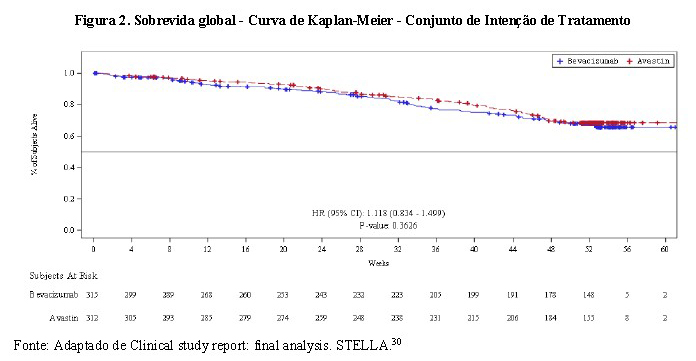
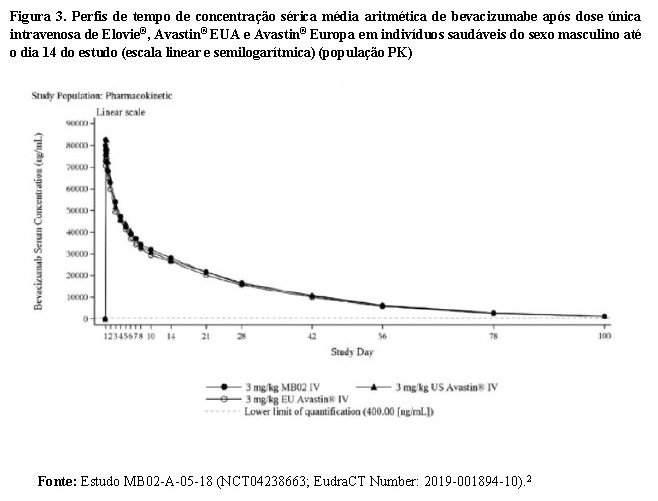
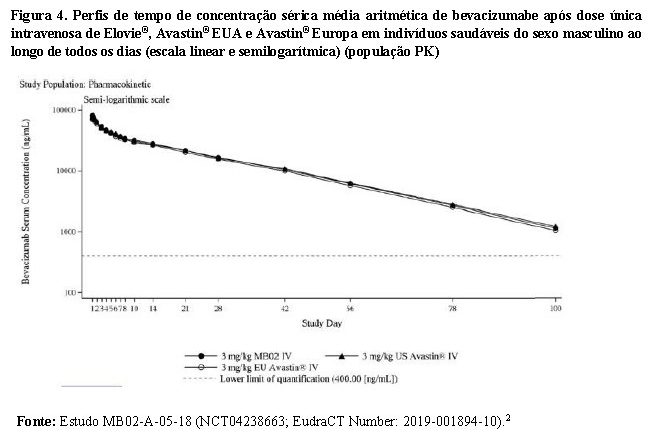
ELOVIE
LIBBS
bevacizumabe
Antineoplásico.
Apresentações.
Solução para diluição para infusão.
Caixa com 1 frasco-ampola de dose única de 100 mg (4 mL) ou 400 mg (16 mL).
VIA INTRAVENOSA
USO ADULTO
Composição.
Elovie® injetável 100 mg
Princípio ativo: bevacizumabe (anticorpo monoclonal anti-VEGF humanizado) 100 mg
(25 mg/mL).
Excipientes: trealose di-hidratada, fosfato de sódio monobásico monoidratado, fosfato de sódio dibásico, polissorbato 20 e água para injetáveis.
Elovie® injetável 400 mg
Princípio ativo: bevacizumabe (anticorpo monoclonal anti-VEGF humanizado) 400 mg
(25 mg/mL).
Excipientes: trealose di-hidratada, fosfato de sódio monobásico monoidratado, fosfato de sódio dibásico, polissorbato 20 e água para injetáveis.
Informações técnicas.
1. INDICAÇÕES
Câncer colorretal metastático (CCRm)
Elovie®, em combinação com quimioterapia à base de fluoropirimidina, é indicado para o tratamento de pacientes com carcinoma colorretal metastático.
Câncer de pulmão de não pequenas células localmente avançado, metastático ou recorrente
Elovie®, em combinação com quimioterapia à base de platina, é indicado para o tratamento de primeira linha de pacientes com câncer de pulmão de não pequenas células, não escamoso, irressecável, localmente avançado, metastático ou recorrente.
Elovie®, em combinação com erlotinibe, é indicado para o tratamento de primeira linha de pacientes com câncer de pulmão de não pequenas células, não escamoso, irressecável, avançado, metastático ou recorrente com mutações ativadoras de EGFR (receptor do fator de crescimento epidérmico).
Câncer de mama metastático ou localmente recorrente (CMM)
Elovie®, em combinação com paclitaxel, é indicado para o tratamento em primeira linha de pacientes com câncer de mama localmente recorrente ou metastático que não tenham recebido quimioterapia prévia para doença metastática ou localmente recorrente.
Elovie®, em combinação com capecitabina, é indicado para o tratamento em primeira linha de pacientes com câncer de mama localmente recorrente ou metastático para os quais o tratamento com outras opções de quimioterapia, incluindo taxanos e antraciclinas, não seja considerado apropriado. Pacientes que tenham recebido regimes de tratamento adjuvante contendo taxanos e antraciclinas nos últimos 12 meses não são elegíveis ao tratamento com Elovie® em combinação com capecitabina.
Câncer de células renais metastático e/ou avançado (mRCC)
Elovie®, em combinação com alfainterferona 2a, é indicado para o tratamento de primeira linha de pacientes com câncer de células renais avançado e/ou metastático.
Câncer epitelial de ovário, tuba uterina e peritoneal primário
Elovie®, em combinação com carboplatina e paclitaxel, é indicado para o tratamento de primeira linha de pacientes com câncer epitelial de ovário, tuba uterina e peritoneal primário avançados (International Federation of Gynecology and Obstetrics - FIGO - III B, III C e IV).
Elovie®, em combinação com carboplatina e gencitabina, é indicado para o tratamento de pacientes adultos com câncer epitelial de ovário, tuba uterina e peritoneal primário com primeira recorrência e sensível à platina, sem terapia prévia com bevacizumabe ou outros inibidores de VEGF ou agentes direcionados a receptores de VEGF.
Elovie®, em combinação com carboplatina e paclitaxel, é indicado para o tratamento de pacientes adultos com câncer epitelial primário de ovário, tuba uterina e peritônio, recorrente e sensível à platina.
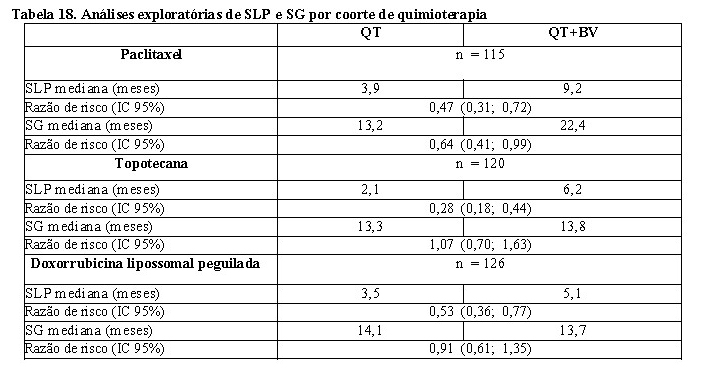
Elovie®, em combinação com paclitaxel, topotecana ou doxorrubicina lipossomal peguilada, é indicado para o tratamento de pacientes com câncer epitelial de ovário, tuba uterina e peritoneal primário, recorrentes e resistentes à platina, que não tenham recebido mais do que dois regimes prévios de quimioterapia e que não receberam terapia prévia com bevacizumabe ou outros inibidores de VEGF ou agentes direcionados a receptores de VEGF.
Câncer de colo do útero
Elovie®, em combinação com paclitaxel e cisplatina ou, alternativamente, paclitaxel e topotecana em pacientes que não podem receber terapia com platina, é indicado para o tratamento de câncer de colo do útero persistente, recorrente ou metastático.
2. RESULTADOS DE EFICÁCIA
O medicamento Elovie® (bevacizumabe) é um medicamento biológico desenvolvido pela via de comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre Elovie® e o medicamento comparador (Avastin®).
Câncer colorretal metastático (CCRm)
A segurança e a eficácia da dose recomendada de Avastin® (5 mg/kg de peso a cada duas semanas), em carcinoma metastático do cólon ou reto, foram observadas em três estudos clínicos randomizados, com controle ativo, avaliando a combinação de Avastin® com quimioterapia à base de fluoropirimidina em primeira linha de câncer colorretal metastático. Avastin® foi combinado com dois esquemas quimioterápicos:
• Estudo AVF2107g: um esquema semanal de irinotecano / 5-fluorouracil e leucovorin em bolus (esquema IFL), durante o total de quatro semanas, dentro de cada ciclo de seis semanas.1
• Estudo AVF0780g: em combinação com 5-fluorouracil / leucovorin (5-FU / LV) em bolus, durante o total de seis semanas, dentro de cada ciclo de oito semanas (esquema Roswell Park).2
• Estudo AVF2192g: em combinação com 5-fluorouracil / leucovorin (5-FU / LV) em bolus, durante o total de seis semanas, dentro de cada ciclo de oito semanas (esquema Roswell Park), em pacientes que não eram candidatos ideais para o tratamento de primeira linha com irinotecano.3
Três estudos clínicos adicionais com Avastin® foram conduzidos para o tratamento de câncer colorretal metastático: primeira linha (NO16966), segunda linha em pacientes que não tenham sido previamente tratados com Avastin® (E3200) e segunda linha em pacientes previamente tratados com Avastin® após a progressão da doença de primeira linha (ML18147). Nesses estudos, Avastin® foi administrado nos regimes a seguir descritos, em combinação com FOLFOX-4 (5-FU / LV / oxaliplatina), XELOX (capecitabina / oxaliplatina), fluoropirimidina / irinotecano e fluoropirimidina / oxaliplatina:
• NO16966: Avastin® 7,5 mg/kg de peso, a cada três semanas, em combinação com capecitabina oral e oxaliplatina intravenosa (XELOX) ou Avastin® 5 mg/kg, a cada duas semanas, em combinação com leucovorin e 5-FU em bolus, seguido de 5-FU infusional com oxaliplatina intravenosa (FOLFOX-4).4
• E3200: Avastin® 10 mg/kg de peso, a cada duas semanas, em combinação com leucovorin e 5-FU em bolus, seguido de 5-FU infusional com oxaliplatina intravenosa (FOLFOX-4), em pacientes que não receberam tratamento prévio com Avastin®.5
• ML18147: Avastin® 5,0 mg/kg de peso a cada duas semanas ou Avastin® 7,5 mg/kg de peso a cada três semanas em combinação com fluoropirimidina / irinotecano ou fluoropirimidina / oxaliplatina em pacientes com progressão da doença após a primeira linha de tratamento com Avastin®. Os regimes contendo irinotecano ou oxaliplatina foram trocados dependendo da utilização em primeira linha de oxaliplatina ou irinotecano21.
AVF2107g: estudo fase III, randomizado, duplo-cego, com controle ativo, que avaliou Avastin® em combinação com IFL como tratamento de primeira linha para carcinoma colorretal metastático.1 Oitocentos e treze (813) pacientes foram randomizados para receber IFL + placebo (Braço 1) ou IFL + Avastin® (5 mg/kg a cada duas semanas, Braço 2). Um terceiro grupo de 110 pacientes recebeu 5-FU em bolus / LV + Avastin® (Braço 3). A inclusão no Braço 3 foi interrompida, conforme predeterminado, depois de estabelecida e considerada aceitável a segurança de Avastin® combinado ao esquema IFL.
O desfecho primário de eficácia do estudo foi a sobrevida global. A adição de Avastin® ao IFL resultou em aumento estatisticamente significativo da sobrevida global, sobrevida livre de progressão (SLP) e taxa de resposta global (Tabela 1). O benefício clínico de Avastin®, medido pela sobrevida, foi observado em todos os subgrupos predeterminados de pacientes, incluindo os definidos por idade, sexo, estado de desempenho (performance status), localização do tumor primário, número de órgãos envolvidos e duração da doença metastática.
Os resultados de eficácia de Avastin® em combinação com quimioterapia IFL estão mostrados na Tabela 1.
Antes da descontinuação do Braço 3 (5-FU / LV + Avastin®), entre os 110 pacientes randomizados, a mediana de sobrevida global foi de 18,3 meses, e a de sobrevida livre de progressão foi de 8,8 meses.
AVF2192g: estudo clínico fase II, randomizado, duplo-cego, com controle ativo, que avaliou os efeitos de Avastin® em combinação com 5-FU / leucovorin como tratamento de primeira linha para câncer colorretal metastático em pacientes que não eram candidatos ideais para o tratamento de primeira linha com irinotecano. Cento e cinco (105) pacientes foram randomizados para 5-FU / LV + placebo, e 104 foram randomizados para 5-FU / LV + Avastin® (5 mg/kg a cada duas semanas). Todos os tratamentos foram mantidos até a progressão da doença.
A adição de Avastin®, 5 mg/kg a cada duas semanas, ao 5-FU / LV resultou em maiores taxas de resposta objetiva, sobrevida livre de progressão significativamente mais prolongada e tendência a sobrevida mais prolongada, em comparação à quimioterapia 5-FU / LV apenas.
NO16966: estudo clínico fase III, randomizado, duplo-cego (para bevacizumabe), que investigou Avastin®, 7,5 mg/kg, em combinação com capecitabina oral e oxaliplatina IV (XELOX), administrado em um esquema de três semanas; ou Avastin®, 5 mg/kg, em combinação com leucovorin e 5-FU em bolus, seguido de 5-FU infusional com oxaliplatina IV (FOLFOX-4), administrado em esquema de duas semanas. Esse estudo continha duas partes: uma inicial, de dois braços (parte I), na qual os pacientes foram divididos em dois braços de tratamento diferentes (XELOX e FOLFOX-4); e outra, subsequente 2x2 fatorial (parte II), na qual os pacientes foram divididos em quatro braços de tratamento (XELOX + placebo, FOLFOX-4 + placebo, XELOX + Avastin®, FOLFOX-4 + Avastin®). Na parte II, a inclusão ao tratamento foi duplo-cego em relação a Avastin®.
Aproximadamente 350 pacientes foram randomizados para cada um dos quatro braços, na parte II, desse estudo.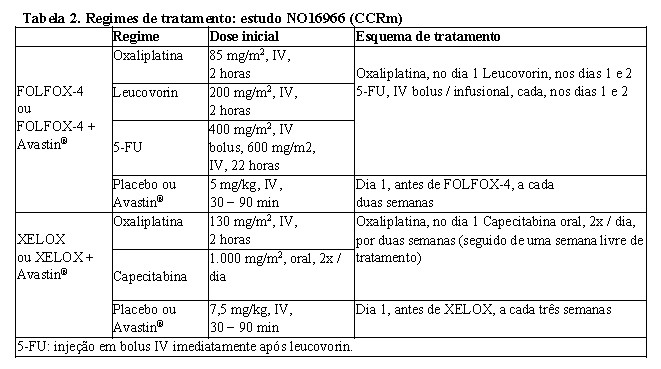
O desfecho primário de eficácia desse estudo foi a duração da sobrevida livre de progressão. Existiam dois desfechos primários: demonstrar que XELOX não era inferior a FOLFOX-4 e que Avastin® em combinação com FOLFOX-4 ou XELOX era superior à quimioterapia somente. Os dois objetivos coprimários foram alcançados:
a) A não inferioridade dos braços que continham XELOX em relação aos braços que continham FOLFOX-4, em comparação global, foi demonstrada em termos de sobrevida livre de progressão e sobrevida global na população elegível por protocolo;
b) A superioridade dos braços que continham Avastin® versus quimioterapia sem adição de Avastin® foi demonstrada em comparação global, em termos de sobrevida livre de progressão, na população com intenção de tratamento (vide Tabela 3).
A análise secundária de SLP, baseada no Comitê de Revisão Independente (CRI) e nas respostas baseadas no subgrupo em tratamento, confirmou, de maneira significativa, o benefício clínico superior para os pacientes tratados com Avastin® (análise de subgrupo na Tabela 3), compatível com o benefício estatisticamente significativo observado na análise combinada.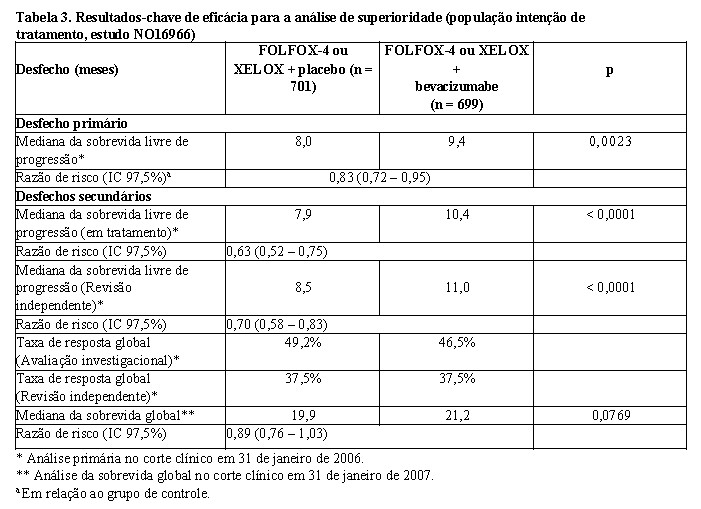
ECOG E3200: estudo clínico fase III, aberto, randomizado, com controle ativo, que investigou Avastin®, 10 mg/kg, em combinação com leucovorin e 5-FU em bolus, seguido de 5-FU infusional com oxaliplatina intravenosa (FOLFOX-4), administrado em um esquema de duas semanas em pacientes previamente tratados (segunda linha) com câncer colorretal avançado. Nos braços de quimioterapia, o regime FOLFOX-4 utilizou as mesmas doses e o mesmo modo de administração do esquema adotado no estudo NO16966, como mostra a Tabela 2.
O desfecho primário de eficácia desse estudo foi a sobrevida global, definida como o tempo entre randomização e óbito por qualquer causa. Oitocentos e vinte e nove (829) pacientes foram selecionados aleatoriamente (292 FOLFOX-4, 293 Avastin® + FOLFOX-4 e 244 monoterapia com Avastin®). A adição de Avastin® a FOLFOX- 4 resultou no prolongamento estatisticamente significativo da sobrevida global. Melhoras estatisticamente significativas em sobrevida livre de progressão e taxa de resposta objetiva também foram observadas (vide Tabela 4).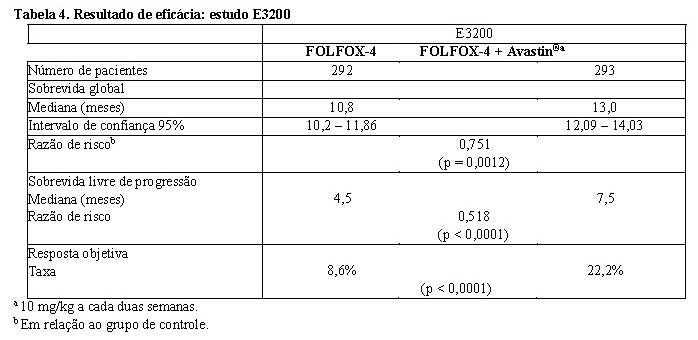
Não foi observada diferença significativa na duração de sobrevida global entre pacientes que receberam Avastin® em monoterapia comparados aos pacientes tratados com FOLFOX-4.
A sobrevida livre de progressão e a taxa de resposta objetiva foram inferiores no braço de Avastin® em monoterapia, em comparação ao braço com FOLFOX-4.
ML1814721: Estudo clínico de fase III, randomizado, controlado, aberto, que investigou Avastin® 5 mg/kg a cada 2 semanas ou 7,5 mg/kg a cada 3 semanas em combinação com quimioterapia à base de fluoropirimidina versus quimioterapia isolada à base de fluoropirimidina em pacientes com câncer colorretal metastático que progrediram após o regime de tratamento de primeira linha contendo Avastin®.
Pacientes com confirmação histológica de câncer colorretal metastático e progressão da doença foram randomizados 1:1 em até 3 meses após a descontinuação do tratamento de primeira linha com Avastin® para receber quimioterapia à base de fluoropirimidina/oxaliplatina ou fluoropirimidina/irinotecano (a quimioterapia foi substituída dependendo da quimioterapia utilizada na primeira linha), com ou sem Avastin®. O tratamento foi mantido até a progressão da doença ou toxicidade inaceitável. O desfecho primário foi a sobrevida global (SG) definido como o tempo desde a randomização até a morte por qualquer causa.
Um total de 820 pacientes foi randomizado. A adição de Avastin® à quimioterapia à base de fluoropirimidina resultou em aumento estatisticamente significativo de sobrevida em pacientes com câncer colorretal metastático que tenham progredido após o regime de tratamento de primeira linha contendo Avastin® (ITT = 819) (vide Tabela 5).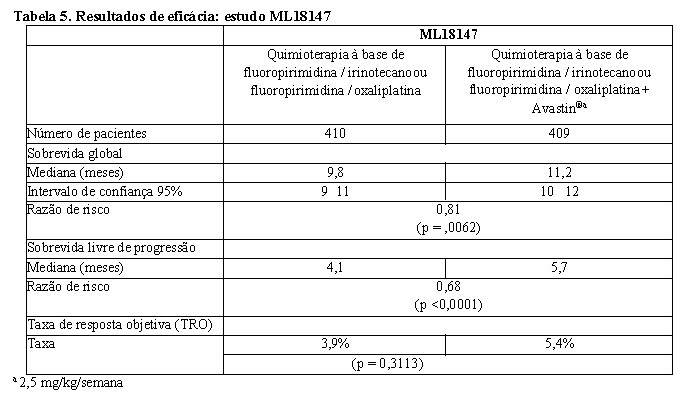
Também foi observada melhora estatisticamente significativa na sobrevida livre de progressão. A taxa de resposta objetiva foi baixa em ambos os braços de tratamento e não atingiu significância estatística.
Câncer de pulmão de não pequenas células localmente avançado, metastático ou recorrente
A segurança e a eficácia de Avastin® no tratamento de primeira linha de pacientes com câncer de pulmão de não pequenas células, excluindo histologia predominantemente escamosa, foram estudadas em associação com quimioterapia à base de platina nos estudos E4599 e BO17704.
E45996: estudo multicêntrico, aberto, randomizado, controlado para avaliação de Avastin® no tratamento de primeira linha de pacientes com câncer de pulmão de não pequenas células localmente avançado, metastático ou recorrente, excluindo histologia predominantemente escamosa.
Os pacientes foram randomizados para receber quimioterapia à base de platina (paclitaxel 200 mg/m2 e carboplatina AUC = 6,0), ambos administrados por infusão IV no dia 1 de cada ciclo de três semanas, até o total de seis ciclos; ou para o braço com carboplatina e paclitaxel em associação com Avastin®, na dose de 15 mg/kg, administrado por infusão IV no dia 1 de cada ciclo de três semanas. Após a conclusão dos seis ciclos de quimioterapia com carboplatina-paclitaxel ou após descontinuação prematura da quimioterapia, os pacientes no braço de Avastin® + carboplatina-paclitaxel continuaram a receber Avastin® em monoterapia a cada três semanas até progressão da doença. Foram randomizados para os dois braços de tratamento 878 pacientes.
Durante o estudo, dos pacientes que receberam medicação de ensaio, 32,2% (136 / 422) receberam 7 - 12 administrações de Avastin® e 21,1% (89/ 422) receberam 13 ou mais administrações de Avastin®. O desfecho primário foi a sobrevida global. Os resultados estão apresentados na Tabela 6.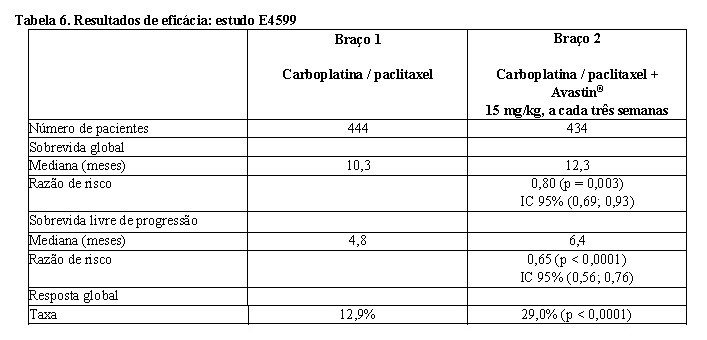
BO177047,8: estudo de fase III, randomizado, duplo-cego, com Avastin® em associação à cisplatina e gencitabina versus placebo, cisplatina e gencitabina, em pacientes com câncer de pulmão de não pequenas células, não escamoso, localmente avançado, metastático ou recorrente e não previamente tratados com quimioterapia. O desfecho primário foi a sobrevida livre de progressão (SLP), e os desfechos secundários incluíram a duração da resposta, sobrevida global e segurança.
Os pacientes foram randomizados para o braço de quimioterapia à base de platina, cisplatina 80 mg/m2, administrada por infusão IV no dia 1, e 1.250 mg/m2 de gencitabina, administrada por infusão IV, nos dias 1 e 8 de cada ciclo, a cada três semanas, até o total de seis ciclos, (CG) com placebo ou para o braço de CG com Avastin®, na dose de 7,5 mg/kg ou 15 mg/kg, administrada por infusão IV, no dia 1 de cada ciclo, a cada três semanas. Nos braços que continham Avastin®, os pacientes receberam Avastin® em monoterapia a cada três semanas até progressão da doença ou toxicidade inaceitável.
Os resultados do estudo mostraram que 94% (277 / 296) dos doentes elegíveis continuaram a receber bevacizumabe em monoterapia no ciclo 7. Uma elevada proporção de pacientes (aproximadamente 62%) continuou a receber uma variedade de terapias antineoplásicas não especificadas no protocolo, e isso pode ter impactado a análise da sobrevida global. Os resultados de eficácia estão apresentados na Tabela 7.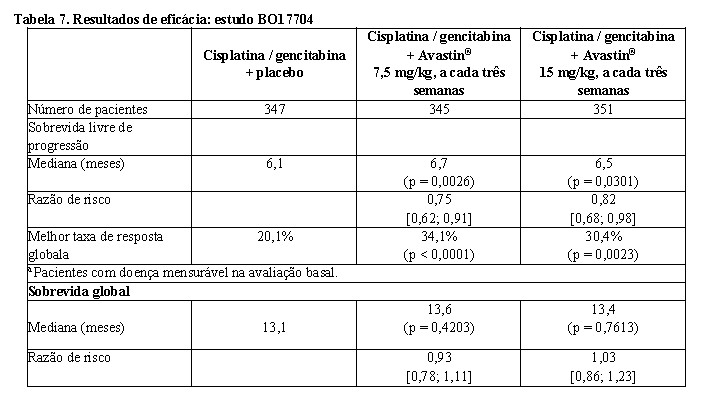
JO2556724
O estudo JO25567 foi um estudo fase II, multicêntrico, aberto, randomizado, conduzido no Japão para avaliar a segurança e eficácia de bevacizumabe utilizado em associação com erlotinibe em pacientes com câncer de pulmão de não pequenas células, não escamoso com mutações ativadoras de EGFR (com deleção do éxon 19 ou mutação L858R do éxon 21), que não receberam terapia sistêmica prévia para o estadio IIIB/IV ou doença recorrente.
O desfecho primário foi a sobrevida livre de progressão (SLP) baseada na avaliação da revisão independente. Os desfechos secundários incluíram sobrevida global, taxa de resposta, taxa de controle da doença, duração da resposta, segurança e qualidade de vida relacionada à saúde (Health Related Quality of Life - HRQoL) baseada no questionário FACT-L (Functional Assessment of Cancer Therapy for Patients with Lung Cancer).
O status da mutação EGFR foi determinado para cada paciente antes da triagem dos pacientes e 154 pacientes foram randomizados para receber ambos erlotinibe + bevacizumabe [150 mg de erlotinibe oral diariamente + bevacizumabe (15 mg/kg IV a cada três semanas)] ou monoterapia de erlotinibe (150 mg oral diariamente) até a progressão da doença (PD) ou toxicidade inaceitável. Na ausência de PD, a descontinuação de um componente do tratamento em estudo no braço erlotinibe + bevacizumabe não levou à descontinuação do outro componente do tratamento em estudo, como especificado no protocolo do estudo. Os resultados de eficácia do estudo estão apresentados na Tabela 8.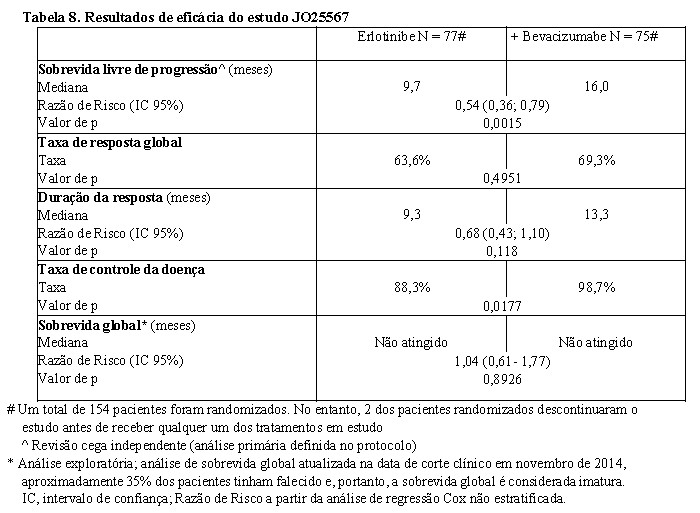
No estudo aberto JO25567, a qualidade de vida relacionada à saúde (Health Related Quality of Life - HRQoL) foi avaliada pelas pontuações FACT-L total, índice de resultado do teste (TOI) e pelos sintomas de câncer de pulmão, como avaliado pela sub-escala de sintomas de câncer de pulmão FACT-L (LCS). Durante o período livre de progressão, as pontuações FACT-L médias da linha de base foram mantidas em ambos braços de tratamento. Não houve diferenças clinicamente significativas na HRQoL FACT-L observadas entre os dois braços de tratamento. É importante notar que pacientes no braço erlotinibe + bevacizumabe foram tratados por mais tempo e receberam administração intravenosa de bevacizumabe, em oposição à monoterapia de erlotinibe oral no braço controle.
Câncer de mama metastático (CMM)9,10
ECOG E2100: estudo clínico multicêntrico, aberto, randomizado, com controle ativo, para avaliação de Avastin® em combinação com paclitaxel para câncer de mama metastático ou localmente recorrente em pacientes que não haviam recebido quimioterapia prévia para doença metastática ou localmente recorrente.
Terapia hormonal prévia para o tratamento de doença metastática foi permitida. Terapia adjuvante prévia com taxano foi permitida apenas se completada, pelo menos, 12 meses antes da inclusão no estudo.
Pacientes foram randomizadas para receber paclitaxel em monoterapia (90 mg/m2, IV, em uma hora, uma vez por semana, por três a cada quatro semanas) ou em combinação com Avastin® (10 mg/kg, infusão IV, a cada duas semanas). As pacientes deveriam permanecer no estudo até a progressão da doença. Nos casos em que a quimioterapia foi descontinuada prematuramente, o tratamento com Avastin® em monoterapia foi mantido até a progressão da doença. O desfecho primário foi a sobrevida livre de progressão (SLP), avaliada pelos investigadores. Adicionalmente, uma revisão, independentemente do desfecho primário, foi realizada. Das 722 pacientes do estudo, a maioria tinha doença HER2 negativa (90%). Um pequeno número de pacientes tinha status HER2 desconhecido (8%) ou positivo (2%). Pacientes status positivo para HER2 haviam sido previamente tratadas com trastuzumabe ou foram consideradas inadequadas para trastuzumabe. A maioria, 65%, tinha recebido quimioterapia adjuvante, incluindo 19% que receberam terapia prévia com taxanos e 49% que receberam terapia prévia com antraciclinas. As características das pacientes eram similares entre os braços de estudo.
Os resultados desse estudo estão apresentados na Tabela 9.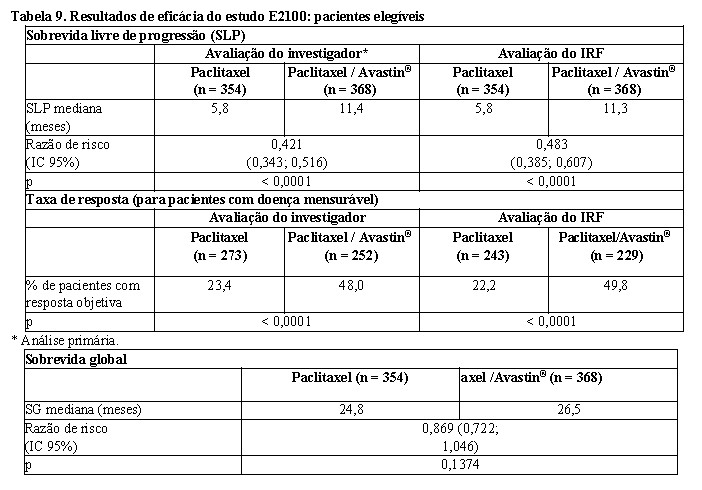
Avastin® é indicado exclusivamente em combinação com paclitaxel e capecitabina para o tratamento de primeira linha em pacientes com câncer de mama localmente recorrente ou metastático. No contexto dessa indicação, a combinação de Avastin® com docetaxel não demonstrou a mesma magnitude de efeito, não sendo aprovada para uso.
AVF3694g22,23: estudo de fase III, multicêntrico, randomizado, placebo controlado, desenhado para avaliar a eficácia e segurança de Avastin® em combinação com quimioterapia, comparado com quimioterapia mais placebo como tratamento de primeira linha para pacientes com câncer de mama metastático ou localmente recorrente HER2 negativo.
A quimioterapia foi escolhida a critério do investigador, antes da randomização na proporção de 2:1 para receber quimioterapia + Avastin® ou quimioterapia + placebo. As escolhas de quimioterapia incluíram taxanos (paclitaxel, paclitaxel peguilhado ou docetaxel), os agentes antraciclínicos (doxorrubicina/ciclofosfamida, epirrubicina/ciclofosfamida, 5-fluorouracil/doxorrubicina/ciclofosfamida, 5-fluorouracil/epirrubicina/ ciclofosfamida) ou capecitabina administrados a cada três semanas. Avastin® ou placebo foram administrados na dose de 15 mg/kg a cada três semanas.
Esse estudo incluiu uma fase de tratamento cego, uma fase aberta opcional pós-progressão e uma nova fase para acompanhamento de sobrevida. Durante a fase de tratamento cego, os pacientes receberam quimioterapia e a droga do estudo (Avastin® ou placebo), a cada 3 semanas, até progressão da doença, toxicidade limitante do tratamento ou óbito.
Na progressão documentada da doença, os pacientes que entraram na fase aberta opcional poderiam receber Avastin® juntamente com uma ampla variedade de terapias de segunda linha. A porcentagem de pacientes em cada braço que recebeu Avastin® de forma aberta foi a seguinte: taxano/antraciclina + placebo: 43,0%; taxano/ antraciclina + Avastin®: 29,6% e cap + placebo: 51,9%; cap + Avastin®: 34,7%.
Os pacientes foram analisados em duas coortes, dependendo do tratamento recebido, da seguinte forma:
• Pacientes recebendo taxano/antraciclina + Avastin®/placebo - Coorte 1
• Pacientes recebendo capecitabina + Avastin®/placebo - Coorte 2
O desfecho primário foi a sobrevida livre de progressão (SLP), baseada na avaliação do investigador para:
1) pacientes recebendo terapia com taxano ou baseada em antraciclina (Coorte 1), e
2) pacientes que recebendo terapia com capecitabina (Coorte 2).
Cada coorte teve poder independente. Além disso, uma análise independente do desfecho primário também foi realizada.
Os resultados desse estudo, a partir das análises definidas no protocolo final para a sobrevida livre de progressão e taxas de resposta, são apresentados na Tabela 10 (Coorte 1) e Tabela 11 (Coorte 2). Resultados de uma análise exploratória de sobrevida global, que inclui um acompanhamento adicional de sete meses, também são apresentados para ambas as coortes. Nesse ponto, cerca de 45% dos pacientes em todos os braços de tratamento haviam ido a óbito.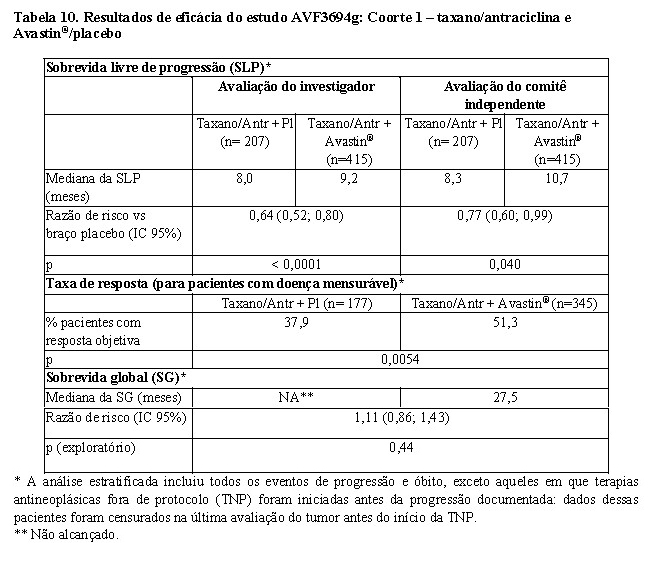
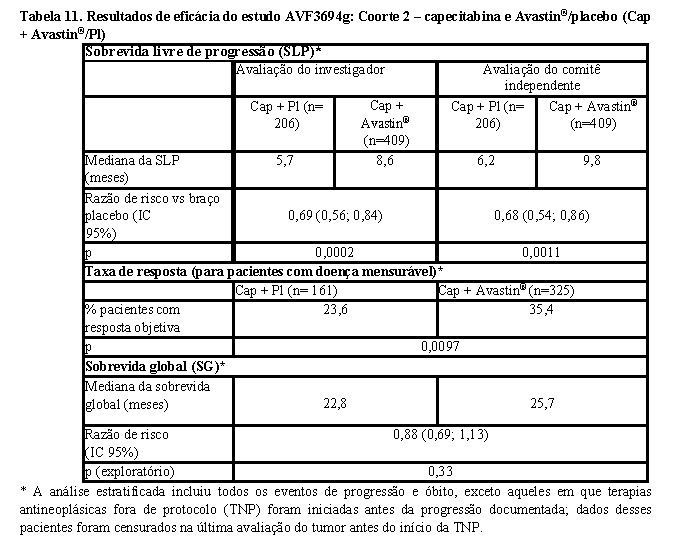
Para ambas as coortes, foram realizadas análises não estratificadas de SLP (avaliação do investigador), que não censuraram terapias fora do protocolo iniciadas previamente à progressão da doença. Os resultados dessas análises foram muito semelhantes aos resultados preliminares de SLP.
Câncer de células renais metastático e / ou avançado (mRCC)11,12
BO17705 foi um estudo de fase III, multicêntrico, randomizado, duplo-cego, conduzido para avaliar a eficácia e a segurança de Avastin® em combinação com alfainterferona 2a (Roferon®) versus alfainterferona 2a em monoterapia como tratamento de primeira linha em mRCC. Os 649 pacientes randomizados (641 tratados) tinham RCC de células claras metastático, desempenho Karnofsky ≥ 70%, ausência de metástases no sistema nervoso central e função orgânica adequada. O tratamento fornecido incluiu alfainterferona 2a (9 MUI, três vezes por semana) mais Avastin® (10 mg/kg a cada duas semanas) ou placebo, administrados até a progressão da doença. Os pacientes foram estratificados de acordo com o país e escore de Motzer, e os braços de tratamento mostraram ser equilibrados para os fatores de prognóstico.
O desfecho primário foi a sobrevida global, com desfechos secundários para o estudo, que incluíam sobrevida livre de progressão (SLP). A adição de Avastin® à alfainterferona 2a aumentou significativamente a SLP e a taxa de resposta objetiva do tumor. Esses resultados foram confirmados por meio de revisão radiológica independente. No entanto, o aumento no desfecho primário de sobrevida global em dois meses não foi significativo (HR = 0,91). Uma alta proporção de pacientes (aproximadamente 63% IFN / placebo, 55% Avastin® / IFN) recebeu uma série de terapias anticâncer pós-protocolo não especificadas, incluindo agentes antineoplásicos, que pode ter impactado na análise da sobrevida global.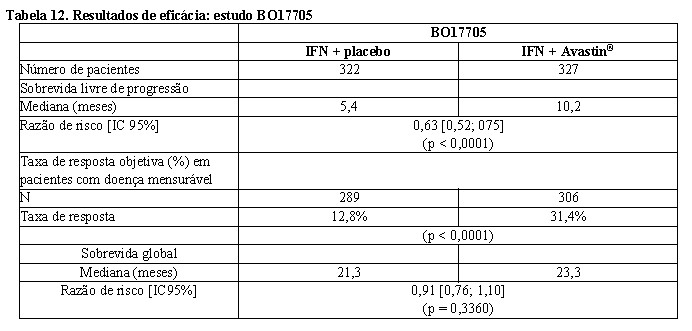
Um modelo de regressão de Cox multivariada exploratória que utilizou seleção retrógada indicou que os seguintes fatores prognósticos basais estavam fortemente associados à sobrevida, independentemente do tratamento: sexo, contagem de glóbulos brancos, plaquetas, perda de peso nos seis meses antecedentes à entrada no estudo, número de sítios metastáticos, soma do maior diâmetro das lesões-alvo e escore de Motzer. O ajuste para esses fatores basais resultou em razão de risco de tratamento de 0,78 (95% IC [0,63; 0,96], p = 0,0219), indicando redução de 22% no risco de morte para os pacientes do braço de Avastin® + alfainterferona 2a, em comparação com o braço de alfainterferona 2a.
Noventa e sete (97) pacientes do braço de alfainterferona 2a e 131 pacientes do braço de Avastin® reduziram a dose de alfainterferona 2a de 9 MUI para 6 ou 3 MUI, três vezes por semana, como pré-especificado no protocolo. A redução da dose de alfainterferona 2a pareceu não afetar a eficácia do uso combinado de Avastin® e alfainterferona 2a, com base nas taxas de SLP livres de eventos ao longo do tempo, como mostrado por uma análise de subgrupo. Os 131 pacientes do braço de Avastin® + alfainterferona 2a que reduziram e mantiveram a dose de alfainterferona 2a em 6 ou 3 MUI durante o estudo exibiram, em 6, 12 e 18 meses, taxas de SLP livres de eventos de 73%, 52% e 21%, respectivamente, em comparação a 61%, 43% e 17% na população total dos pacientes tratados com Avastin® + alfainterferona 2a.
AVF2938: estudo clínico de fase II, randomizado, duplo-cego, para avaliar Avastin®, 10 mg/kg, em um esquema a cada duas semanas, com a mesma dose de Avastin® em combinação com 150 mg/dia de erlotinibe, em pacientes com RCC de células claras metastático. Foram randomizados 104 pacientes para o tratamento nesse estudo, 53 para 10 mg/kg de Avastin® a cada duas semanas, mais placebo e 51 para 10 mg/kg de Avastin® a cada duas semanas, mais erlotinibe 150 mg/dia. A análise do desfecho primário não mostrou diferença entre o braço de Avastin® + placebo e o de Avastin® + erlotinibe (mediana de SLP de 8,5 versus 9,9 meses). Sete pacientes em cada braço apresentaram uma resposta objetiva.
Câncer epitelial de ovário, tuba uterina e peritoneal primário13,14,15,16,17
Tratamento de primeira linha de câncer de ovário13,14,15
A segurança e a eficácia de Avastin® no tratamento de primeira linha de pacientes com câncer epitelial de ovário, tuba uterina e peritoneal primário foram avaliadas em dois estudos clínicos fase III (GOG-0218 e BO17707) que compararam o efeito de Avastin® associado à carboplatina e paclitaxel em relação ao regime quimioterápico isolado.
GOG-021813: foi um estudo de fase III, multicêntrico, randomizado, duplo-cego, controlado por placebo, de três braços, para avaliar o efeito da adição de Avastin® a um regime quimioterápico padrão (carboplatina e paclitaxel) em pacientes com câncer epitelial de ovário, tuba uterina e peritoneal primário estadio III ou IV com citorredução ótima ou subótima.
Um total de 1.873 pacientes foi randomizado em proporções iguais para os três braços descritos a seguir:
• Braço CPP: placebo em combinação com carboplatina (AUC 6) e paclitaxel (175 mg/m2) durante seis ciclos, seguido de placebo isolado, num total de até 15 meses de terapia;
• Braço CPB15: cinco ciclos de Avastin® (15 mg/kg, a cada três semanas) em combinação com carboplatina (AUC 6) e paclitaxel (175 mg/m2) por seis ciclos (Avastin® iniciado no ciclo dois da quimioterapia), seguido de placebo isolado, num total de até 15 meses de terapia;
• Braço CPB15+: cinco ciclos de Avastin® (15 mg/kg, a cada três semanas) em combinação com carboplatina (AUC 6) e paclitaxel (175 mg/m2) por seis ciclos (Avastin® iniciado no ciclo dois da quimioterapia), seguido pelo uso continuado de Avastin® (15 mg/kg, a cada três semanas) em monoterapia, num total de até 15 meses de terapia.
O desfecho primário foi sobrevida livre de progressão (SLP) baseada na avaliação radiológica pelo investigador. Adicionalmente, uma revisão independente do desfecho primário foi realizada.
Os resultados desse estudo são apresentados na Tabela 13.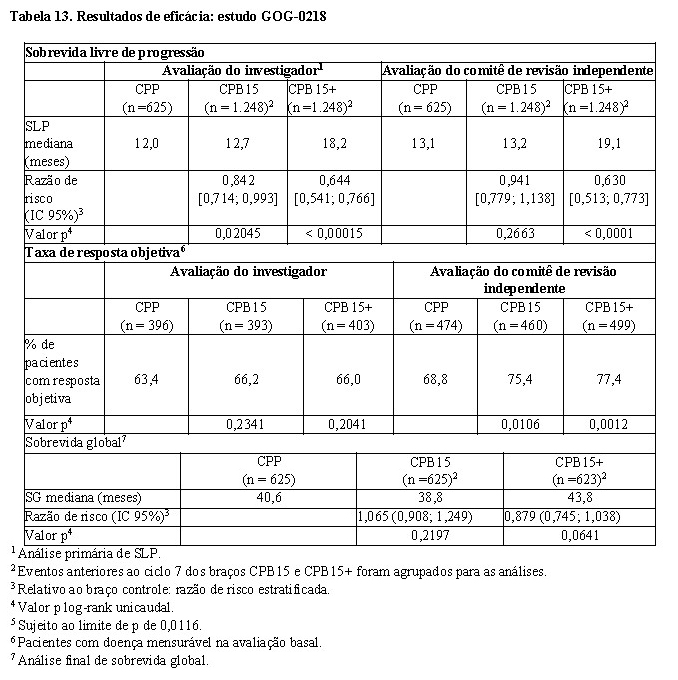
O estudo atingiu o seu desfecho primário de melhora na SLP. Comparado aos pacientes tratados com quimioterapia isolada (carboplatina e paclitaxel), os pacientes que receberam bevacizumabe na primeira linha na dose de 15 mg/kg, a cada três semanas, em combinação com quimioterapia, seguido pelo uso continuado de bevacizumabe em monoterapia, apresentaram aumento clinicamente relevante e estatisticamente significativo da SLP. Embora os pacientes que receberam bevacizumabe na primeira linha em combinação com quimioterapia isolada, mas que não mantiveram o seu uso continuado, tenham apresentado aumento na SLP, este não foi clinicamente relevante nem estatisticamente significativo, em comparação aos pacientes que receberam apenas quimioterapia.
BO17707 (ICON7)14,15: foi um estudo fase III, de dois braços, multicêntrico, randomizado, controlado, aberto comparando os efeitos da associação de Avastin® à carboplatina mais paclitaxel em pacientes com câncer epitelial de ovário, tuba uterina e peritoneal primário estadios FIGO I ou IIA (grau 3 ou histologia de células claras somente), ou estádios FIGO IIB - IV (todos os graus e todos os tipos histológicos), após cirurgia, ou naquelas pacientes em que nenhuma cirurgia estava planejada antes da progressão.
Um total de 1.528 pacientes foi randomizado em proporções iguais para os dois braços descritos a seguir:
• Braço CP: carboplatina (AUC 6) e paclitaxel (175 mg/m2) por seis ciclos;
• Braço CPB7,5+: carboplatina (AUC 6) e paclitaxel (175 mg/m2) durante seis ciclos, mais Avastin® (7,5 mg, a cada três semanas) por até 18 ciclos.
O desfecho primário foi sobrevida livre de progressão (SLP) avaliada pelo investigador. Os resultados desse estudo estão resumidos na Tabela 14.
O estudo atingiu o seu desfecho primário de aumento da SLP. Em comparação aos pacientes tratados com quimioterapia isolada (carboplatina e paclitaxel), os pacientes tratados com bevacizumabe na dose de 7,5 mg/kg, a cada três semanas, em combinação com quimioterapia, seguido pelo uso continuado de bevacizumabe por até 18 ciclos, tiveram melhora estatisticamente significativa da SLP.
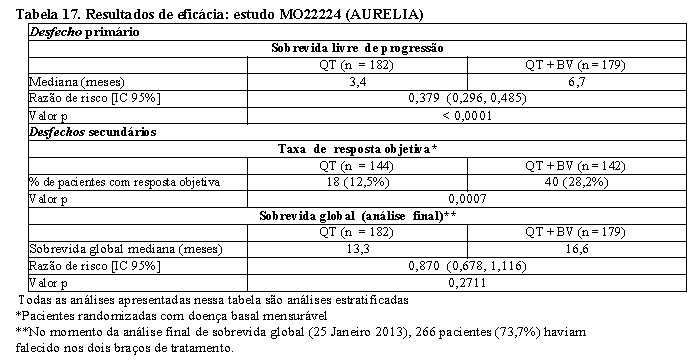
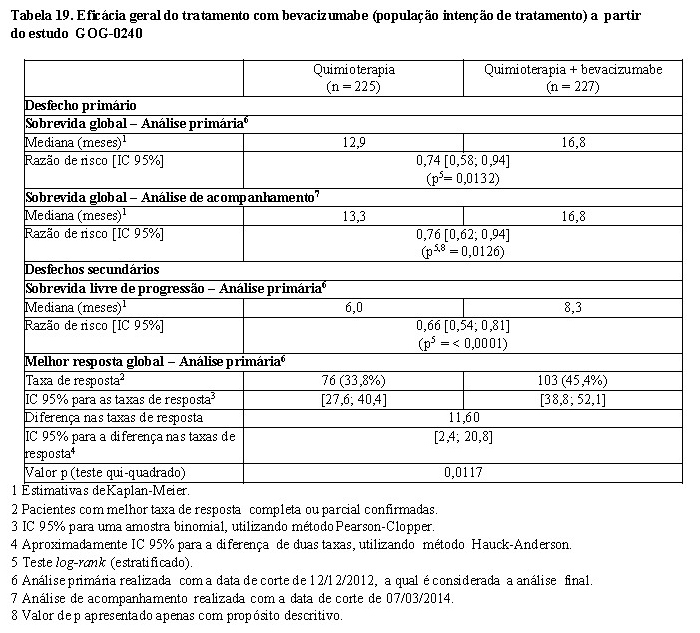
Câncer de ovário recorrente16,17,25
GOG-0213: foi o estudo controlado fase III, que avaliou a segurança e eficácia de Avastin® no tratamento de pacientes com câncer epitelial de ovário, tuba uterina ou peritoneal primário, recorrente e sensível à platina que não receberam quimioterapia prévia para a doença recorrente. Não houve critério de exclusão para terapia prévia com anti-angiogênico.
O estudo avaliou o efeito da combinação de Avastin® com carboplatina e paclitaxel e a continuação de Avastin® como agente único comparado somente com carboplatina e paclitaxel.
Um total de 673 pacientes foram randomizados em proporções iguais para os seguintes dois braços de tratamento. 2
• Braço CP: carboplatina (AUC5) e paclitaxel (175 mg/m2 IV no decorrer de 3 horas) a cada 3 semanas por 6 e até 8 ciclos.
• Braço CPB: carboplatina (AUC5) e paclitaxel (175 mg/m2 IV no decorrer de 3 horas) e Avastin® (15 mg/kg) concomitante a cada 3 semanas por 6 e até 8 ciclos, seguido por Avastin® (15 mg/kg a cada 3 semanas) como agente único até a progressão da doença ou toxicidade inaceitável.
O desfecho primário de eficácia do estudo foi a sobrevida global (SG). O desfecho secundário de eficácia foi a sobrevida livre de progressão (SLP). As taxas de resposta objetiva (TRO) foram também examinadas. Resultados são apresentados na Tabela 15.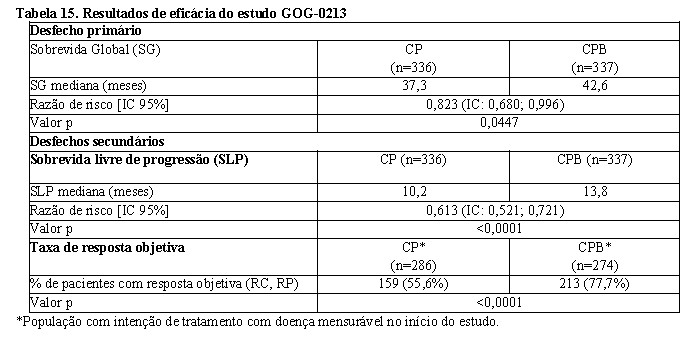
Tratamento com Avastin® 15 mg/kg a cada 3 semanas em combinação com quimioterapia (carboplatina e paclitaxel) por 6 a até 8 ciclos então seguidos por Avastin® como agente único, resultou em uma melhora estatisticamente significante na SG comparado ao tratamento somente com carboplatina e paclitaxel.
No estudo GOG-0213 foram incluídos pacientes que receberam terapia anti-angiogênica prévia, incluindo o tratamento com bevacizumabe. A inclusão de pacientes previamente tratados com um regime contendo bevacizumabe no estudo GOG-0213 permitiu que as análises de subgrupos fossem realizadas para explorar se os pacientes tratados com um regime contendo bevacizumabe na primeira linha apresentariam benefício clínico (ou seja, SG, SLP e TRO) quando tratados com outro regime contando bevacizumabe após recorrência.
Houve 69 pacientes com câncer de ovário recorrente e sensível a platina que já haviam recebido tratamento com bevacizumabe anteriormente. Este subgrupo de pacientes em que bevacizumabe foi adicionado à combinação quimioterápica carboplatina + paclitaxel também apresentou melhora na SG e SLP em comparação com aqueles que receberam o tratamento de carboplatina + paclitaxel sem bevacizumabe, em linha com a população geral com intenção de tratamento (ITT). A razão de risco não estratificada para SG foi 0,764 (IC 95% [0,436; 1,340]). A duração mediana da SG foi de 32,0 meses no braço de carboplatina + paclitaxel, e de 36,8 meses no braço de carboplatina + paclitaxel + bevacizumabe.
Para SLP, a razão de risco não estratificada foi de 0,841 (IC 95%: [0,516; 1,373]). A duração mediana da SLP foi de 9,8 meses no braço de carboplatina + paclitaxel e de 10,7 meses no braço de carboplatina + paclitaxel + bevacizumabe.
Uma melhora na TRO também foi observada nesses pacientes. A TRO no braço de carboplatina + paclitaxel + bevacizumabe foi de 82,1% e de 53,6% no braço de carboplatina + paclitaxel e a diferença na TRO foi de 28,6% (IC 95% [5,3; 51,9]).
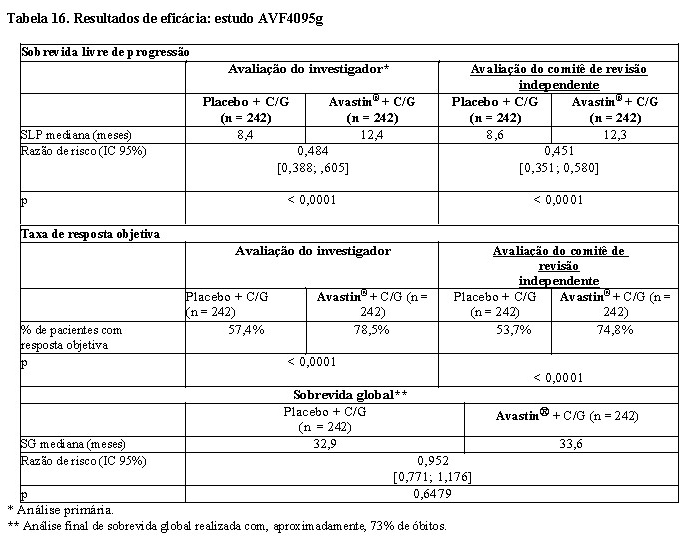
AVF4095g: a segurança e a eficácia de Avastin® no tratamento de pacientes com câncer epitelial de ovário, tuba uterina e peritoneal primário, recorrente e sensível à platina que não receberam quimioterapia prévia para a doença recorrente e não receberam tratamento com bevacizumabe prévio foram avaliadas em um estudo fase III, randomizado, duplo-cego, placebo controlado (AVF4095g). O estudo comparou o efeito da associação de Avastin® à quimioterapia com carboplatina e gencitabina e a continuação de Avastin® como agente único até a progressão da doença a somente carboplatina e gencitabina.
Um total de 484 pacientes com doença mensurável foi randomizado em porções iguais:
• Carboplatina (AUC4, dia 1) e gencitabina (1.000 mg/m2 nos dias 1 e 8) e placebo concomitantemente a cada três semanas por 6 e até 10 ciclos, seguido de somente placebo até a progressão da doença ou toxicidade inaceitável.
• Carboplatina (AUC4, dia 1) e gencitabina (1.000 mg/m2 nos dias 1 e 8) e Avastin® (15 mg/kg, dia
1) concomitantemente a cada três semanas por 6 e até 10 ciclos, seguido de somente Avastin® (15 mg/kg, a cada três semanas) até a progressão da doença ou toxicidade inaceitável.
O