ELREXFIO
PFIZER
elranatamabe
Antineoplásico.
Apresentações.
Elrexfio® 44 mg (40 mg/mL) em embalagem contendo 1 frasco-ampola com 1,1 mL de solução injetável. Elrexfio® 76 mg (40 mg/mL) em embalagem contendo 1 frasco-ampola com 1,9 mL de solução injetável.
VIA DE ADMINISTRAÇÃO: VIA SUBCUTÂNEA
USO ADULTO
Composição.
Cada frasco-ampola de Elrexfio® com 1,1 mL contém o equivalente a 44 mg (40 mg/mL) de elranatamabe. Cada frasco-ampola de Elrexfio® com 1,9 mL contém o equivalente a 76 mg (40 mg/mL) de elranatamabe. Excipientes: sacarose, histidina, edetato dissódico di-hidratado, polissorbato 80, cloridrato de histidina monoidratado e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Elrexfio® é indicado como monoterapia para o tratamento de pacientes adultos com mieloma múltiplo recidivante ou refratário, que receberam pelo menos três terapias prévias, incluindo um inibidor de proteassoma, um agente imunomodulador e um anticorpo monoclonal anti-CD38, e que demonstraram progressão da doença na última terapia.
Esta indicação foi aprovada com base em resultados de eficácia do Estudo de Fase 2 (MagnetismMM-3), com desfechos de taxa de resposta objetiva (TRO) e duração da resposta (DdR) (ver item 2. Resultados de Eficácia). A manutenção da aprovação para esta indicação, depende da verificação e descrição do benefício clínico em estudos confirmatórios.
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
• Mieloma múltiplo recidivante ou refratário
A eficácia da monoterapia com Elrexfio® foi avaliada em pacientes com mieloma múltiplo recidivante ou refratário em um estudo aberto, não randomizado, multicêntrico, Fase 2 (MagnetisMM-3). O estudo incluiu pacientes refratários a pelo menos um inibidor de proteossoma (IP), um agente imunomodulador (IMiD) e um anticorpo monoclonal anti-CD38. MagnetisMM-3 incluiu 123 pacientes virgens de terapia prévia direcionada ao BCMA (Coorte A central) e 64 pacientes com anticorpo droga conjugado (ADC) prévio direcionado a BCMA ou terapia de células T de receptor de antígeno quimérico (CAR-T) (Coorte B de apoio). Os pacientes apresentavam doença mensurável pelos critérios do International Myeloma Working Group (IMWG) na inclusão. O estudo incluiu pacientes com pontuação ECOG ≤2, medula óssea adequada na avaliação inicial (contagem absoluta de neutrófilos ≥1,0 x 109/L, contagem de plaquetas ≥25 x 109/L, nível de hemoglobina ≥8 g/dL), função renal (CrCL ≥30 mL/min) e hepática (AST e ALT ≤2,5 x LSN, bilirrubina total ≤2 x LSN) e fração de ejeção do ventrículo esquerdo ≥40%. Pacientes com transplante de células-tronco dentro de 12 semanas antes da inclusão e infecções ativas foram excluídos do estudo.
Os pacientes elegíveis receberam administração subcutânea de Elrexfio® em doses de aumento gradual de 12 mg no Dia 1 e 32 mg no Dia 4 de tratamento, seguidas pela primeira dose de tratamento completo de Elrexfio® (76 mg) no Dia 8 de tratamento. A partir de então, os pacientes receberam 76 mg uma vez por semana. Após 24 semanas, em pacientes que alcançaram uma categoria de resposta IMWG de PR ou melhor, com respostas persistindo por pelo menos 2 meses, o intervalo da dose foi alterado de semanal para a cada 2 semanas (vide item 8. Posologia e Modo de Usar).
Entre os 123 pacientes tratados na Coorte A central, a idade mediana foi 68 (faixa de variação: 36 a 89) anos, com 20% dos pacientes ≥75 anos de idade. 44,7% eram do sexo feminino; 58,5% eram brancos, 13,0% asiáticos, 8,9% hispânicos/latinos e 7,3% negros. O estágio da doença (R-ISS) na entrada no estudo foi 22,8% no Estágio I, 55,3% no Estágio II e 15,4% no Estágio III. O tempo mediano desde o diagnóstico inicial de mieloma múltiplo até a inclusão foi 72,9 (faixa de variação: 16 a 228) meses. Os pacientes receberam uma mediana de 5 linhas de terapia prévias (faixa de variação: 2 a 22); com 96,0% que haviam recebido ≥3 linhas de terapia prévias. 96,7% eram triplo-refratários e 95,9% refratários à última linha de terapia. 68,3% receberam transplante autólogo de células-tronco e 5,7% receberam transplante alogênico de células-tronco prévio. Citogenética de alto risco (t(4;14), t(14;16) ou del(17p)) estava presente em 25,2% dos pacientes. 31,7% dos pacientes apresentavam doença extramedular (presença de qualquer plasmocitoma (extramedular e/ou paramedular)) com um componente de tecido mole) na avaliação inicial, de acordo com a Revisão Central Cega Independente (BICR).
Os resultados de eficácia foram baseados na taxa de resposta e na duração da resposta (DDR), conforme avaliados pela BICR com base nos critérios do IMWG. Os resultados de eficácia da Coorte A central são mostrados na Tabela 1. O acompanhamento mediano (faixa de variação) para respondedores foi 10,9 (3,6 -20,1) meses.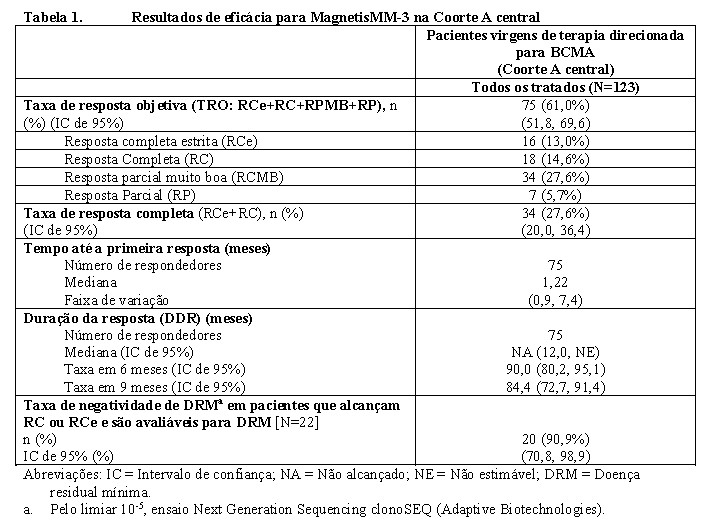
Entre os 64 pacientes tratados na Coorte B de apoio (pacientes expostos a BCMA: ADC direcionada a BCMA e/ou terapia com células CAR-T), a idade mediana foi 67 (faixa de variação: 41 a 84) anos, com 18,8% dos pacientes ≥75 anos de idade. 53,1% eram do sexo feminino; 68,8% eram brancos, 10,9% eram hispânicos/latinos, 3,1% eram negros e 1,6% eram asiáticos. O estágio da doença (R-ISS) na entrada no estudo foi 17,2% no Estágio I, 56,3% no Estágio II e 23,4% no Estágio III. O tempo mediano desde o diagnóstico inicial de mieloma múltiplo até a inclusão foi 102,6 (faixa de variação: 23 a 219) meses. Os pacientes receberam uma mediana de 7,5 linhas de terapia prévias (faixa de variação: 3 a 19); 96,9% eram triplo-refratários e 51,6% eram penta-refratários (refratários a pelo menos 2 PIs, 2 IMiDs e 1 anticorpo anti-CD38); 87,5% eram refratários à última linha de terapia. 71,9% e 32,8% receberam terapia prévia com ADC e CAR-T, respectivamente. 82,8% receberam transplante autólogo de células-tronco e 3,1% receberam transplante alogênico de células-tronco prévio. Citogenética de alto risco (t(4;14), t(14;16) ou del(17p)) estava presente em 20,3% dos pacientes. 57,8% dos pacientes apresentavam doença extramedular na avaliação inicial, por BICR.
Os resultados de eficácia na Coorte B de apoio incluem TRO confirmada por BICR de 34,4% (IC de 95%: 22,9, 47,3); 7,8% dos pacientes alcançaram RC ou melhor e 32,8% alcançaram RPMB ou melhor. A TTR mediana foi 1,92 (faixa de variação: 0,92; 6,74) meses. Após um acompanhamento mediano (faixa de variação) de 10,2 (6,4, 12,3) meses nos respondedores, a DDR mediana não foi alcançada e a taxa de DDR de Kaplan Meier foi 85,1% (ICde 95%: 60,5, 95,0) em 6 meses e 85,1% (IC de 95%: 60,5, 95,0) em 9 meses.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Mecanismo de ação
Elrexfio® é um anticorpo biespecífico de envolvimento de células T direcionado ao antígeno de maturação de células B (BCMA) que se liga ao BCMA em células plasmáticas, plasmablastos e células de mieloma múltiplo e CD3-epsilon em células T, levando à citólise seletiva das células que expressam BCMA. A atividade anticancerígena de Elrexfio® envolve o direcionamento terapêutico seletivo e a ativação de células T redirecionadas contra células plasmáticas malignas que expressam BCMA. Elrexfio® ativa células T, causando a liberação de citocinas pró-inflamatórias, resultando em lise de células do mieloma múltiplo.
Efeitos farmacodinâmicos
• Relação de resposta à exposição
As concentrações séricas de citocinas (IL-2, IL-6, IL-8, IL-10, TNF-a e IFN-c) foram medidas antes e depois da administração da dose 1 de aumento gradual, dose 2 de aumento gradual e as primeiras três doses de tratamento completo de Elrexfio®. O tempo da concentração máxima de citocinas geralmente ocorreu durante a administração de aumento gradual e as concentrações continuam a diminuir ao longo do primeiro mês de tratamento.
Propriedades Farmacocinéticas
A Cmáx e a ASCtau de elranatamabe após a primeira dose subcutânea aumentaram de modo proporcional à dose ao longo da faixa de variação de doses avaliada, via administração SC (~ 6 a 76 mg). A razão média de acúmulo após 24 semanas de administração semanal, em relação à primeira dose subcutânea de elranatamabe 76 mg para Cmáx e ASCtau foi 4,8 vezes e 8,0 vezes, respectivamente. A Cmáx, Cvale e ASCtau de elranatamabe são apresentadas na Tabela 2.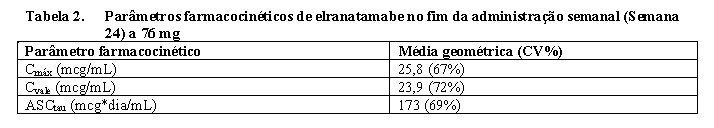
Absorção
A biodisponibilidade média de elranatamabe foi 56,2% quando administrado via subcutânea. O Tmáx mediano após a administração SC de elranatamabe entre todos os níveis de dose variou de 3 a 7 dias para as concentrações séricas total e livre.
Distribuição
O volume central médio (coeficiente de variação [CV]%) de distribuição de elrantamabe foi 4,78 L (69%).
Eliminação
A média geométrica (CV%) da depuração aparente de elranatamabe foi de 0,44 L/dia (69%). É esperado que os pacientes que descontinuarem elranatamabe após a Semana 24 tenham uma redução de 50% da Cmáx em um tempo mediano (5o ao 95o percentil) de 25 (9,6 a 70) dias após o Tmáx e uma redução de 97% da Cmáx em um tempo mediano de 130 (43 a 275) dias após o Tmáx.
Populações especiais
Não foram observadas diferenças clinicamente relevantes na farmacocinética de elranatamabe para idade (36 a 89 anos), sexo (167 homens, 154 mulheres), raça (193 brancos, 49 asiáticos, 29 negros) e peso corporal (37 a 160 kg).
• Insuficiência renal
Não foram realizados estudos formais de Elrexfio® em pacientes com insuficiência renal. Os resultados das análises farmacocinéticas populacionais indicam que a insuficiência renal leve (60 mL/min/1,73 m2 ≤ taxa de filtração glomerular estimada (TFGe) < 90 mL/min/1,73 m2) ou insuficiência renal moderada (30 mL/min/1,73 m2 ≤TFGe < 60 mL/min/1,73 m2) não influenciaram significativamente a farmacocinética de elranatamabe. Dados limitados estão disponíveis para pacientes com insuficiência renal grave (TFGe menor que 30 mL/min/1,73 m2).
• Insuficiência hepática
Não foram realizados estudos formais de Elrexfio® em pacientes com insuficiência hepática. Os resultados das análises farmacocinéticas populacionais indicam que a insuficiência hepática leve (bilirrubina total > 1 a 1,5 vezes o limite superior da normalidade (LSN) e qualquer aspartato aminotransferase (AST) ou bilirrubina total ≤LSN e AST > LSN) não influenciaram significativamente a farmacocinética de elranatamabe. Não há dados disponíveis em pacientes com insuficiência hepática moderada (bilirrubina total > 1,5 a 3,0 × LSN e qualquer AST) ou grave (bilirrubina total > 3,0 × LSN e qualquer AST).
Dados de segurança pré-clínicos
Carcinogenicidade e mutagenicidade
Não foram realizados estudos em animais para avaliar o potencial carcinogênico e genotóxico de elranatamabe.
Toxicologia reprodutiva e fertilidade
Não foram realizados estudos em animais para avaliar os efeitos de elranatamabe na fertilidade, reprodução e
desenvolvimento fetal. Em um estudo de toxicidade de dose repetida de 13 semanas em macacos-cinomolgos sexualmente maduros, não houve efeitos adversos nos órgãos reprodutores masculinos e femininos após doses subcutâneas de até 6 mg/kg/semana (aproximadamente 6,5 vezes a dose humana máxima recomendada, com base na exposição ASC) via subcutânea.
4. CONTRAINDICAÇÕES
Elrexfio® é contraindicado a pacientes com hipersensibilidade à elranatamabe ou a qualquer componente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Síndrome de liberação de citocinas (CRS)
A CRS, incluindo reações potencialmente fatais ou fatais, pode ocorrer em pacientes tratados com Elrexfio®.
Os sinais e sintomas clínicos da CRS podem incluir, mas não estão limitados à, febre, hipóxia, calafrios, hipotensão, taquicardia, cefaleia e elevação das enzimas hepáticas.
A terapia deve ser iniciada de acordo com o esquema de administração de aumento gradual de Elrexfio® para reduzir o risco de CRS, e os pacientes devem ser monitorados após a administração de Elrexfio® de acordo com isso. Medicamentos pré-tratamento devem ser administrados antes das três primeiras doses de Elrexfio® no esquema de administração para reduzir o risco de CRS (vide item 8. Posologia e Modo de Usar).
Os pacientes devem ser aconselhados a procurar atendimento médico caso ocorram sinais ou sintomas de CRS.
• Manejo da síndrome de liberação de citocinas A CRS deve ser identificada com base na apresentação clínica. Os pacientes devem ser avaliados e tratados para outras causas de febre, hipóxia e hipotensão.
Ao primeiro sinal de CRS, Elrexfio® deve ser suspenso e os pacientes devem ser imediatamente avaliados para hospitalização. A CRS deve ser manejada de acordo com as recomendações da Tabela 3 e um manejo adicional deve ser considerado de acordo com as diretrizes institucionais locais (vide item 8. Posologia e Modo de Usar). A terapia de suporte para CRS (incluindo, mas não limitado a, agentes antipiréticos, apoio com fluido intravenoso, vasopressores, inibidores de IL-6 ou receptor de IL-6, oxigênio suplementar etc.) deve ser administrada conforme apropriado. Testes laboratoriais para monitorar a coagulação intravascular disseminada (CIVD), parâmetros hematológicos, bem como função pulmonar, cardíaca, renal e hepática, devem ser considerados.
Toxicidades neurológicas, incluindo ICANS
Toxicidades neurológicas sérias ou potencialmente fatais, incluindo ICANS, podem ocorrer após o tratamento com Elrexfio®.
Os pacientes devem ser monitorados quanto aos sinais e sintomas de toxicidades neurológicas durante o tratamento com Elrexfio®.
Os pacientes devem ser aconselhados a procurar atendimento médico caso ocorram sinais ou sintomas de toxicidade neurológica.
Devido ao potencial para ICANS, os pacientes devem ser aconselhados a não dirigir ou operar máquinas pesadas ou potencialmente perigosas durante o esquema de administração de aumento gradual de Elrexfio® e por 48 horas após concluir cada uma das 2 doses de aumento gradual dentro do esquema de administração de Elrexfio®, e no caso de novo início de quaisquer sintomas neurológicos (vide item 8. Posologia e Modo de Usar e item 5. Advertências e Precauções - Efeitos na capacidade de dirigir e operar máquinas).
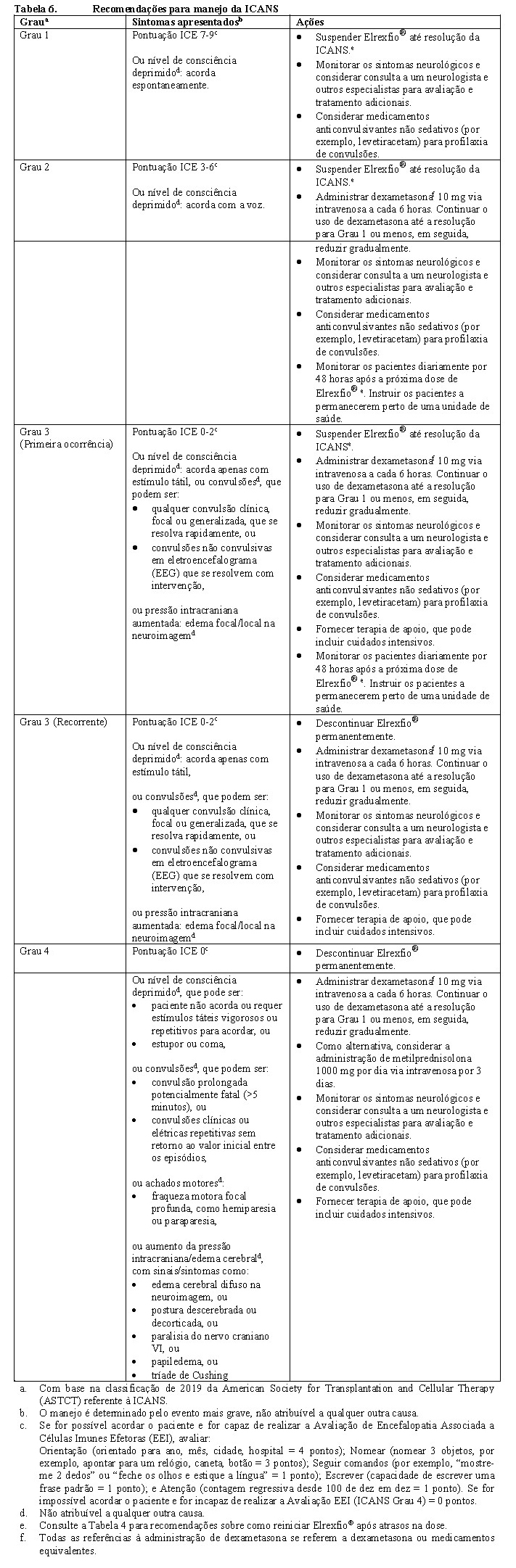
• Manejo de toxicidades neurológicas, incluindo ICANS Ao primeiro sinal de toxicidade neurológica, incluindo ICANS, Elrexfio® deve ser suspenso e a avaliação neurológica considerada. Outras causas de sintomas neurológicos devem ser descartadas. Os pacientes devem ser imediatamente avaliados e tratados com base na gravidade. Deve ser fornecida terapia de apoio, que pode incluir cuidados intensivos, para toxicidades neurológicas graves ou potencialmente fatais. O manejo geral para a toxicidade neurológica (por exemplo, ICANS) está resumido na Tabela 4 (vide item 8. Posologia e Modo de Usar). Os pacientes que apresentarem ICANS Grau 2 ou mais alto com a dose prévia de Elrexfio® devem ser instruídos a permanecer nas proximidades de uma unidade de saúde e ser monitorados quanto a sinais e sintomas diariamente por 48 horas após a próxima dose.
Infecções
Infecções graves, potencialmente fatais ou fatais foram relatadas em pacientes tratados com Elrexfio® (vide item 9. Reações Adversas).
O tratamento com Elrexfio® não deve ser iniciado em pacientes com infecções ativas. Os pacientes devem ser monitorados quanto a sinais e sintomas de infecção antes e durante o tratamento com Elrexfio® e tratados adequadamente. Elrexfio® deve ser suspenso com base na gravidade, conforme indicado na Tabela 5 (vide item 8. Posologia e Modo de Usar). Antimicrobianos e antivirais profiláticos devem ser administrados de acordo com as diretrizes institucionais locais. O tratamento com imunoglobulina subcutânea ou intravenosa (IVIG) deve ser considerado, conforme apropriado.
Neutropenia
Neutropenia e neutropenia febril foram relatadas em pacientes tratados com Elrexfio® (vide item 9. Reações Adversas).
As contagens completas de células sanguíneas devem ser monitoradas na avaliação inicial e periodicamente durante o tratamento. A terapia de apoio deve ser fornecida de acordo com as diretrizes da instituição local. Os pacientes com neutropenia devem ser monitorados quanto a sinais de infecção.
O tratamento com Elrexfio® deve ser suspenso conforme indicado na Tabela 5 (vide item 8. Posologia e Modo de Usar).
Hipogamaglobulinemia
Hipogamaglobulinemia foi relatada em pacientes tratados com Elrexfio® (vide item 9. Reações Adversas).
Os níveis de imunoglobulina devem ser monitorados durante o tratamento com Elrexfio®. A terapia subcutânea ou intravenosa ou imunoglobulina (IVIG) deve ser considerada se os níveis de IgG caírem abaixo de 400 mg/dL e os pacientes devem ser tratados de acordo com as diretrizes institucionais locais, incluindo precauções contra infecções e profilaxia antimicrobiana.
Uso concomitante de vacinas virais vivas
A segurança da imunização com vacinas virais vivas durante ou após o tratamento com Elrexfio® não foi estudada. A vacinação com vacinas virais vivas não é recomendada dentro de 4 semanas antes da primeira dose de Elrexfio® e durante o tratamento com Elrexfio®.
Fertilidade, gravidez e lactação
Mulheres com capacidade de engravidar/Contracepção em homens e mulheres
Mulheres com capacidade de engravidar devem usar contracepção efetiva durante o tratamento com Elrexfio® e por 5 meses após a última dose.
Gravidez
Não há dados em humanos ou animais para avaliar o risco do uso de elranatamabe durante a gravidez. Sabe-se que a imunoglobulina humana (IgG) atravessa a placenta após o primeiro trimestre da gravidez. Com base no mecanismo de ação, elranatamabe pode causar danos fetais quando administrado a mulheres grávidas e, portanto, Elrexfio® não é recomendado para uso durante a gravidez.
Elrexfio® está associado à hipogamaglobulinemia e, portanto, a avaliação dos níveis de imunoglobulina em recém-nascidos de mães tratadas com Elrexfio® deve ser considerada.
O estado de gravidez de mulheres com capacidade de engravidar deve ser verificado antes de iniciar o tratamento com Elrexfio®.
Elrexfio® é um medicamento classificado na categoria C de risco de gravidez. Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgiãodentista.
Lactação
Não se sabe se elranatamabe é excretado no leite humano ou animal, afeta os lactentes amamentados ou afeta a produção de leite. As IgGs humanas são conhecidas por serem excretadas no leite materno. Não pode ser excluído um risco para a criança amamentada e, portanto, a amamentação não é recomendada durante o tratamento com Elrexfio® e por 5 meses após a última dose.
O uso deste medicamento no período da lactação depende da avaliação e acompanhamento do seu médico ou cirurgião-dentista.
Fertilidade
Não há dados sobre o efeito de elranatamabe na fertilidade humana. Os efeitos de elranatamabe na fertilidade masculina e feminina não foram avaliados em estudos com animais.
Efeitos na Habilidade de Dirigir e Operar Máquinas
Elrexfio® pode ter grande influência na habilidade de dirigir e operar máquinas.
Devido ao potencial para ICANS, os pacientes que recebem Elrexfio® estão em risco de nível de consciência deprimido (vide item 9. Reações Adversas). Os pacientes devem ser instruídos a abster-se de dirigir ou operar máquinas pesadas ou potencialmente perigosas durante e por 48 horas após concluir cada uma das 2 doses de aumento gradual dentro do esquema de administração de Elrexfio®, e no caso de novo início de toxicidade neurológica até a resolução de qualquer sintoma neurológico (vide item 8. Posologia e Modo de Usar).
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo de interação foi realizado com Elrexfio®.
A liberação inicial de citocinas associada ao início de Elrexfio® pode suprimir as enzimas do citocromo P450 (CYP). É esperado que o risco mais alto de interação ocorra durante o esquema de administração de aumento gradual para Elrexfio® e até 7 dias após a CRS. Durante esse período, a toxicidade ou as concentrações do medicamento (por exemplo, ciclosporina) devem ser monitoradas em pacientes que estão recebendo concomitantemente substratos sensíveis de CYP com um índice terapêutico restrito. A dose do medicamento concomitante deve ser ajustada conforme necessário.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Elrexfio® deve ser armazenado em geladeira (de 2 °C a 8 °C). Manter na embalagem original para proteger da luz.
Não congelar. Não agitar.
Pode ser utilizado por 24 meses a partir da data de fabricação.
Uma vez perfurado, o frasco-ampola e a seringa de administração devem ser usados imediatamente. Do ponto de vista físico-químico, se a seringa de administração preparada não for usada imediatamente, poderá ser armazenada entre 2 e 30°C no máximo por 24 horas. Quanto ao aspecto microbiológico, a responsabilidade é do profissional de saúde que manipulará o medicamento. Elrexfio® está disponível como um frasco-ampola de dose única. Qualquer solução remanescente no frasco-ampola deve ser descartada após a retirada única.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características físicas e organolépticas: solução líquida transparente a ligeiramente opalescente, incolor a acastanhada pálida, com um pH de 5,8 e osmolaridade de aproximadamente 301 mOsm/L (solução para injeção de 40 mg/mL).
8. POSOLOGIA E MODO DE USAR
O tratamento com Elrexfio® deve ser iniciado e supervisionado por médicos experientes no tratamento de mieloma múltiplo.
Elrexfio® deve ser administrado por um profissional de saúde com pessoal médico adequadamente treinado e equipamento médico adequado para manejar reações graves, incluindo síndrome de liberação de citocinas (CRS) e síndrome de neurotoxicidade associada a células imunes efetoras (ICANS) (vide item 5. Advertências e Precauções).
Posologia
• Esquema de administração recomendado
O esquema de administração recomendado para Elrexfio® é fornecido na Tabela 3. As doses recomendadas de Elrexfio® por injeção subcutânea (SC) são doses de aumento gradual de 12 mg no Dia 1 e 32 mg no Dia 4, seguidas por uma dose de tratamento completo de 76 mg semanalmente, da Semana 2 até a Semana 24.
Para pacientes que receberam pelo menos 24 semanas de tratamento com Elrexfio® e alcançaram uma resposta, o intervalo entre as doses deve ser transferido para um esquema a cada duas semanas.
O tratamento com Elrexfio® deve ser continuado até progressão da doença ou toxicidade inaceitável.
Medicamentos pré-tratamento devem ser administrados antes das três primeiras doses de Elrexfio® no esquema de administração, que inclui a dose 1 de aumento gradual (12 mg), a dose 2 de aumento gradual (32 mg) e a primeira dose de tratamento completo (76 mg), conforme descrito na Tabela 3 (consulte abaixo).
Elrexfio® deve ser administrado por via subcutânea de acordo com o esquema de administração com aumento gradual na Tabela 3 para reduzir a incidência e a gravidade da CRS e da ICANS. Devido ao risco de CRS e ICANS, os pacientes devem ser monitorados quanto a sinais e sintomas por 48 horas após a administração de cada uma das 2 doses de aumento gradual dentro do esquema de administração do Elrexfio®, e instruídos a permanecer perto de uma unidade de saúde (vide item 5. Advertências e Precauções).
• Doses esquecidas Se uma dose de Elrexfio® for perdida, deverá ser administrada assim que possível, e o esquema de administração deverá ser ajustado conforme necessário para manter o intervalo entre as doses (consulte a Tabela 3).
• Medicamentos pré-tratamento recomendados Os seguintes medicamentos pré-tratamento devem ser administrados aproximadamente 1 hora antes das três primeiras doses de Elrexfio® no esquema de administração, que inclui a dose 1 de aumento gradual, a dose 2 de aumento gradual e a primeira dose de tratamento completo, conforme descrito na Tabela 3, para reduzir o risco de CRS (vide item 5. Advertências Precauções):
• paracetamol 500 mg via oral (ou equivalente)
• dexametasona 20 mg via oral ou intravenosa (ou equivalente)
• difenidramina 25 mg via oral (ou equivalente)
Reiniciar Elrexfio® após atraso na dose
Se uma dose de Elrexfio® for atrasada, a terapia deverá ser reiniciada com base nas recomendações listadas na Tabela 4, e o Elrexfio® reiniciado de acordo com o esquema de administração (consulte a Tabela 3). Medicamentos pré-tratamento devem ser administrados conforme indicado na Tabela 4.
Modificações de dose para Elrexfio®
Não são recomendadas reduções de dose de Elrexfio®.
Atrasos na dose podem ser exigidos para manejar as toxicidades relacionadas ao Elrexfio® (vide item 5. Advertências e Precauções). As recomendações para reiniciar Elrexfio® após um atraso na dose são fornecidas na Tabela 4.
Consulte as Tabelas 5 e 6 para as ações recomendadas para as reações adversas de CRS e ICANS, respectivamente. Consulte a Tabela 7 para as ações recomendadas para outras reações adversas após a administração de Elrexfio®.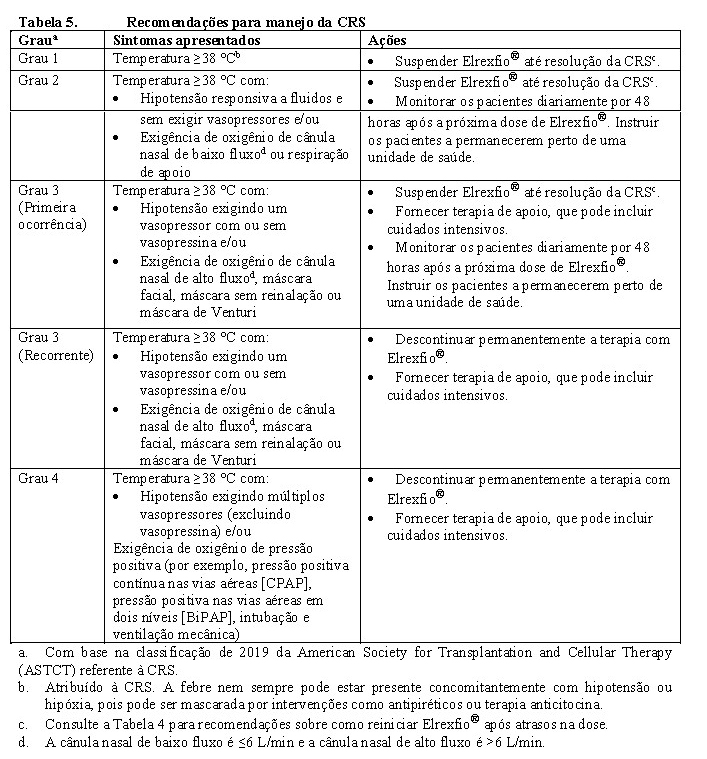
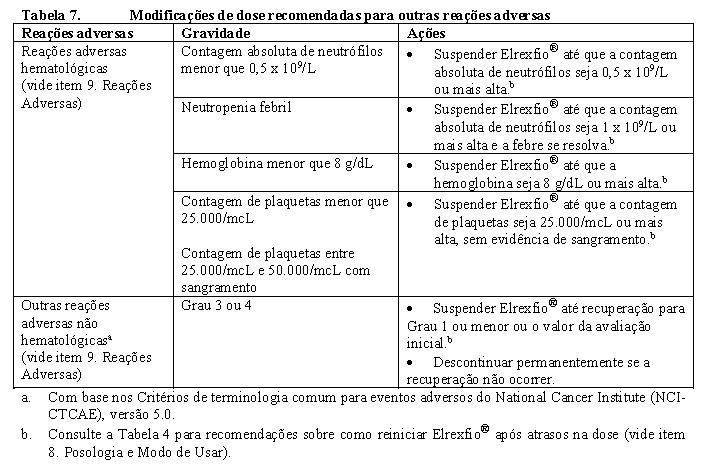
Populações especiais
• Idosos (65 anos de idade ou mais) Nenhum ajuste de dose é necessário (vide item 3. Características Farmacológicas - Propriedades Farmacodinâmicas e Propriedades Farmacocinéticas).
• Insuficiência renal Nenhum ajuste de dose é recomendado para pacientes com insuficiência renal leve a moderada. Elrexfio® não foi estudado em pacientes com insuficiência renal grave (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas).
• Insuficiência hepática Nenhum ajuste de dose é exigido para insuficiência hepática leve. Os efeitos da insuficiência hepática moderada a grave na farmacocinética de elranatamabe não foram estudados (vide item 3. Características Farmacológicas - Propriedades Farmacocinéticas).
• População pediátrica Não há uso relevante de Elrexfio® na população pediátrica (abaixo de 18 anos de idade) para o tratamento de mieloma.
Método de administração
Elrexfio® destina-se apenas para injeção subcutânea.
Para instruções sobre o manuseio do medicamento antes da administração, vide item abaixo.
Precauções especiais para descarte e manuseio
Elrexfio® destina-se ao uso subcutâneo apenas por um profissional de saúde.
Elrexfio® deve ser administrado por um profissional de saúde com pessoal médico adequado e equipamento médico adequado para manejar reações graves, incluindo CRS e toxicidade neurológica, incluindo ICANS (vide item 5. Advertências e Precauções).
O frasco-ampola de Elrexfio® 76 mg/1,9 mL (40 mg/mL) e o frasco-ampola de 44 mg/1,1 mL (40 mg/mL) são fornecidos como solução pronta para uso, que não precisa de diluição antes da administração.
Elrexfio® é uma solução líquida transparente a ligeiramente opalescente, incolor a acastanhada pálida. Elrexfio® deve ser inspecionado visualmente para detectar matéria particulada e descoloração antes da administração, sempre que a solução e o recipiente permitirem. A solução não deverá ser administrada se estiver descolorida ou contiver material particulado.
Técnica asséptica deve ser usada para preparar e administrar Elrexfio®.
Instruções de preparação
Os frascos-ampola de Elrexfio® são de dose única e não contêm conservantes.
Elrexfio® deve ser preparado seguindo as instruções abaixo (consulte a Tabela 8), dependendo da dose exigida. É sugerido usar um frasco-ampola de dose única de 44 mg/1,1 mL (40 mg/mL) para a dose 1 de aumento gradual ou a dose 2 de aumento gradual.
Uma vez perfurado, o frasco-ampola e a seringa de administração devem ser usados imediatamente. Se a seringa de administração preparada não for usada imediatamente, armazene a seringa entre 2 e 30°C no máximo por 24 horas. Elrexfio® está disponível como um frasco-ampola de dose única. Qualquer solução remanescente no frasco-ampola deve ser descartada após a retirada única.
Instruções de administração
Elrexfio® deve ser administrado por um profissional de saúde.
A dose exigida de Elrexfio® deve ser injetada no tecido subcutâneo do abdômen (local de injeção preferencial). Como alternativa, Elrexfio® pode ser injetado no tecido subcutâneo em outros locais (por exemplo, coxa).
Compatibilidade
Na ausência de estudos de compatibilidade, este medicamento não deve ser misturado com outros.
Descarte
O frasco-ampola e qualquer conteúdo remanescente após a retirada de uma dose única devem ser descartados. Os medicamentos não utilizados ou os resíduos devem ser descartados de acordo com as exigências locais.
9. REAÇÕES ADVERSAS
Resumo do perfil de segurança
A segurança de Elrexfio® foi avaliada no MagnetisMM-3 (vide item 3. Características Farmacológicas - Propriedades Farmacodinâmicas), que incluiu 183 pacientes adultos com mieloma múltiplo que receberam o esquema de administração recomendado de Elrexfio®. A duração mediana do tratamento com Elrexfio® foi 4,1 (faixa de variação: 0,03 a 14,9) meses.
As reações adversas mais frequentes de qualquer grau nos pacientes foram: CRS (57,9%), anemia (53,6%), neutropenia (44,3%), fadiga (42,6%), diarreia (35,5%), trombocitopenia (35,0%), linfopenia (29,5%), diminuição do apetite (26,2%), erupção cutânea (25,7%), artralgia (21,9%), náusea (21,3%), hipocalemia (21,3%), pirexia (21,3%), reação no local da injeção (21,3%) e pele seca (20,8%).
Reações adversas sérias foram relatadas em 57,9% dos pacientes que receberam Elrexfio®, incluindo pneumonia (25,1%), sepse (13,1%), CRS (12,6%), anemia (5,5%), infecção das vias aéreas superiores (4,4%), infecção do trato urinário (3,3%), dispneia (2,2%) e neutropenia febril (2,2%).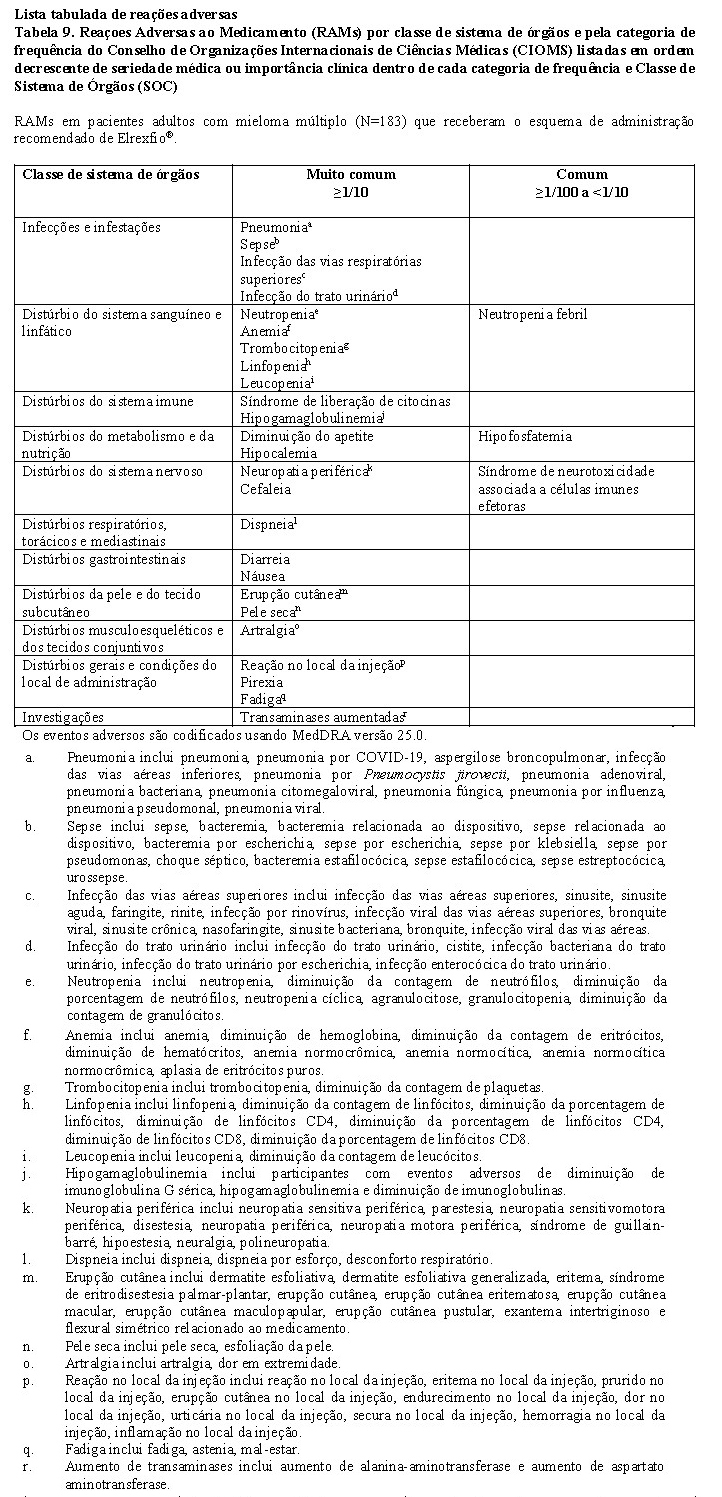
Descrição das reações adversas selecionadas
• Síndrome de liberação de citocinas (CRS) A CRS ocorreu em 57,9% dos pacientes que receberam Elrexfio® no esquema de administração recomendado (vide item 8. Posologia e Modo de Usar), com CRS Grau 1 em 43,7% dos pacientes, CRS Grau 2 em 13,7% dos pacientes e CRS Grau 3 em 0,5% dos pacientes. A maioria dos pacientes apresentou CRS após a primeira dose de aumento gradual (43,2%) ou a segunda dose de aumento gradual (19,1%), com 7,1% dos pacientes apresentando CRS após a primeira dose de tratamento completo e 1,6% dos pacientes após uma dose subsequente. A CRS recorrente ocorreu em 13,1% dos pacientes. O tempo mediano até o início da CRS foi 2 (faixa de variação: 1 a 9) dias após a dose mais recente, com uma duração mediana de 2 (faixa de variação: 1 a 19) dias.
Entre os pacientes que desenvolveram CRS, os sintomas associados incluíram febre (99,0%), hipóxia (11,4%) e hipotensão (21,0%). Entre os pacientes que receberam Elrexfio® no esquema de dose recomendada, 19,1% receberam tocilizumabe (ou siltuximabe) e 8,7% receberam corticosteroides para tratamento de CRS.
• Síndrome de neurotoxicidade associada a células imunes efetoras (ICANS) ICANS ocorreu em 3,3% dos pacientes após o tratamento com Elrexfio® no esquema de administração recomendado (vide item 8. Posologia e Modo de Usar). A maioria dos pacientes apresentou ICANS após a primeira dose de aumento gradual (2,7%), 1 (0,5%) paciente apresentou ICANS após a segunda dose de aumento gradual e 1 (0,5%) paciente apresentou ICANS após uma dose subsequente. A ICANS recorrente ocorreu em 1,1% dos pacientes. O tempo mediano até o início foi 3 (faixa de variação: 1 a 4) dias após a dose mais recente, com uma duração mediana de 2 (faixa de variação: 1 a 18) dias.
O início da ICANS pode ser concomitante com a CRS, após a resolução da CRS ou na ausência de CRS. Os sintomas de ICANS incluíram um nível deprimido de consciência e alterações Grau 1 ou Grau 2 na pontuação de Encefalopatia Associada a Células Imunes Efetoras (ICE). Entre os pacientes que receberam Elrexfio® no esquema de dose recomendada, 2,2% receberam corticosteroides, 1,1% receberam tocilizumabe (ou siltuximabe) e 0,5% receberam anakinra para tratamento de ICANS.
Imunogenicidade
Durante o tratamento nos dados de segurança agrupados (até 24 meses), 20 dos 240 participantes avaliáveis para imunogenicidade (8,3%) tratados com Elrexfio® na dose recomendada desenvolveram anticorpos antielranatamabe. Não foi identificado qualquer efeito clinicamente significativo de anticorpos antidroga (ADA) na farmacocinética, segurança ou efetividade de elranatamabe.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo ao Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Nenhum participante relatou superdose de elranatamabe no programa de estudo clínico e a dose máxima tolerada não foi determinada. Em estudos clínicos, foram administradas doses de até 76 mg uma vez por semana.
Tratamento
Em caso de superdose, o paciente deve ser monitorado quanto a quaisquer sinais ou sintomas de reações adversas e o tratamento de apoio adequado deve ser instituído imediatamente.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
Registro: 1.2110.0493
VENDA SOB PRESCRIÇÃO
USO RESTRITO A ESTABELECIMENTOS DE SAÚDE