EMGALITY
LIBBS
galcanezumab
Profilaxia da enxaqueca.
Apresentações.
EMGALITY é disponibilizado como uma solução injetável contendo 120 mg de galcanezumabe em 1 mL (120 mg/mL).
Cada embalagem contém 1 auto-injetor pré-preenchido, para dose única, com 1 mL de solução contendo 120 mg de galcanezumabe.
USO SUBCUTÂNEO
USO ADULTO
Composição.
Cada 1 mL contém: galcanezumabe 120 mg. Excipientes: histidina, cloridrato de histidina monoidratado, polissorbato 80, cloreto de sódio em água para injetáveis.
Informações técnicas.
INDICAÇÕES
EMGALITY é indicado para a profilaxia da enxaqueca em adultos que apresentam pelo menos quatro dias de enxaqueca por mês.
RESULTADOS DE EFICÁCIA
A eficácia de galcanezumabe foi avaliada como um tratamento preventivo para enxaqueca episódica ou crônica em três estudos randomizados, multicêntricos, duplo-cegos e controlados por placebo: dois estudos de 6 meses em pacientes com enxaqueca episódica (estudos 1 e 2) e um estudo de 3 meses em pacientes com enxaqueca crônica (estudo 3).
Enxaqueca episódica:
O estudo 1 (NCT02614183) e o estudo 2 (NCT02614196) incluíram adultos com histórico de enxaqueca episódica (4 a 14 dias de enxaqueca por mês). Todos os pacientes foram randomizados na proporção 1:1:2 para receber injeções subcutâneas mensais de galcanezumabe 120 mg, galcanezumabe 240 mg, ou placebo. Todos os pacientes no grupo de galcanezumabe 120 mg receberam uma dose de ataque inicial de 240 mg. Os pacientes foram autorizados a usar tratamentos para cefaleia aguda, incluindo medicamentos específicos para enxaqueca (isto é, triptanos, derivados da ergotamina), anti-inflamatórios não-esteroidais (AINEs) e paracetamol durante o estudo.
Os estudos excluíram pacientes em qualquer outro tratamento preventivo de enxaqueca, pacientes com cefaleia por uso excessivo de medicação, pacientes com anomalias no eletrocardiograma (ECG) compatíveis com um evento cardiovascular agudo e pacientes com histórico de acidente vascular cerebral, infarto do miocárdio, angina instável, intervenção coronariana percutânea, revascularização do miocárdio, trombose venosa profunda, ou embolia pulmonar dentro de 6 meses da triagem.
O desfecho primário de eficácia dos estudos 1 e 2 foi a alteração média em relação ao período basal do número de dias de enxaqueca por mês durante o período de seis meses de tratamento. Os desfechos secundários principais incluíram a taxa de resposta (porcentagem média de pacientes com redução em relação ao período basal de pelo menos 50%, 75% e 100% do número de dias de enxaqueca por mês, durante o período de seis meses de tratamento), a alteração média em relação ao período basal no número de dias de enxaqueca por mês nos quais qualquer medicação para cefaleia aguda foi utilizada durante o período de seis meses de tratamento, e o impacto da enxaqueca nas atividades diárias, conforme avaliado pela alteração média em relação ao período basal, no domínio de Restrição Funcional do Questionário de Qualidade de Vida Específico para Enxaqueca versão 2.1 (MSQ v2.1) durante os últimos três meses de tratamento (meses 4 a 6). As pontuações são escalonadas de 0 a 100, com pontuações mais altas indicando menor impacto da enxaqueca nas atividades diárias.
No estudo 1, um total de 858 pacientes (718 mulheres, 140 homens) com idade variando de 18 a 65 anos, foram randomizados. Um total de 703 pacientes completaram a fase duplo-cega de 6 meses. No estudo 2, um total de 915 pacientes (781 mulheres, 134 homens) com idade variando de 18 a 65 anos, foram randomizados. Um total de 703 pacientes completaram a fase duplo-cega de 6 meses. No estudo 2, um total de 915 pacientes (781 mulheres, 134 homens) com idade variando de 18 a 65 anos, foram randomizados. Um total de 785 pacientes completaram a fase duplo-cega de 6 meses. No estudo 1 e no estudo 2, a frequência média de enxaqueca no período basal foi de aproximadamente 9 dias de enxaqueca por mês, e foi semelhante entre os grupos de tratamento.
Galcanezumabe 120 mg demonstrou melhorias estatisticamente significantes nos desfechos de eficácia em comparação com placebo no período de 6 meses, conforme resumido na Tabela 1. O tratamento com galcanezumabe com a dose de 240 mg por mês não demonstrou benefício adicional em relação à dose de galcanezumabe 120 mg uma vez por mês.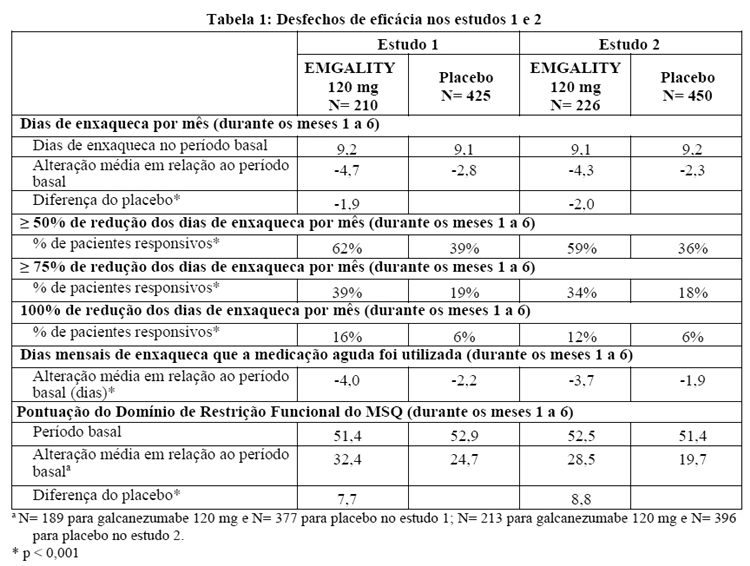
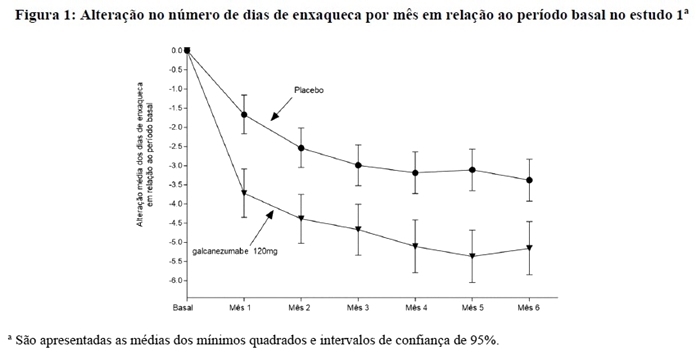
A Figura 3 mostra a distribuição da alteração em relação ao período basal no número médio de dias de enxaqueca por mês em intervalos de 2 dias, por grupo de tratamento, no estudo 1. Um benefício do tratamento com galcanezumabe em relação ao placebo é observado ao longo de uma série de alterações nos dias de enxaqueca por mês em relação ao período basal.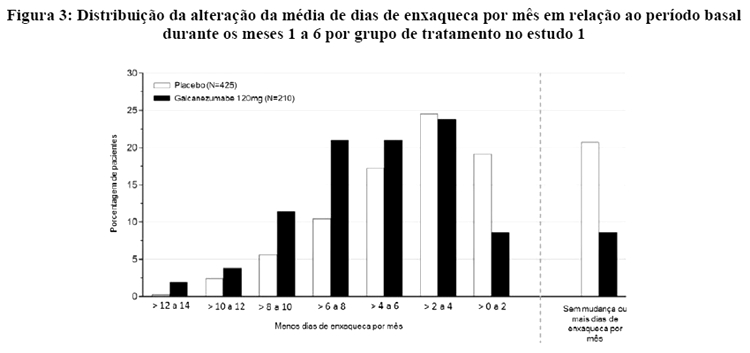
A Figura 4 mostra a distribuição da alteração em relação ao período basal no número médio de dias de enxaqueca por mês em intervalos de 2 dias, por grupo de tratamento, no estudo 2. Um benefício do tratamento com galcanezumabe em relação ao placebo é observado ao longo de uma série de alterações nos dias de enxaqueca por mês em relação ao período basal.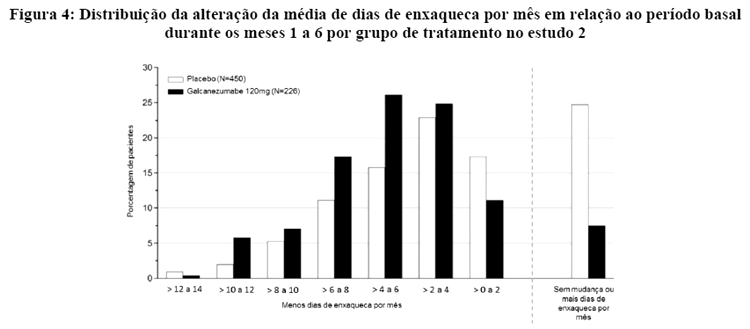
Enxaqueca crônica:
O estudo 3 (NCT02614261) incluiu adultos com histórico de enxaqueca crônica (≥ 15 dias de cefaleia por mês com ≥ 8 dias de enxaqueca por mês). Todos os pacientes foram randomizados na proporção 1:1:2 para receber injeções subcutâneas mensais de galcanezumabe 120 mg, galcanezumabe 240 mg, ou placebo, durante um período de tratamento de 3 meses. Todos os pacientes no grupo de galcanezumabe 120 mg receberam uma dose de ataque inicial de 240 mg.
Os pacientes foram autorizados a usar tratamentos para cefaleia aguda, incluindo medicamentos específicos para enxaqueca (isto é, triptanos, derivados da ergotamina), anti-inflamatórios não-esteroidais (AINEs) e paracetamol. Um subconjunto de pacientes (15%) foi autorizado a usar uma medicação preventiva de enxaqueca concomitantemente. Pacientes com cefaleia por uso excessivo de medicação foram autorizados a se inscrever.
O estudo excluiu pacientes com anomalias no eletrocardiograma (ECG) compatíveis com um evento cardiovascular agudo e pacientes com histórico de acidente vascular cerebral, infarto do miocárdio, angina instável, intervenção coronariana percutânea, revascularização do miocárdio, trombose instável, intervenção coronariana percutânea, revascularização do miocárdio, trombose venosa profunda, ou embolia pulmonar dentro de 6 meses da triagem.
O desfecho primário foi a alteração média em relação ao período basal no número de dias de enxaqueca por mês durante o período de 3 meses de tratamento. Os desfechos secundários foram as taxas de resposta (porcentagem média de pacientes com redução em relação ao período basal de pelo menos 50%, 75% e 100% de dias de enxaqueca no mês, durante o período de 3 meses de tratamento), a alteração média em relação ao período basal no número de dias de enxaqueca por mês nos quais qualquer medicação para cefaleia aguda foi utilizada durante o período de 3 meses de tratamento, e o impacto da enxaqueca nas atividades diárias, conforme avaliado pela alteração média em relação ao período basal, no domínio de Restrição Funcional do Questionário de Qualidade de Vida Específico para Enxaqueca versão 2.1 (MSQ v2.1) no mês 3. As pontuações são escalonadas de 0 a 100, com pontuações mais altas indicando menor impacto da enxaqueca nas atividades diárias.
No estudo 3, um total de 1113 pacientes (946 mulheres, 167 homens) com idade variando de 18 a 65 anos, foram randomizados. Um total de 1037 pacientes completou a fase duplo-cega de 3 meses. O número médio de dias de enxaqueca por mês no período basal era de aproximadamente 19.
Galcanezumabe 120 mg demonstrou melhora estatisticamente significante para a alteração média em relação ao período basal no número de dias de enxaqueca por mês ao longo do período de tratamento de 3 meses, e na porcentagem média de pacientes atingindo pelo menos 50% de redução em relação ao período basal do número de dias de enxaqueca por mês ao longo do período de 3 meses de tratamento, conforme resumido na Tabela 2. O tratamento com galcanezumabe com a dose mensal de 240 mg não demonstrou qualquer benefício adicional em relação à dose de galcanezumabe 120 mg uma vez por mês.
O estudo 3 utilizou um procedimento de teste sequencial para controlar a taxa de erro do Tipo-I para os múltiplos desfechos secundários. Uma vez que um desfecho secundário não atingiu o nível requerido de significância estatística, o teste formal de hipóteses foi terminado para desfechos subsequentes, e os valores-p foram considerados apenas nominais. No estudo 3, galcanezumabe 120 mg não foi significativamente melhor do que o placebo na proporção de pacientes com redução ≥ 75% ou 100% dos dias de enxaqueca. Pacientes tratados com galcanezumabe 120 mg apresentaram uma redução nominalmente maior no número de dias de enxaqueca por mês em que a medicação aguda foi tomada (-4,7 para galcanezumabe 120 mg versus. -2,2 para placebo; valor-p nominal < 0,001), e a alteração média em relação ao período basal no domínio de Restrição Funcional do MSQ no mês 3 foi nominalmente maior em pacientes tratados com galcanezumabe 120 mg do que em pacientes com placebo (21,8 para galcanezumabe 120 mg versus 16,8 para placebo; valor-p nominal < 0,001).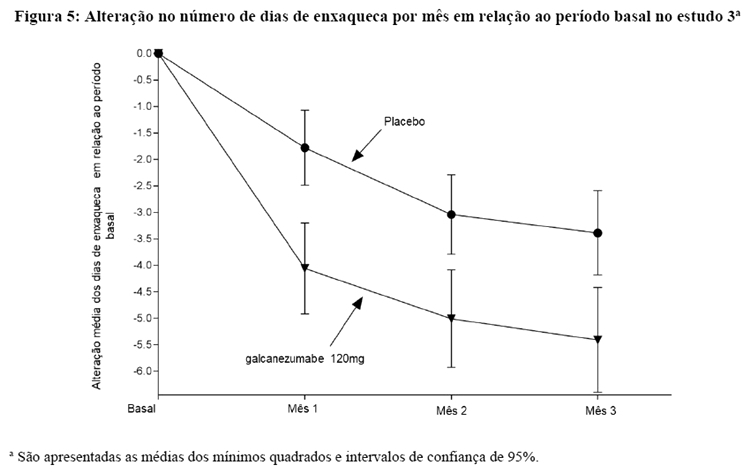
A Figura 6 mostra a distribuição da alteração em relação ao período basal no número médio de dias de enxaqueca por mês durante o período de 3 meses de estudo, em intervalos de 3 dias, por grupo de tratamento. Um benefício do tratamento com galcanezumabe em relação ao placebo é observado ao longo de uma série de alterações nos dias de enxaqueca por mês em relação ao período basal.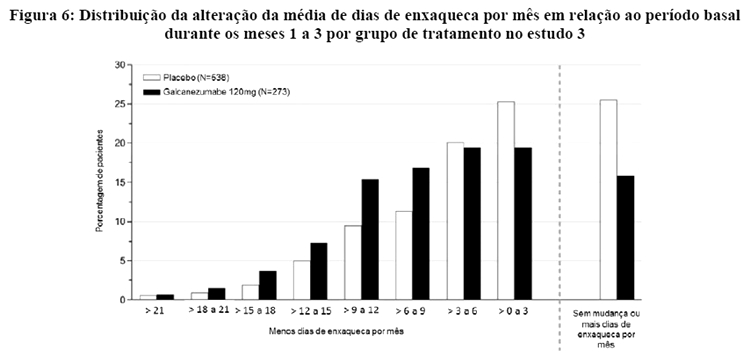
CARACTERÍSTICAS FARMACOLÓGICAS
Descrição: galcanezumabe é um anticorpo monoclonal humanizado de IgG4, que se liga ao peptídeo relacionado ao gene da calcitonina (CGRP) e impede sua atividade biológica sem bloquear o receptor do CGRP. Galcanezumabe é produzido em células de ovário de Hamster Chinês (CHO) por tecnologia de DNA recombinante. Galcanezumabe é composto por duas cadeias kappa leves idênticas de imunoglobulina e duas cadeias gama pesadas idênticas de imunoglobulina, e tem um peso molecular de 144.084 Da, não glicosilado, com ligação dissulfeto.
Propriedades farmacodinâmicas:
Mecanismo de ação: galcanezumabe é um anticorpo monoclonal humanizado de IgG4, que se liga ao peptídeo relacionado ao gene da calcitonina (CGRP) e impede sua atividade biológica, sem bloquear o receptor do CGRP. Concentrações séricas elevadas de CGRP foram associadas à enxaqueca. Além disso, infusões de CGRP podem induzir ataques semelhantes à enxaqueca em alguns indivíduos com histórico de enxaqueca. Galcanezumabe tem como alvo o CGRP e se liga com alta afinidade (KD= 31 pM) e elevada especificidade ( > 10.000 vezes versus os peptídeos relacionados adrenomedulina, amilina, calcitonina e intermedina).
O CGRP é um mediador importante do fluxo sanguíneo cutâneo induzido por capsaicina. Doses únicas de galcanezumabe (75, 200 ou 600 mg) resultaram na atenuação do fluxo sanguíneo cutâneo induzido por capsaicina no dia 3. A administração de galcanezumabe 150 mg a cada 2 semanas durante 6 semanas (total de 4 doses) resultou na inibição do fluxo sanguíneo cutâneo induzido por capsaicina por ao menos 134 dias depois que a última dose foi administrada.
Propriedades farmacocinéticas:
Absorção: com base em uma análise da farmacocinética (PK) da população, após uma dose de ataque de 240 mg, a concentração sérica máxima (Cmáx) de galcanezumabe foi de aproximadamente 30 mg/mL [coeficiente de variação (CV) de 27%]. Doses mensais de 120 mg ou 240 mg alcançaram o steady-state Cmáx (Cmáx,ss) de aproximadamente 28 mg/mL (CV de 35%) ou 54 mg/mL (CV de 31%), respectivamente. O local da injeção não influenciou a absorção de galcanezumabe de modo significativo.
Distribuição: com base em uma análise de PK da população, o volume de distribuição aparente (V/F) de galcanezumabe foi de 7,3 L.
Metabolismo: por ser um anticorpo monoclonal de IgG4 humanizado, espera-se que galcanezumabe seja degradado em pequenos peptídeos e aminoácidos por meio de vias catabólicas, do mesmo modo que a IgG endógena.
Eliminação: com base em uma análise de PK da população, o clearance aparente (CL/F) de galcanezumabe foi de aproximadamente 0,008 L/h e a meia-vida de galcanezumabe foi de 27 dias.
Linearidade da dose: a exposição a galcanezumabe aumenta proporcionalmente com a dose. Com base em uma análise de PK da população que incluiu doses que variaram de 5 - 300 mg, a taxa de absorção, o CL/F e o V/F foram independentes da dose.
Farmacocinética em populações especiais:
Idade, sexo, peso, raça e etnia: não é necessário nenhum ajuste da dose com base na idade, sexo, peso, raça ou etnia, já que não houve efeito clinicamente significativo desses fatores no CL/F ou no V/F de galcanezumabe.
Comprometimento renal ou hepático: não foram realizados estudos de farmacologia clínica específicos para avaliar os efeitos do comprometimento renal e do comprometimento hepático na PK de galcanezumabe. A eliminação renal de anticorpos monoclonais de IgG é baixa. Do mesmo modo, os anticorpos monoclonais de IgG são eliminados principalmente via catabolismo intracelular, e não se espera que o comprometimento hepático influencie no clearance de galcanezumabe. Com base em uma análise de PK da população, a concentração de bilirrubina ou o clearance de creatinina não influenciaram significativamente o CL/F de galcanezumabe.
CONTRAINDICAÇÕES
EMGALITY é contraindicado para pacientes com hipersensibilidade grave conhecida a galcanezumabe ou a qualquer um de seus excipientes.
ADVERTÊNCIAS E PRECAUÇÕES
Hipersensibilidade grave: reações graves de hipersensibilidade, incluindo casos de anafilaxia, angioedema e urticária foram relatados. Se ocorrer uma reação grave de hipersensibilidade, descontinuar EMGALITY imediatamente e iniciar a terapia apropriada. As reações graves de hipersensibilidade podem ocorrer dias após a administração e podem se prolongar.
Imunogenicidade: do mesmo modo que com todas as proteínas terapêuticas, existe o potencial de imunogenicidade. A detecção da formação de anticorpos é altamente dependente da sensibilidade e especificidade do ensaio. Além disso, a incidência observada de positividade para anticorpos (incluindo anticorpos neutralizantes) em um ensaio pode ser influenciada por diversos fatores, incluindo a metodologia do ensaio, o manuseio das amostras, o momento da coleta das amostras, as medicações concomitantes e doença de base. Por essas razões, comparar a incidência de anticorpos contra galcanezumabe à incidência de anticorpos contra outros produtos pode induzir ao erro. A presença de anticorpos anti-medicamento não afetou a farmacocinética, eficácia ou segurança de galcanezumabe. Em 12 meses de tratamento, até 12,5% dos pacientes tratados com galcanezumabe desenvolveram anticorpos anti-medicamento, a maioria dos quais apresentava baixa titulação e resultado positivo para atividade neutralizante in vitro.
Toxicologia não-clínica:
Carcinogênese e mutagênese: estudos não clínicos não foram realizados para avaliar o potencial carcinogênico ou mutagênico de galcanezumabe. Não há evidências que indiquem que o tratamento crônico com galcanezumabe aumentaria o risco de carcinogênese com base em dados de estudos de farmacologia e de toxicologia crônica com galcanezumabe, bem como em uma avaliação da literatura a respeito de CGRP.
Comprometimento da fertilidade: não foram observados efeitos nos parâmetros de fertilidade, tais como órgãos reprodutores, cio, análise do esperma ou acasalamento e fertilidade, em ratos que receberam galcanezumabe em doses subcutâneas de 250 mg/Kg (exposições de 4 a 20 vezes a dose humana máxima recomendada).
Uso durante a gestação, lactação e em pessoas com potencial reprodutivo:
Uso durante a gestação (categoria B): estudos de toxicidade no desenvolvimento realizados em coelhas e ratas prenhes não revelaram nenhuma evidência de dano ao feto. Há dados insuficientes em humanos para estabelecer a segurança de galcanezumabe durante a gestação. Sabe-se que a IgG humana atravessa a barreira placentária. Desse modo, galcanezumabe pode ser transmitido da mãe para o feto em desenvolvimento. EMGALITY deve ser utilizado na gestação somente se o benefício potencial justificar o possível risco à mãe ou ao feto.
Uso durante a lactação: não há dados sobre a presença de galcanezumabe no leite humano, sobre os efeitos no bebê amamentado ou sobre os efeitos na produção de leite. Sabe-se que a IgG humana é excretada no leite materno. Desse modo, galcanezumabe pode ser transmitido da mãe para o bebê amamentado. Os benefícios da amamentação para o desenvolvimento e a saúde devem ser considerados, junto à necessidade clínica da mãe por EMGALITY e qualquer potencial efeito adverso no bebê amamentado.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Uso pediátrico: a segurança e a eficácia em pacientes pediátricos menores de 18 anos de idade não foram estabelecidas.
Uso geriátrico: a segurança e a eficácia em pacientes geriátricos maiores de 65 anos de idade não foram estabelecidas.
Efeitos na capacidade de dirigir e utilizar máquinas: EMGALITY pode ter uma pequena influência sobre a capacidade de conduzir e utilizar máquinas. Vertigem pode ocorrer após a administração de EMGALITY (ver REAÇÕES ADVERSAS).
INTERAÇÕES MEDICAMENTOSAS
Não foi realizado nenhum estudo de interação medicamentosa. Interações medicamentosas farmacocinéticas não são esperadas, com base nas características de EMGALITY.
Nenhum estudo foi conduzido para investigar possível interação entre EMGALITY e plantas medicinais, álcool, nicotina e exames laboratoriais e não laboratoriais.
CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Proteger EMGALITY da luz até a sua utilização. Armazenar refrigerado de 2°C a 8°C. Não congelar. Não agitar.
EMGALITY pode ser armazenado sem refrigeração por até 7 dias, desde que armazenado em temperatura de até 30°C. Se estas condições não forem cumpridas, EMGALITY deve ser descartado.
O prazo de validade de EMGALITY é de 24 meses após a data de fabricação, quando respeitadas as condições de armazenamento acima.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
EMGALITY é uma solução estéril, livre de conservantes, límpida e incolor a ligeiramente amarela.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
POSOLOGIA E MODO DE USAR
Posologia: a dose recomendada é de 120 mg, injetada pela via subcutânea uma vez por mês, com uma dose de ataque de 240 mg como dose inicial.
Orientar os pacientes a aplicar, assim que possível, a dose eventualmente esquecida. Subsequentemente, retomar a administração mensal.
O benefício do tratamento deve ser avaliado em até 3 meses após o seu início. A decisão de continuar o tratamento deverá ser tomada individualmente para cada paciente. Posteriormente, é recomendada a avaliação da necessidade de continuar o tratamento regularmente.
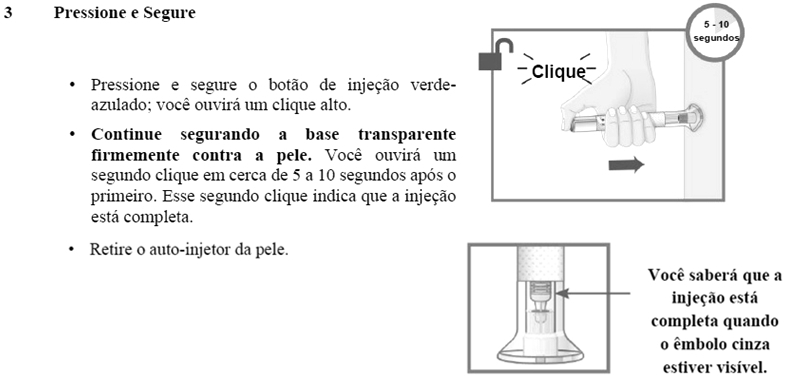
Método de administração: EMGALITY é para administração subcutânea. O paciente pode se auto-injetar EMGALITY seguindo as instruções de uso. Os locais para injeção incluem abdome, coxa, parte posterior do braço e nádegas.
Antes da administração e sempre que a solução e o recipiente permitirem, medicamentos parenterais devem ser inspecionados visualmente quanto a material particulado e descoloração. Não utilizar EMGALITY caso o medicamento esteja turvo ou se houver partículas visíveis.
As INSTRUÇÕES DE USO que acompanham o produto contêm informações mais detalhadas sobre a preparação e administração de EMGALITY. Em caso de problemas de funcionamento do dispositivo injetor de EMGALITY, consulte o folheto informativo e entre em contato com o Lilly SAC 0800 701 0444.


REAÇÕES ADVERSAS
Dados de estudos clínicos: 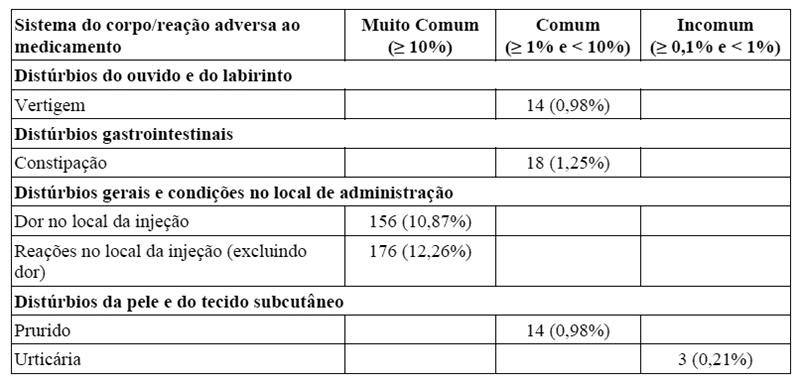
Reações no local da injeção: dor no local da injeção foi o evento mais frequentemente relatado (≥ 10%). Outras reações adversas no local da injeção, relatadas em ≥ 1% dos casos, foram: reação local, eritema, prurido, hematoma e edema. A maioria dos eventos foi de intensidade leve a moderada e não levou à descontinuação de galcanezumabe.
Dados pós-comercialização:
Os seguintes efeitos indesejáveis (reações adversas ao medicamento) são baseados em relatos espontâneos pós-comercialização:
Distúrbios do sistema imunológico:
Reação rara (≥ 0,01% e < 0,1%): anafilaxia e angioedema.
Distúrbios da pele e do tecido subcutâneo:
Reação comum (≥ 1% e < 10%): rash.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificação de Eventos Adversos a Medicamentos - VIGIMED, disponível em http://portal.anvisa.gov.br/vigimed, ou para a Vigilância Sanitária Estadual ou Municipal.
SUPERDOSE
Doses de até 600 mg foram administradas pela via subcutânea em humanos, sem toxicidade limitante da dose. No caso de superdosagem, recomenda-se que o paciente seja monitorado quanto a quaisquer sinais ou sintomas de reações adversas e que o tratamento sintomático apropriado seja instituído imediatamente.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
Registro MS - 1.1260.0200
Venda sob prescrição médica.