibrados entre os dois braços de tratamento. A idade mediana no momento da randomização era de 69 anos no grupo de enzalutamida e de 68 anos no grupo de AANE (tratados com bicalutamida, nilutamida ou flutamida). A maioria dos pacientes apresentou no estado de desempenho do ECOG um escore 0 (72%) e um escore de Gleason 2 8 (58%). Quarenta e oito por cento (48%) dos pacientes apresentaram um baixo volume de doença e 52% dos pacientes apresentaram alto volume de doença. O alto volume de doença é definido como metástases com envolvimento das vísceras ou, na ausência de lesões viscerais, quatro ou mais lesões ósseas, sendo que pelo menos uma delas deve estar presente em estrutura óssea além da coluna vertebral e ossos pélvicos. Dez por cento (10%) dos pacientes estavam em uso de terapia reabsortiva concomitante, 75% não apresentaram qualquer comorbidade ou apresentaram apenas comorbidades leves (pontuação ACE-27 de 0 a 1) e 45% foram submetidos a um total de 6 ciclos de docetaxel, sendo que foram autorizados até 2 ciclos de 0 a 2 antes da randomização.
No momento da análise, o acompanhamento mediano para a SG foi de 33,8 meses. A análise interina demonstrou uma redução estatisticamente significativa no risco de óbito para pacientes tratados com enzalutamida em comparação ao tratamento convencional com AANE [HR de 0,67 (IC de 95%: 0,52, 0,86; p=0,0018)] (Figura 9).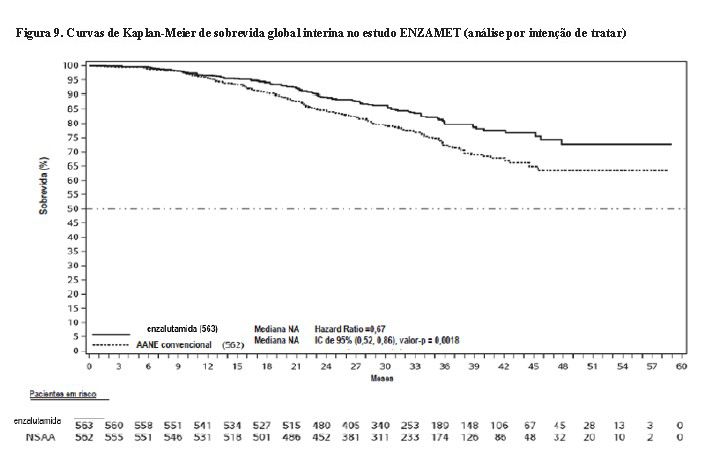
A melhora de SG foi consistente, com HRs < 1 para a maioria dos subgrupos pré-especificados (Figura 10). O subgrupo de pacientes sem planejamento antecipado do uso concomitante de docetaxel teve resultado favorável para o tratamento com enzalutamida + TPA versus antiandrogênio não esteroidal (AANE) + TPA. Esta é a população que mais se assemelha à população do estudo ARCHES. Importante notar que este efeito foi observado tanto para o subgrupo de pacientes com baixo volume de doença (HR: 0,377 [IC 95% 0,200; 0,675]), quanto para alto volume de doença (HR: 0,645 [IC 95% 0,417; 0,986]).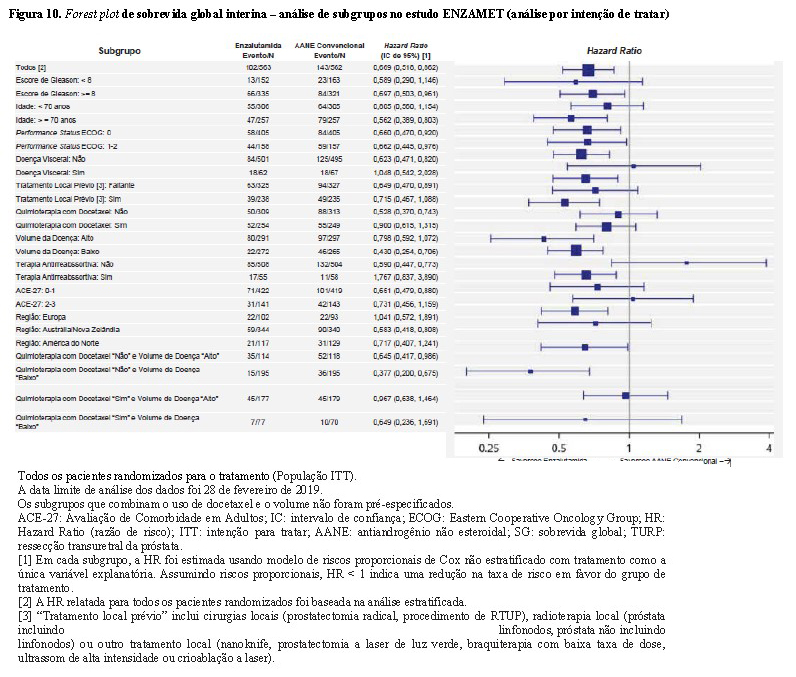
No momento da análise final, o acompanhamento mediano para SG foi de 68,2 meses. A análise demonstrou uma redução estatisticamente significativa de 30% no risco de morte para pacientes tratados com enzalutamida em comparação com o tratamento convencional com AANEs [HR de 0,70 (IC 95%: 0,58; 0,83; p < 0,0001)] (Figura 11).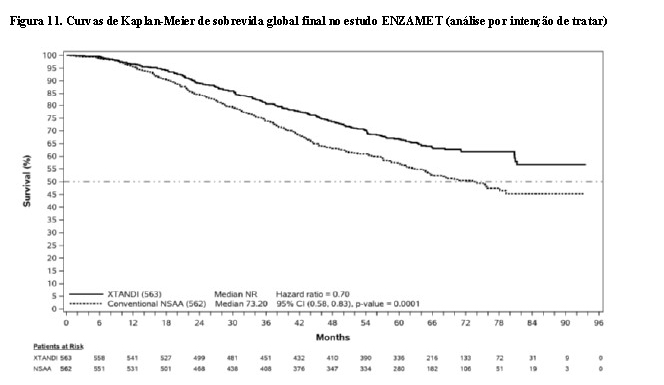
A análise final do subgrupo de SG na população com intenção de tratar é apresentada como um gráfico em forest plot na Figura 12. Embora o efeito do tratamento com enzalutamida mais TPA versus AANE mais TPA nas análises de subgrupos tenha variado entre os subgrupos, o limite superior das estimativas pontuais de HR para enzalutamida mais TPA versus AANE mais TPA caíram abaixo de 1, com exceção dos seguintes subgrupos com um tamanho de amostra geralmente menor: doença visceral, terapia reabsortiva, região, uso inicial planejado de docetaxel combinado com um volume de doença baixo ou alto; em todos os outros subgrupos, os resultados foram consistentes com um efeito favorável para pacientes no grupo enzalutamida mais TPA em comparação com pacientes no grupo de AANE mais TPA, inclusive para o subgrupo de pacientes sem o uso inicial planejado de docetaxel combinado com um volume de doença baixo ou alto. De modo geral, os resultados desses subgrupos corroboram a consistência do benefício da SG demonstrado na população.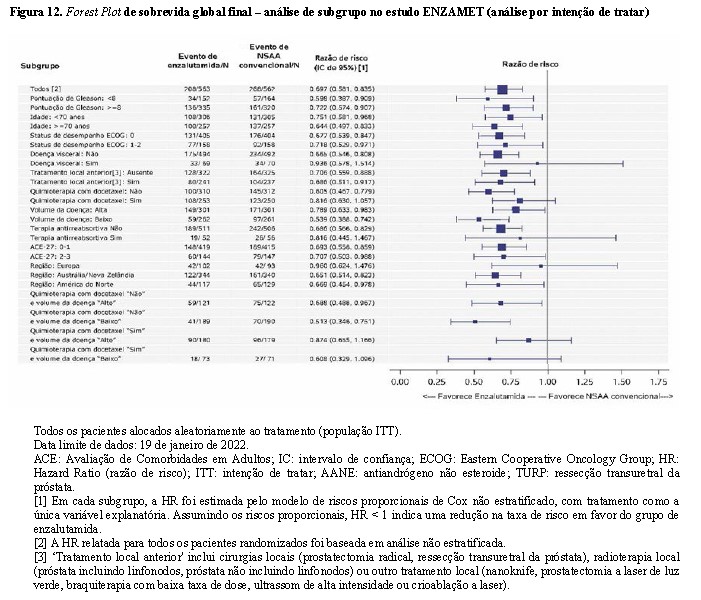
Estudo MDV3100-14 (PROSPER) (pacientes com CPRC não metastático)
O estudo PROSPER incluiu 1.401 pacientes com CPRC não metastático que continuaram com terapia de privação androgênica (TPA; definida como análogo do LHRH ou orquiectomia bilateral anterior). Os pacientes foram randomizados em proporção 2:1 para receber enzalutamida, em uma dose de 160 mg uma vez ao dia (N = 933), ou placebo (N = 468).4
Os dados demográficos e as características basais foram equilibrados entre os dois grupos de tratamento. A idade mediana foi 74 anos no grupo de enzalutamida e 73 anos no grupo de placebo. A maioria dos pacientes (aproximadamente 71%) no estudo eram caucasianos; 16% eram asiáticos e 2% eram negros.
A sobrevida livre de metástase (SLM) foi o desfecho primário definido como o momento desde a randomização até a progressão radiográfica ou morte em até 112 dias da descontinuação do tratamento sem evidências de progressão radiográfica, o que ocorrer primeiro. Os principais desfechos secundários avaliados no estudo foram tempo até a progressão do PSA, tempo até o primeiro uso de nova terapia antineoplásica (TTA) e sobrevida global (SG). Os desfechos secundários adicionais incluíam tempo até o primeiro uso de quimioterapia citotóxica e sobrevida livre de quimioterapia. Veja os resultados abaixo (Tabela 3).
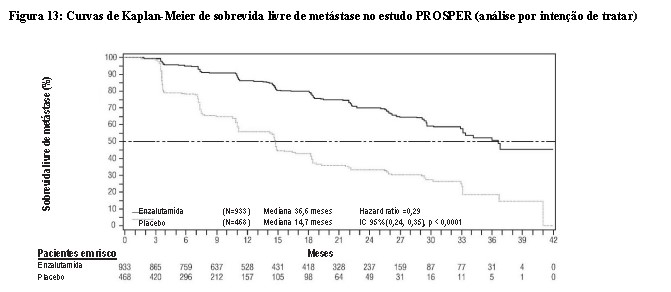
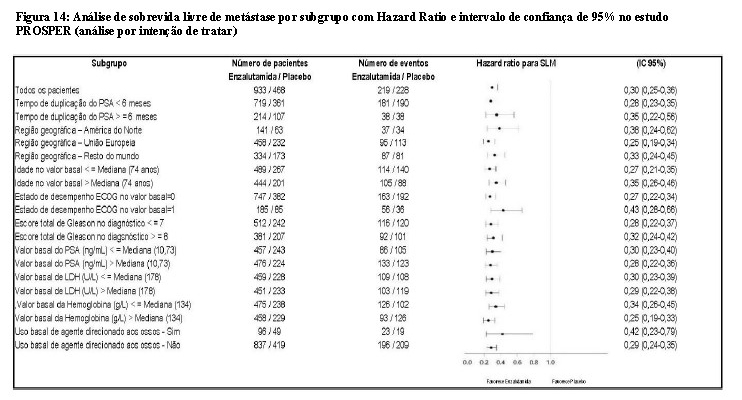
Enzalutamida demonstrou uma redução estatisticamente significativa de 71% no risco relativo de progressão radiográfica ou morte em comparação ao placebo [HR = 0,29 (IC 95%: 0,24, 0,35), p < 0,0001]. A SLM mediana foi de 36,6 meses (IC 95%: 33,1, NR) no grupo de enzalutamida versus 14,7 meses (IC 95%: 14,2, 15,0) no grupo de placebo.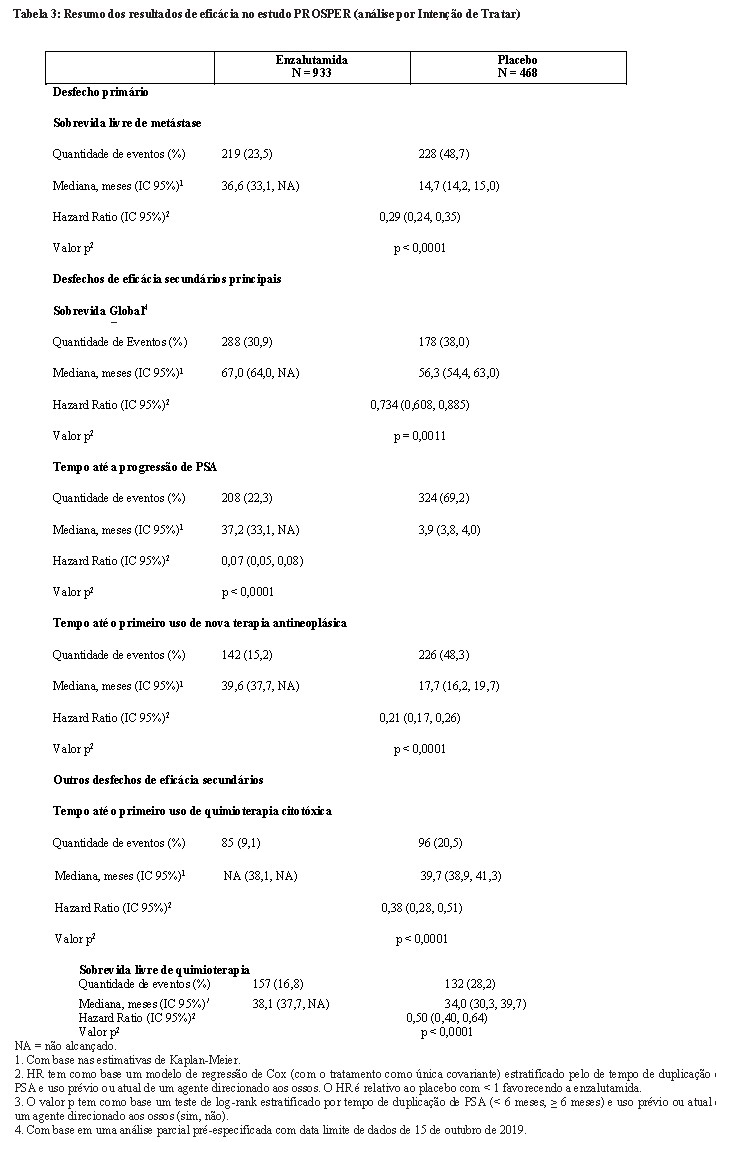
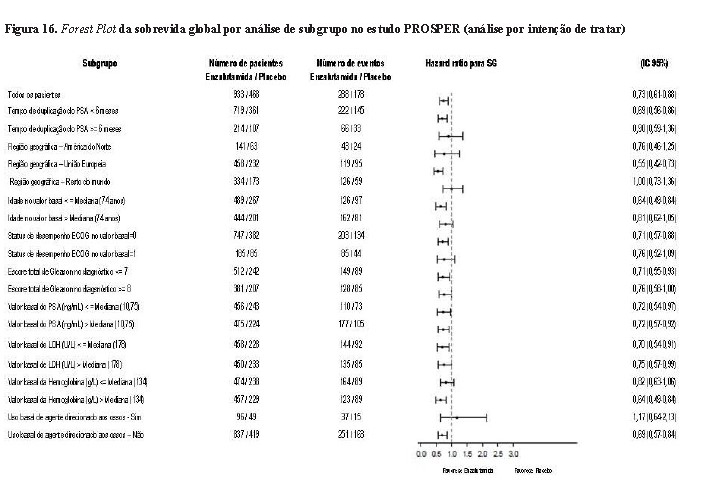
Em uma análise parcial pré-especificada de sobrevida global, conduzida quando 466 mortes foram observadas, uma melhoria estatisticamente significativa na sobrevida global foi demonstrada em pacientes randomizados para receber XTANDI em comparação com pacientes randomizados para receber placebo com uma redução de 26,6% em risco de morte [Hazard Ratio (HR) = 0,734, (IC 95%: 0,608; 0,885), p = 0,0011].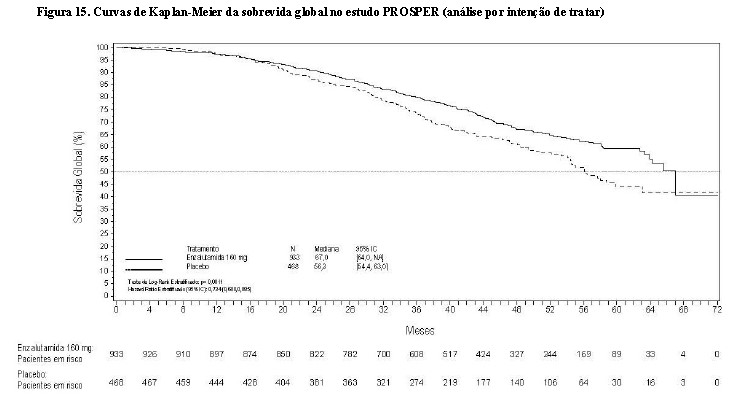
Enzalutamida demonstrou uma redução de 93% estatisticamente significativa no risco relativo de progressão de PSA em comparação com placebo [HR = 0,07 (IC 95%: 0,05, 0,08), p < 0,0001]. O tempo mediano até a progressão de PSA foi de 37,2 meses (IC 95%: 33,1, NA) no grupo de enzalutamida versus 3,9 meses (IC 95%: 3,8, 4,0) no grupo de placebo.
Enzalutamida demonstrou um retardo estatisticamente significativo no tempo até o primeiro uso de nova terapia antineoplástica em comparação ao placebo [HR = 0,21 (IC 95%: 0,17, 0,26), p < 0,0001]. Tempo mediano até o primeiro uso de nova terapia antineoplástica foi de 39,6 meses (IC 95%: 37,7, NA) no grupo de enzalutamida versus 17,7 meses (IC 95%: 16,2, 19,7) no grupo de placebo.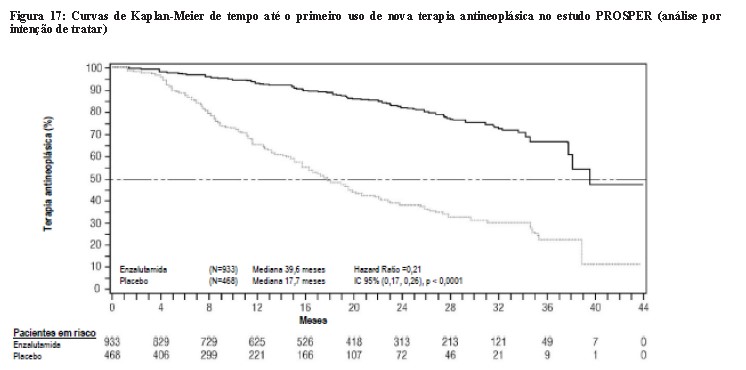
Enzalutamida demonstrou um atraso estatisticamente significativo no tempo até o início do primeiro uso de quimioterapia citotóxica [HR: 0,38 (IC 95%: 0,28, 0,51), p < 0,0001]. O tempo mediano até o primeiro uso de quimioterapia citotóxica foi NA (IC 95%: 38,1, NA) no grupo de enzalutamida versus 39,7 meses (IC 95%: 38,9, 41,3) no grupo de placebo.
Enzalutamida demonstrou uma melhora estatisticamente significativa na sobrevida livre de quimioterapia em comparação ao tratamento com placebo [HR: 0,50 (IC 95%: 0,40, 0,64), p < 0,0001]. A sobrevida livre de quimioterapia mediana foi 38,1 meses (IC 95% 37,7, NA) no grupo de enzalutamida versus 34,0 meses (IC 95%: 30,3, 39,7) no grupo de placebo.
Estudo MDV3100-09 (STRIVE) (pacientes com CPRC metastático/não metastático não tratados previamente com quimioterapia)
O estudo STRIVE incluiu 396 pacientes com CPRC metastático ou não metastático que apresentaram progressão bioquímica ou radiográfica da doença apesar da terapia de privação androgênica primária, que foram randomizados para receber enzalutamida em uma dose de 160 mg uma vez ao dia (N = 198) + TPA, ou bicalutamida em uma dose de 50 mg uma vez ao dia (N = 198) + TPA. A sobrevida livre de progressão (SLP) foi o desfecho primário definido como o tempo desde a randomização até a primeira evidência objetiva de progressão radiográfica, progressão do PSA ou morte no estudo. A SLP mediana foi de 19,4 meses (IC 95%: 16,5, NA) no grupo de enzalutamida versus 5,7 meses (IC 95%: 5,6, 8,1) no grupo de bicalutamida [HR = 0,24 (IC 95%: 0,18, 0,32), p < 0,0001]. Foi observado benefício consistente de enzalutamida sobre bicalutamida em todos os subgrupos de pacientes pré-especificados. Para o grupo não metastático (N = 139), um total de 19 de 70 (27,1%) pacientes tratados com enzalutamida e 49 de 69 (71,0%) pacientes tratados com bicalutamida apresentou eventos de SLP (total de 68 eventos). Hazard Ratio foi 0,24 (IC 95%: 0,14, 0,42) e o tempo mediano até o evento de SLP não foi alcançado no grupo de enzalutamida versus 8,6 meses no grupo de bicalutamida. 5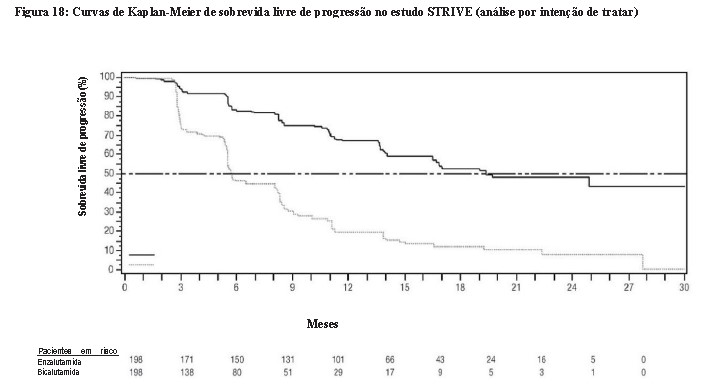
Estudo 9785-CL-0222 (TERRAIN) (pacientes com CPRC metastático, virgens de quimioterapia)
O estudo TERRAIN incluiu 375 pacientes com CRPC metastático virgens de quimioterapia e terapia antiandrogênica que foram randomizados para receber enzalutamida 160 mg uma vez ao dia + TPA (N = 184) ou bicalutamida 50 mg uma vez ao dia + TPA (N = 191). A sobrevida livre de progressão (SLP) mediana foi de 15,7 meses nos pacientes em uso de enzalutamida versus 5,8 meses nos pacientes em uso de bicalutamida [HR = 0,44 (IC 95%: 0,34; 0,57), p < 0,0001]. Sobrevida livre de progressão foi definida como evidência objetiva de progressão radiográfica da doença de acordo com uma revisão central independente, eventos relacionados ao esqueleto, início de nova terapia antineoplásica ou morte devido a qualquer causa, o que ocorrer primeiro. Foi observado benefício consistente quanto à SLP em todos os subgrupos de pacientes pré-especificados.6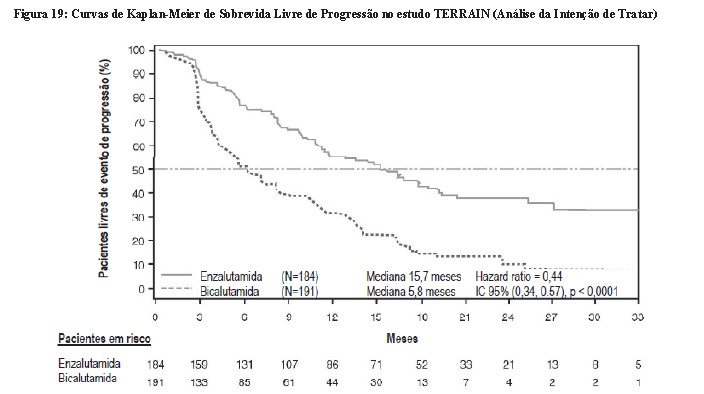
Estudo MDV3100-03 (PREVAIL) (pacientes com CPRC metastático não tratados previamente com quimioterapia)
Foi randomizado, em distribuição de 1:1, um total de 1.717 pacientes assintomáticos ou ligeiramente sintomáticos, não tratados previamente com quimioterapia, para receber enzalutamida por via oral na dose de 160 mg, uma vez ao dia + TPA (N = 872), ou placebo por via oral, uma vez ao dia + TPA (N = 845). Foi permitida a inclusão de pacientes com doença visceral, pacientes com histórico de insuficiência cardíaca leve a moderada (NYHA Classes I ou II) e pacientes em uso de medicações associadas à diminuição do limiar convulsivo. 7
Foram excluídos pacientes com histórico de convulsão ou alguma condição que pudesse predispor à convulsão e pacientes com dor moderada ou grave decorrente do câncer de próstata. O tratamento do estudo continuou até a progressão da doença (evidência de progressão radiográfica, evento relacionado ao esqueleto ou progressão clínica) e o início de quimioterapia citotóxica ou de algum agente em investigação, ou até toxicidade inaceitável. Os dados demográficos dos pacientes e as características basais da doença foram equilibrados entre os grupos de tratamento. A idade mediana foi 71 anos (variação 42-93) e a distribuição racial foi de 77% caucasianos; 10% asiáticos; 2% negros e 11% outras raças ou desconhecido. Sessenta e oito por cento (68%) dos pacientes apresentava índice de desempenho ECOG 0 e 32% dos pacientes apresentavam um ECOG de 1. A avaliação basal da dor foi de 0-1 (assintomático) em 67% dos pacientes e de 2-3 (ligeiramente sintomático) em 32% dos pacientes, conforme definido pelo Inventário Breve de Dor - Forma Abreviada (pior dor relatada pelo paciente ao longo das 24 horas anteriores, em escala de 0 a 10). Aproximadamente 45% dos pacientes apresentaram doença mensurável nos tecidos moles ao ingressar no estudo, e 12% dos pacientes tinham metástases viscerais (pulmão e/ou fígado).
Os desfechos coprimários de eficácia foram a sobrevida global e a sobrevida livre de progressão radiográfica (SLPr). Além dos desfechos coprimários, o benefício também foi avaliado usando o tempo até o início de quimioterapia citotóxica, melhor resposta global nos tecidos moles, tempo até o primeiro evento relacionado ao esqueleto, resposta do PSA (diminuição 2 50% em relação ao valor basal), tempo até progressão do PSA e tempo até a degradação do escore total do questionário de qualidade de vida FACT-P (Avaliação Funcional de Terapia de Câncer - Próstata).
A progressão radiográfica foi avaliada com uso de estudos de imagens sequenciais de acordo com os critérios definidos pelo Grupo de Trabalho dos Ensaios Clínicos de Câncer da Próstata 2 (PCWG2 - Prostate Cancer Clinical Trials Working Group 2) (para lesões ósseas) e/ou com os critérios dos Critérios de Avaliação de Resposta em Tumores Sólidos (Response Evaluation Criteria in Solid Tumors - RECIST v 1.1) (para lesões em tecidos moles). A análise de SLPr utilizou avaliação radiográfica da progressão revisada centralmente.
Durante a análise interina pré-especificada para sobrevida global, quando foram observadas 540 mortes, o tratamento com enzalutamida demonstrou melhora estatisticamente significativa na sobrevida global comparada com o placebo, com redução do risco de morte em 29,4% [HR = 0,706; (IC 95%: 0,60; 0,84), p < 0,0001]. Uma análise de sobrevida atualizada foi realizada quando foram observadas 784 mortes. Os resultados dessa análise foram consistentes com os da análise interina (Tabela 3, Figura 18). Na análise atualizada, 52% dos pacientes tratados com enzalutamida e 81% dos pacientes tratados com placebo haviam recebido terapias subsequentes para câncer de próstata metastático resistente à castração (CPRCm) que podem prolongar a sobrevida global.
Uma análise final dos dados de 5 anos do estudo PREVAIL mostrou que um aumento estatisticamente significativo na sobrevida global foi mantido em pacientes tratados com enzalutamida em comparação ao placebo [HR = 0,835 (IC 95%: 0,75, 0,93); valor p = 0,0008], embora 28% dos pacientes que receberam placebo tenham passado para enzalutamida. A taxa de SG em 5 anos foi de 26% no grupo de enzalutamida em comparação a 21% no grupo placebo.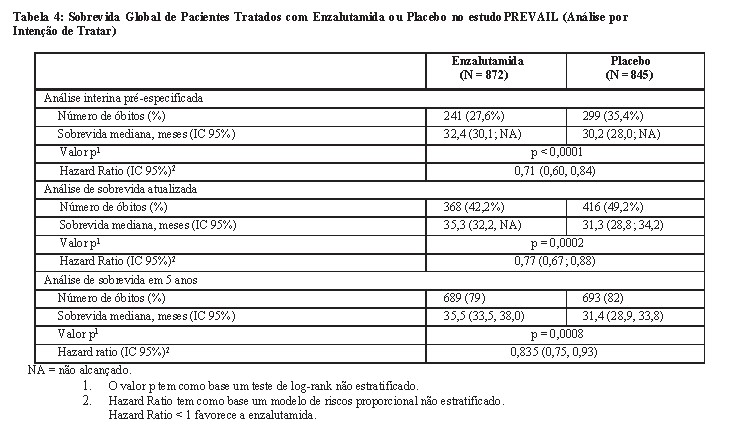
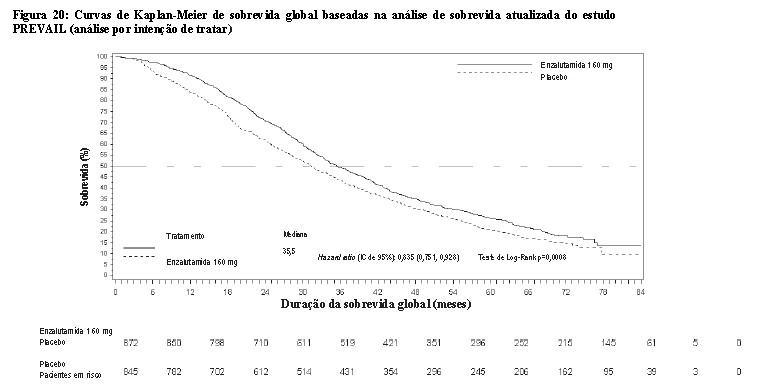
Durante a análise pré-especificada de SLPr, foi demonstrada melhora estatisticamente significativa entre os grupos de tratamento com redução de 81,4% do risco de progressão radiográfica ou morte [HR = 0,19 (IC 95%: 0,15; 0,23), p < 0,0001]. Cento e dezoito (14%) pacientes tratados com enzalutamida e 321 (40%) dos pacientes tratados com placebo apresentaram um evento. A mediana de SLPr não foi alcançada (IC 95%: 13,8, NA) no grupo de pacientes tratados com enzalutamida e foi de 3,9 meses (IC 95%: 3,7; 5,4) no grupo de pacientes com placebo (Figura 20). Foi observado um benefício consistente quanto a SLPr em todos os subgrupos pré-especificados de pacientes (ex., idade, desempenho do ECOG basal, PSA e LDH basais, escore de Gleason ao diagnóstico e doença visceral na triagem). Uma análise do seguimento pré-especificado de SLPr com base na avaliação do investigador da progressão radiográfica demonstrou melhora estatisticamente significativa entre os grupos de tratamento, com redução de 69,3% no risco de progressão radiográfica ou morte [HR = 0,31 (IC 95%: 0,27; 0,35), p < 0,0001]. A SLPrmediana foi de 19,7 meses no grupo enzalutamida e de 5,4 meses no grupo placebo.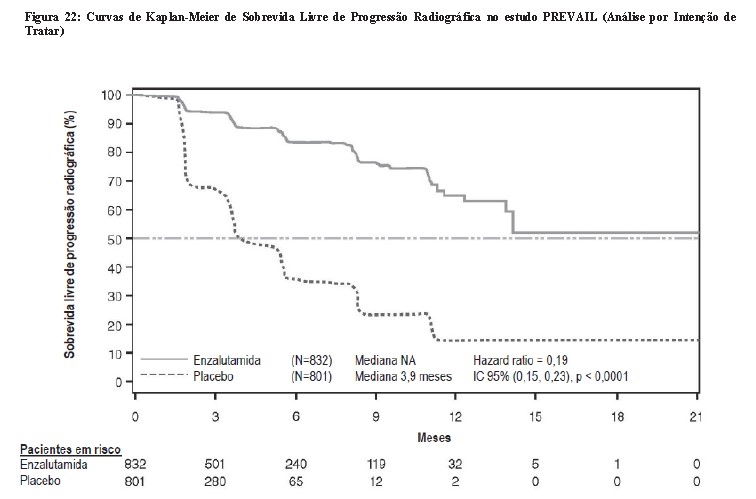
No momento da análise primária havia 1.633 pacientes randomizados.
Além dos desfechos coprimários de eficácia, também foram demonstradas melhoras estatisticamente significativas nos seguintes desfechos prospectivamente definidos.
O tempo mediano até início da quimioterapia citotóxica foi de 28,0 meses para pacientes recebendo enzalutamida e de 10,8 meses para pacientes recebendo placebo [HR = 0,35 (IC 95%: 0,30; 0,40);p < 0,0001].
A proporção de pacientes tratados com enzalutamida com doença mensurável na avaliação basal do estudo que tiveram uma resposta objetiva dos tecidos moles foi de 58,8% (IC 95%: 53,8; 63,7) em comparação com 5,0% (IC 95%: 3,0; 7,7) dos pacientes que receberam placebo. A diferença absoluta na resposta objetiva dos tecidos moles entre enzalutamida e placebo foi de [53,9% (IC 95%: 48,5; 59,1);p < 0,0001]. Respostas completas foram relatadas em 19,7% dos pacientes tratados com enzalutamida, em comparação com 1,0% dos pacientes tratados com placebo, e respostas parciais foram relatadas em 39,1% dos pacientes tratados com enzalutamida, contra 3,9% dos pacientes tratados com placebo.
A enzalutamida diminuiu significativamente o risco do primeiro evento relacionado ao esqueleto em até 28% [HR = 0,718 (IC 95%: 0,61; 0,84), p < 0,0001]. Um evento relacionado ao esqueleto foi definido como radioterapia ou cirurgia óssea para câncer de próstata, fratura óssea patológica, compressão da medula espinhal ou mudança de terapia antineoplásica para tratar a dor óssea. A análise incluiu 587 eventos relacionados ao esqueleto, dos quais 389 eventos (66,3%) foram de radiação óssea, 79 eventos (13,5%) foram de compressão da medula espinhal, 70 eventos (11,9%) foram de fratura óssea patológica, 45 eventos (7,6%) foram de mudança de terapia antineoplásica para tratar a dor óssea, e 22 eventos (3,7%) foram de cirurgia óssea.
Os pacientes que recebem enzalutamida demonstraram uma superioridade estatisticamente significativa na taxa de resposta total do PSA (definida como uma redução 2 50% desde o basal), em comparação com os pacientes que receberam placebo, 78,0% versus 3,5% (diferença = 74,5%; p < 0,0001).
O tempo mediano para progressão do PSA de acordo com os critérios do PCWG2 foi de 11,2 meses para pacientes tratados com enzalutamida e de 2,8 meses para pacientes que receberam placebo [HR = 0,17, (IC 95%: 0,15; 0,20); p < 0,0001].
O tratamento com enzalutamida diminuiu o risco de degradação da avaliação FACT-P em 37,5% em comparação com o placebo (p < 0,0001). O tempo mediano até a degradação da avaliação FACT-P foi de 11,3 meses no grupo enzalutamida e 5,6 meses no grupo de placebo.
Estudo CRPC2 (AFFIRM) (pacientes com CPRC metastático que receberam quimioterapia prévia)
A eficácia e segurança de enzalutamida em pacientes com câncer de próstata resistente à castração metastático (CPRCm) que tenham recebido docetaxel e estavam usando um análogo do LHRH ou que foram submetidos à orquiectomia, foram avaliados por um ensaio clínico de fase 3, multicêntrico, controlado por placebo, randomizado. Um total de 1.199 pacientes foram randomizados em proporção 2:1 para receber enzalutamida oralmente, em uma dose de 160 mg uma vez ao dia + TPA (N = 800), ou placebo + TPA uma vez ao dia (N = 399). Foi permitido, mas não exigido, que os pacientes tomassem prednisona (dose máxima diária permitida de 10 mg de prednisona ou equivalente). Os pacientes randomizados para cada grupo tinham que continuar o tratamento até a progressão da doença (definida como progressão radiográfica confirmada ou a ocorrência de um evento relacionado ao esqueleto) e iniciação de novo tratamento antineoplásico sistêmico, toxicidade inaceitável ou retirada do consentimento.
Os dados demográficos de pacientes e características de referência da doença a seguir foram equilibrados entre as unidades de tratamento. A idade média foi 69 anos (variação 41-92) e a distribuição racial foi 93% caucasianos; 4% negros; 1% asiáticos e 2% outras raças. O índice de desempenho do ECOG foi 0-1 em 91,5% dos pacientes e 2 em 8,5% dos pacientes; 28% tiveram um índice de Inventário Breve de Dor de 2 4 (média de pior dor relatada por paciente sobre as 24 horas anteriores calculada por 7 dias antes da randomização). A maioria (91%) dos pacientes tinham metástases nos ossos e 23% tinham comprometimento visceral de pulmão e/ou fígado. Ao ingressar no estudo, 41% dos pacientes randomizados apresentaram apenas progressão do PSA, enquanto 59% dos pacientes apresentaram progressão radiográfica. Cinquenta e um (51%) dos pacientes estavam em uso de bisfosfonados no período basal.
O estudo AFFIRM excluiu pacientes com afecções clínicas que predispusessem a crises convulsivas e uso de medicamentos que sabidamente reduzem o limiar convulsivo, bem como doenças cardiovasculares significativas como hipertensão descontrolada, histórico recente de infarto do miocárdio ou angina instável, insuficiência cardíaca classe III ou IV da New York Heart Association (a não ser que a fração de ejeção fosse ≥ 45%), arritmias ventriculares clinicamente significativas ou bloqueio AV (sem marca-passo permanente).
A análise interina pré-especificada do protocolo após 520 óbitos demonstrou uma superioridade estatisticamente significativa na sobrevida global mediana em pacientes tratados com enzalutamida comparado com o placebo (Tabela 5 e Figuras 23 e 24).
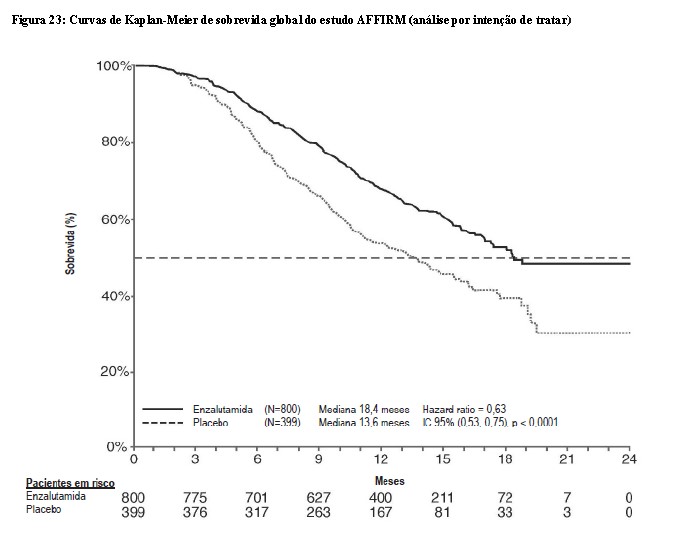
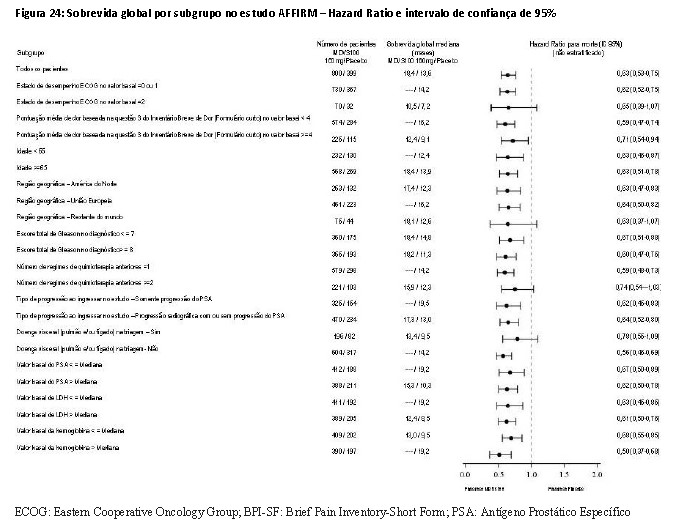
Em adição ao aumento de sobrevida global, os principais desfechos secundários (progressão do PSA, sobrevida livre de progressão radiográfica e tempo até o primeiro evento relacionado ao esqueleto) favoreceram a enzalutamida e foram estatisticamente importantes após o ajuste para testagem múltipla como segue:
A sobrevida livre de progressão radiográfica, conforme avaliada pelo investigador utilizando o RECIST v1.1 para tecidos moles, e o aparecimento de duas ou mais lesões ósseas em exame ósseo foi de 8,3 meses para pacientes tratados com enzalutamida e de 2,9 meses para pacientes que receberam placebo [HR = 0,40; [IC 95%: 0,35; 0,47), p < 0,0001]. A análise envolveu 216 óbitos sem progressão documentada e 645 eventos documentados de progressão, dos quais 303 (47%) foram devido à progressão em tecidos moles, 268 (42%) foram devido à progressão de lesão óssea e 74 (11%) foram devido a ambos, progressão em tecidos moles e lesões ósseas.
A redução de PSA confirmada de 50% ou 90% foi de 54,0% e 24,8%, respectivamente, para pacientes tratados com enzalutamida e 1,5% e 0,9%, respectivamente, para pacientes que receberam placebo (p < 0,0001). O tempo mediano para progressão do PSA foi de 8,3 meses para pacientes tratados com enzalutamida e de 3,0 meses para pacientes que receberam placebo [HR = 0,248; (IC 95%: 0,20; 0,30), p < 0,0001].
O tempo mediano para o primeiro evento relacionado ao esqueleto (SER) foi de 16,7 meses para pacientes tratados com enzalutamida e de 13,3 meses para pacientes que receberam placebo [HR = 0,69; (IC 95%: 0,57; 0,84) p < 0,0001]. Evento relacionado ao esqueleto foi definido como uma terapia de radiação ou cirurgia óssea, fratura patológica de osso, compressão da medula espinhal ou mudança de terapia antineoplásica para tratar a dor óssea. A análise envolveu 448 eventos relacionados ao esqueleto, dos quais 277 eventos (62%) foram de radioterapia óssea, 95 eventos (21%) foram de compressão da medula espinhal, 47 eventos (10%) foram de fratura patológica de osso, 36 eventos (8%) foram de mudança de terapia antineoplásica para tratar a dor óssea e 7 eventos (2%) foram de cirurgia óssea.
A taxa de resposta para qualidade de vida (Avaliação Funcional de Terapia de Câncer - Próstata; FACT-P) foi 43,2% para pacientes tratados com enzalutamida e 18,3% para pacientes que receberam placebo (p < 0,0001).
A taxa de resposta radiográfica objetiva avaliada pelo investigador (definida como a soma das respostas total e parcial) entre pacientes tratados com enzalutamida foi 28,9% comparado com uma taxa de resposta objetiva de 3,8% para pacientes que receberam placebo (p < 0,0001).
O risco de progressão da dor foi reduzido em 44% para pacientes tratados com enzalutamida comparado aos pacientes que receberam placebo (HR = 0,56; [IC 95%: 0,41; 0,78], p = 0,0004). O tempo mediano para progressão da dor foi de 13,8 meses para pacientes que receberam placebo e não foi alcançado nos pacientes tratados com enzalutamida. A progressão da dor foi definida como um aumento acima do valor de referência na avaliação FACT-P da dor, confirmada por uma segunda avaliação consecutiva obtida 3 ou mais semanas depois.
Referências Bibliográficas:
1. [EMBARK]. Freedland, S.,De Giorgi, U., Gleave, M., Rosbrook, B., Shen, Q., Sugg, J., Haas, GP., Shore, N: A phase 3 randomised study of enzalutamide plus leuprolide and enzalutamide monotherapy in high-risk nonmetastatic hormone-sensitive prostate cancer with rising PSA after local therapy: EMBARK study design. BMJ Open 2021;11:e046588. doi:10.1136/bmjopen-2020