EGROTIB
DR. REDDY'S
erlotinibe, cloridrato
Antineoplásico.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Apresentações.
Comprimidos revestidos de 25 mg, 100 mg ou 150 mg em caixa com 30 comprimidos.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de 25 mg contém: erlotinibe (equivalente a 27,32 mg de cloridrato de erlotinibe) 25 mg
Cada comprimido revestido de 100 mg contém: erlotinibe (equivalente a 109,27 mg de cloridrato de erlotinibe) 100 mg
Cada comprimido revestido de 150 mg contém: erlotinibe (equivalente a 163,90 mg de cloridrato de erlotinibe) 150 mg
Excipientes: celulose microcristalina, lactose monoidratada, laurilsulfato de sódio, amidoglicolato de sódio, estearato de magnésio e opadry branco.
Informações técnicas.
1. INDICAÇÕES
Câncer de pulmão de não pequenas células
O EGROTIB® (cloridrato de erlotinibe) é indicado para o tratamento de primeira linha e de manutenção de pacientes com câncer de pulmão do tipo não pequenas células (CPNPC), localmente avançado ou metastático, com mutações ativadoras de EGFR (receptor do fator de crescimento epidérmico).
No tratamento de manutenção, nenhum benefício clinicamente relevante foi demonstrado em pacientes com CPNPC sem mutação ativadora de EGFR.
O EGROTIB® também é indicado para o tratamento de pacientes com câncer de pulmão de não pequenas células (CPNPC), localmente avançado ou metastático (estadios IIIb e IV), após a falha de pelo menos um esquema quimioterápico prévio.
Câncer de pâncreas
O EGROTIB®, em combinação com gencitabina, é indicado para o tratamento de primeira linha de pacientes com câncer de pâncreas localmente avançado, irressecável ou metastático.
2. RESULTADOS DE EFICÁCIA
Eficácia/ estudos clínicos
Câncer de pulmão de não pequenas células (CPNPC)
Terapia de primeira linha para pacientes com mutações ativadoras de EGFR:
A eficácia de cloridrato de erlotinibe no tratamento de primeira linha de pacientes com mutações ativadoras de EGFR em CPNPC foi demonstrada em um estudo clinico randomizado e aberto de fase III (ML20650, EURTAC). Esse estudo foi conduzido em pacientes caucasianos com CPNPC localmente avançado ou metastático (estadios IIIB e IV), os quais não haviam recebido previamente quimioterapia ou terapia sistêmica anti-tumoral para doença avançada e os quais apresentavam mutações no dominio da tirosina quinase do EGFR (deleção do éxon 19 ou mutação do éxon 21). Os pacientes foram randomizados 1:1 para receber cloridrato de erlotinibe 150mg ou quimioterapia baseada em esquemas duplos de platina.
O objetivo primário foi avaliar a sobrevida livre de progressão (SLP), determinada em uma análise interina pré- planejada [n= 153, razão de risco (RR) = 0,42; IC 95% 0,27 a 0,64; p < 0,0001 para o grupo de cloridrato de erlotinibe (n=77) relativamente ao grupo de quimioterapia (n=76)]. Foi observada redução de 58% no risco de progressão da doença ou morte. A mediana da SLP foi 9,4 e 5,2 meses e a taxa de resposta objetiva foi 54,5% e 10,5%, nos braços de cloridrato de erlotinibe e quimioterapia, respectivamente. Os resultados de SLP foram confirmados por uma revisão independente das imagens, a mediana da SLP foi 10,4 meses no grupo de cloridrato de erlotinibe, comparado com 5,4 meses no grupo de quimioterapia (RR=0,47; IC 95% 0,27 a 0,78; p=0,003). Os dados de sobrevida global estavam imaturos quando foi feita a análise interina (RR=0,80; IC 95% 0,47 a 1,37, p=0,4170).
Em uma análise atualizada com 62% de maturidade dos dados de sobrevida global (OS), a razão de risco (RR) foi de 0,93 (IC 95%: 0,64 - 1,36, p = 0,7149). Observou-se uma alta taxa de cruzamento, onde 82% dos pacientes do grupo tratado com quimioterapia receberam, subsequentemente, terapia com um inibidor da tirosina quinase do EGFR e todos, menos dois desses pacientes, receberam cloridrato de erlotinibe subsequentemente. Na análise atualizada, os resultados da SLP permaneceram consistentes com os resultados da análise provisória. A mediana da SLP avaliada pelos investigadores foi de 10,4 e 5,1 meses no braço de cloridrato de erlotinibe e quimioterapia, respectivamente (RR = 0,34, IC 95%, 0,23 - 0,49, p
< 0,0001).5
- Dados adicionais publicados
Em uma análise prospectiva dos pacientes com CPNPC avançado, que apresentavam tumores com mutações ativadoras no domínio TK do EGFR, a SLP mediana para os 113 pacientes tratados com cloridrato de erlotinibe na primeira linha foi 14 meses (IC 95% 9,7 a 18,3 meses), e a mediana da sobrevida global foi 28,0 meses (IC 95%, 22,7 a 33 meses). A análise conjunta dos dados publicados a partir de pacientes com CPNPC demonstrou que pacientes com tumores com mutações ativadoras de EGFR tratados predominantemente com cloridrato de erlotinibe na primeira linha (n=70; 12, 5 meses; IC 95% 10,6 a 16,0) apresentaram uma mediana maior de SLP, comparada com aqueles tratados com quimioterapia (n=359; 6,0 meses; IC 95% 5,4 a 6,7).
Terapia de manutenção de primeira linha
A eficácia e a segurança de cloridrato de erlotinibe como terapia de manutenção de primeira linha em CPNPC foram demonstradas em estudo randomizado, duplo-cego e placebo controlado (BO18192). Esse estudo foi conduzido com 889 pacientes com CPNPC localmente avançado ou metastático que não progrediram durante 4 ciclos de quimioterapia baseada em esquemas duplos com platina. Os pacientes foram randomizados na proporção 1:1 com cloridrato de erlotinibe, 150 mg, ou placebo, por via oral, uma vez ao dia. O objetivo primário do estudo foi avaliar a sobrevida livre de progressão (SLP) em todos os pacientes e naqueles com tumor EGFR (receptor do fator de crescimento epidérmico) IHC (imunohistoquímica) positivo. As características demográficas e as características da doença foram bem equilibradas entre os dois braços de tratamento.
No estudo BO18192 (SATURN), a população geral demonstrou benefício para o desfecho primário de SLP (RR = 0,71 p < 0,0001) e para o desfecho secundário de SG (RR = 0,81 p=0,0088). No entanto, o maior benefício foi observado em uma análise exploratória pré definida em pacientes com mutações ativadoras de EGFR (n=49), demonstrando um benefício substancial na SLP (RR = 0,10, IC 95%, 0,04 a 0,25; p < 0,0001) e uma razão de risco de SG de 0,83 (IC 95%, 0,34 a 2,02). No subgrupo de mutações positivas de EGFR, 67% dos pacientes com placebo receberam segunda linha de tratamento ou linhas adicionais com inibidores de EGFR. Nos pacientes com tumores do tipo EGFR selvagem (n = 388), a razão de risco de SLP foi 0,78 (IC 95%, 0,63 a 0,96; p = 0,0185) e razão de risco de SG foi de 0,77 (IC 95%, 0,61 a 0,97; p = 0,0243).
O estudo BO25460 (IUNO) foi conduzido em 643 pacientes com câncer de pulmão de não pequenas células avançado cujos tumores não possuíam mutação ativadora de EGFR (deleção do éxon 19 ou mutação no éxon 21 L858R) e que não apresentaram progressão da doença após 4 ciclos de quimioterapia baseada em platina.
O objetivo do estudo foi comparar a SG da terapia de manutenção de primeira linha com erlotinibe versus erlotinibe administrado no momento da progressão da doença. O estudo não atingiu seu desfecho primário. A SG de cloridrato de erlotinibe na manutenção de primeira linha não foi superior à cloridrato de erlotinibe no tratamento de segunda linha
em pacientes cujo tumor não possuía mutação ativadora de EGFR (RR = 1,02, IC 95%, 0,85 a 1,22, p = 0,82). O desfecho secundário de SLP não apresentou diferença entre cloridrato de erlotinibe e placebo no tratamento de manutenção (RR = 0,94, IC 95%, 0,80 a 1,11; p = 0,48).
Com base nos dados do estudo BO25460 (IUNO), o cloridrato de erlotinibe não é recomendado para o tratamento de manutenção de primeira linha em pacientes sem mutação ativadora de EGFR.
Terapia de segunda/ terceira linha
A eficácia e a segurança de cloridrato de erlotinibe como terapia de segunda/ terceira linha foram demonstradas em estudo randomizado, duplo-cego, controlado com placebo (BR.21). Esse estudo foi conduzido em 17 países, incluindo 731 pacientes com CPNPC localmente avançado ou metastático, após falha de pelo menos um esquema quimioterápico. Os pacientes foram randomizados na proporção de 2:1 para receber cloridrato de erlotinibe 150 mg ou placebo via oral diariamente. Os objetivos do estudo incluíram avaliar a sobrevida global, o tempo até deterioração de sintomas relacionados ao câncer de pulmão (tosse, dispneia e dor), taxa de resposta, duração da resposta, sobrevida livre de progressão (SLP) e segurança. O objetivo primário foi a sobrevida global.
Devido à randomização 2:1, 488 pacientes foram randomizados para cloridrato de erlotinibe e 243 pacientes para placebo. Os pacientes não foram selecionados por expressão imunohistoquímica HER1/EGFR, sexo, raça, história de tabagismo ou classificação histológica.
As características demográficas foram bem equilibradas entre os dois braços de tratamento. Aproximadamente dois terços dos pacientes eram homens, um terço apresentava estado de desempenho ECOG basal de 2, e 9% dos pacientes apresentavam um ECOG basal de 3. Noventa e três por cento e 92% de todos os pacientes nos grupos cloridrato de erlotinibe e placebo, respectivamente, tinham recebido um esquema prévio com platina, e 36% e 37% de todos os pacientes, respectivamente, tinham recebido uma terapia prévia com taxano. Cinquenta por cento dos pacientes tinham recebido apenas um esquema prévio de quimioterapia.
A sobrevida foi avaliada na população ITT. A mediana da sobrevida global aumentou em 42,5% e foi de 6,7 meses no grupo cloridrato de erlotinibe (IC 95%, 5,5 a 7,8 meses), em comparação com 4,7 meses no grupo placebo (IC 95%, 4,1 a 6,3 meses), resultando em uma diferença de dois meses entre os grupos. A análise de sobrevida primária foi ajustada para fatores de estratificação, como descrito no momento da randomização (ECOG PS, melhor resposta à terapia prévia, número de esquemas prévios e exposição prévia à platina) e expressão imunohistoquímica HER1/EGFR. Nessa análise primária, a razão de risco ajustada para óbito no grupo cloridrato de erlotinibe em relação ao grupo placebo foi de 0,73 (IC 95%, 0,60 a 0,87; p = 0,001). A porcentagem de pacientes vivos em 12 meses foi de 31,2% e 21,5%, respectivamente.
O benefício de sobrevida com o tratamento com cloridrato de erlotinibe foi observado na maioria dos subgrupos. Uma série de subgrupos de pacientes formada pela estratificação das diferentes características do estado basal dos pacientes, tais como expressão imunohistoquímica HER1/EGFR, exposição prévia a taxanos, história de tabagismo, sexo, idade, histologia, perda de peso prévia, tempo entre o diagnóstico inicial e a randomização e localização geográfica, foi examinada por análises univariadas exploratórias para avaliar a consistência do resultado de sobrevida global. Praticamente todas as razões de risco (RR) no grupo cloridrato de erlotinibe, em relação ao grupo placebo, foram menores que 1,0, sugerindo que o benefício de cloridrato de erlotinibe foi consistente entre os subgrupos. O benefício de sobrevida de cloridrato de erlotinibe foi comparável em pacientes com performance status (PS) ECOG basal de 2 - 3 (RR = 0,77) ou um PS de 0 - 1 (RR
= 0,73) e pacientes que receberam um regime de quimioterapia (RR = 0,76) ou dois ou mais regimes (RR=0,76).
O benefício de sobrevida de cloridrato de erlotinibe também foi observado em pacientes que não atingiram uma resposta objetiva do tumor (critério RECIST). Isso foi evidenciado por uma razão de risco para óbito de 0,83 entre pacientes cuja melhor resposta foi doença estável e 0,85 entre pacientes cuja melhor resposta foi doença progressiva.
Em uma análise detalhada de alguns desses subgrupos, podemos explicitar as diferenças em meses de sobrevida entre os pacientes que receberam cloridrato de erlotinibe ou placebo. No grupo de pacientes com ECOG 0 - 1, essa diferença foi de 1,45 mês (RR = 0,73) e com ECOG 2 - 3 de 0,39 mês (RR = 0,77); no grupo de pacientes que havia recebido um tratamento quimioterápico prévio, a diferença foi de 0,83 mês (RR = 0,76) e de 2,18 meses (RR=0,76) no grupo que havia recebido dois ou mais tratamentos prévios.
Entre os pacientes que nunca fumaram e que foram tratados com cloridrato de erlotinibe, o ganho de sobrevida foi mais que seis meses: a sobrevida mediana no grupo de pacientes não fumantes tratados com cloridrato de erlotinibe foi de 12,25 meses e no grupo de pacientes não fumantes que receberam placebo foi de 5,62 meses.
Não se pode afirmar que houve benefício entre pacientes fumantes ou ex-fumantes: a sobrevida mediana obtida no grupo de pacientes fumantes ou ex-fumantes que foram tratados com cloridrato de erlotinibe foi de 5,52 meses e no grupo de pacientes fumantes ou ex-fumantes que receberam placebo foi de 4,63 meses, resultando em diferença de 0,89 mês.
Houve benefício de sobrevida, e esse foi ainda superior em alguns grupos de pacientes: asiáticos, mulheres, pacientes com câncer de pulmão do tipo adenocarcinoma.
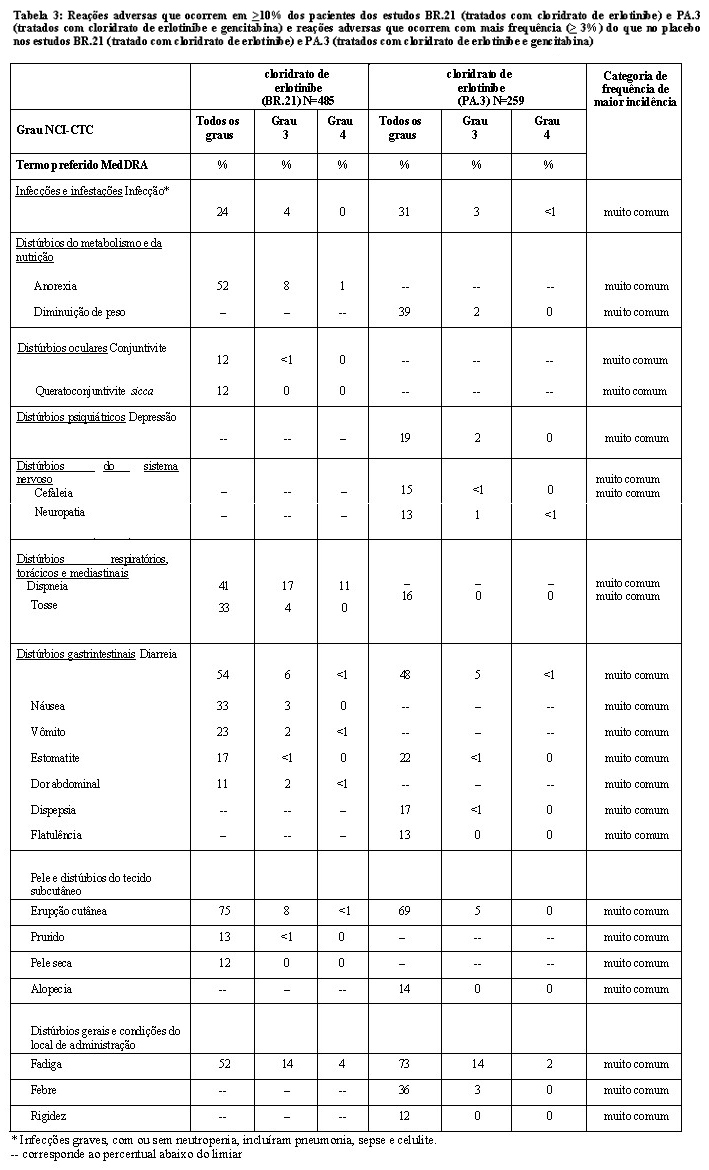
A Tabela 1 resume os resultados para o estudo, incluindo sobrevida, tempo até deterioração de sintomas relacionados ao câncer de pulmão e sobrevida livre de progressão (SLP).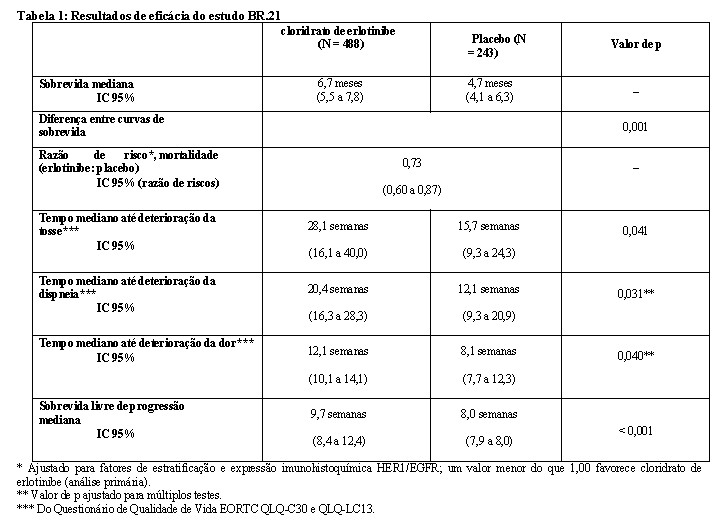
A deterioração do sintoma foi medida usando os questionários de qualidade de vida EORTC QLQ-C30 e QLQ- LC13. Os escores basais dos sintomas: tosse, dispneia e dor foram semelhantes nos dois grupos de tratamento. O grupo de pacientes tratados com cloridrato de erlotinibe mostrou melhora nos sintomas por meio do aumento do tempo para deterioração da tosse de 12,4 semanas (RR = 0,75), da dispneia de 8,3 semanas (RR = 0,72) e da dor de quatro semanas (RR = 0,77), quando comparado ao placebo. Esses benefícios sintomáticos não foram devidos a uso aumentado de radioterapia paliativa ou medicação concomitante no grupo cloridrato de erlotinibe.
A mediana da sobrevida livre de progressão foi de 9,7 semanas no grupo cloridrato de erlotinibe (IC 95%, 8,4 a 12,4 semanas), em comparação com 8,0 semanas no grupo placebo (IC 95%, 7,9 a 8,1 semanas), resultando em diferença de 1,7 semana entre os grupos. A RR para progressão, ajustada para fatores de estratificação e expressão imunohistoquímica HER1/EGFR, foi de 0,61 (IC 95% 0,51 a 0,73; p < 0,001). A porcentagem de pacientes livres de progressão aos 6 meses foi de 24,5% e 9,3%, respectivamente, para braços cloridrato de erlotinibe e placebo.
A taxa de resposta objetiva por RECIST no grupo cloridrato de erlotinibe foi de 8,9% (IC 95% 6,4 a 12,0%). A duração mediana da resposta foi de 34,3 semanas, variando de 9,7 a 57,6+ semanas. Duas respostas (0,9%; IC 95% 0,1 a 3,4) foram relatadas no grupo placebo. A proporção de pacientes que apresentaram resposta completa, resposta parcial ou doença estável foi de 44,0% e 27,5%, respectivamente, para os grupos cloridrato de erlotinibe e placebo (p = 0,004).
Em um estudo randomizado, duplo-cego, fase III, (MO22162, CURRENTS) foram comparadas duas doses de cloridrato de erlotinibe (300 mg versus 150 mg) em fumantes ativos (média de 38 pacotes anos) com CPNPC localmente avançado ou metastático no tratamento de segunda linha após falha de quimioterapia. A dose de 300 mg de cloridrato de erlotinibe não demonstrou nenhum benefício na sobrevida livre de progressão (SLP) em relação à dose recomendada (7,00 versus 6,86 semanas, respectivamente). Os pacientes neste estudo não foram selecionados com base no estado da mutação EGFR8.
Os dados de segurança foram comparáveis entre as doses de 300 mg e de 150 mg, no entanto houve um aumento numérico na incidência de erupção cutânea, de doença pulmonar intersticial e de diarreia nos pacientes que receberam a dose mais elevada de cloridrato de erlotinibe.
Câncer de pâncreas (cloridrato de erlotinibe administrado simultaneamente com gencitabina)
A eficácia e a segurança de cloridrato de erlotinibe em combinação com gencitabina como tratamento de primeira linha foram avaliadas em um estudo randomizado, duplo-cego, placebo controlado, em 569 pacientes com câncer de pâncreas localmente avançado, irressecável ou metastático. Os pacientes foram randomizados na proporção de 1:1 para receber cloridrato de erlotinibe (100 ou 150 mg) ou placebo, uma vez ao dia, em esquema contínuo, mais gencitabina IV (1.000 mg/m2, Ciclo 1 - Dias 1, 8, 15, 22, 29, 36 e 43 de um ciclo de oito semanas, Ciclo 2 e ciclos subsequentes - Dias 1, 8 e 15 de um ciclo de 4 semanas < dose aprovada e esquema para câncer de pâncreas, vide bula de gencitabina). O cloridrato de erlotinibe ou placebo foi ingerido uma vez ao dia até a progressão da doença ou toxicidade inaceitável. O objetivo do estudo incluiu sobrevida global, taxa de resposta e sobrevida livre de progressão (SLP). A duração da resposta também foi avaliada. O objetivo primário foi sobrevida. Um total de 285 pacientes foi randomizado para receber gencitabina mais cloridrato de erlotinibe (261 pacientes do grupo de 100 mg e 24 pacientes do grupo de 150 mg) e 284 pacientes foram randomizados para receber gencitabina mais placebo (260 pacientes do grupo de 100 mg e 24 pacientes no grupo de 150 mg). O pequeno número de pacientes na dose de 150 mg não permitiu conclusões.
Características basais demográficas e da doença dos pacientes foram similares entre os dois grupos de tratamento, cloridrato de erlotinibe 100 mg mais gencitabina ou placebo mais gencitabina, exceto para uma proporção levemente aumentada de mulheres no braço cloridrato de erlotinibe (51%) comparado com o braço placebo (44%). O tempo mediano do diagnóstico inicial para a randomização foi aproximadamente um mês. Aproximadamente metade dos pacientes teve performance status (PS) ECOG basal de 1, e 17% tiveram ECOG PS basal de 2. A maioria dos pacientes apresentou-se com doença metastática na entrada do estudo como manifestação inicial do câncer de pâncreas (77% no braço cloridrato de erlotinibe, 76% no braço placebo).
A sobrevida foi avaliada na população com intenção de tratamento com base nos dados de acompanhamento de sobrevida, incluindo 551 óbitos. Os resultados são apresentados para o grupo de dose de 100 mg (504 óbitos). A razão de risco ajustada para óbito no grupo cloridrato de erlotinibe relativo ao grupo placebo foi 0,82 (IC 95% 0,69 a 0,98; p = 0,028). A porcentagem de pacientes vivos aos 12 meses foi de 23,8% no grupo cloridrato de erlotinibe comparado aos 19,4% no grupo placebo. O tratamento com cloridrato de erlotinibe esteve associado a discreto ganho de sobrevida global, estatisticamente significante. A sobrevida global mediana foi de 6,4 meses no grupo cloridrato de erlotinibe comparado com 6 meses no grupo placebo.
A Tabela 2 resume os resultados do estudo.
A duração mediana de resposta foi de 23,9 semanas, intervalo de 3,71 a 56+ semanas. A taxa de resposta objetiva (resposta completa e resposta parcial) foi de 8,6% no grupo cloridrato de erlotinibe e 7,9% no grupo placebo. A
proporção de pacientes que apresentaram resposta completa, resposta parcial ou doença estável foi de 59% e 49,4%, respectivamente, para os grupos cloridrato de erlotinibe e placebo (p = 0,036).
Referências bibliográficas
1. Sheperd FA, Pereira JR, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 2005;353:123-132.
2. Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. JCO 2007;26(15):1960-66.
3. Clinical Study Report - BO18192 - A multi-centre, double-blind randomized, placebo-controlled Phase III study to evaluate the efficacy of Tarceva® following 4 cycles of standard platinum-based chemotherapy in patients with histologically documented, advanced, recurrent or metastatic (Stage IV) NSCLC who have not experienced disease progression or unacceptable toxicity during chemotherapy. Report No. 1031460, February 2009.
4. Clinical Study Report Addendum - Protocol BO18192 - A multi-centre, double-blind randomized, Phase III study to evaluate the efficacy of Tarceva® or placebo following 4 cycles of platinum-based chemotherapy in patients with histologically documented, advanced or recurrent (Stage IIIB and not amenable for combined modality treatment) or metastatic (Stage IV) non-small cell lung cancer (NSCLC) who have not experienced disease progression or unacceptable toxicity during chemotherapy. Report No. 1033732, August 2009.
5. Clinical Study Report - ML20650 (EURTAC) Phase III, multicenter, open-label, randomized study of erlotinib (Tarceva®) treatment versus chemotherapy in patients with advanced non-small-cell carcinoma of the lung who present mutations in the tyrosine kinase (TK) domain of epidermal growth factor receptor (EGFR) / Report Number 1050832 / October, 2012
6. Paz-Ares, L et al. Clinical outcome in non-small-cell lung cancer patients with EGFR mutations: pooled analysis. J Cell. Mol. Med. 2009 XX;XX:1-19
7. Data Summary: Summary of Clinical Efficacy and Summary of Clinical Safety - BO25460 (IUNO) A randomized, double-blind, placebo controlled Phase III study of first-line maintenance Tarceva vs. Tarceva at the time of disease progression in patients with advanced non?small cell lung cancer (NSCLC) who have not progressed following 4 cycles of platinum-based chemotherapy.
8. Clinical Study Report - Protocol MO22162 - A prospective double-blind randomized phase III study of 300 mg versus 150 mg erlotinib in current smokers with locally advanced or metastatic NSCLC in second-line setting after failure on chemotherapy (CURRENTS). Report n° 1062520. July 9, 2015.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Erlotinibe potencialmente inibe a fosforilação intracelular do receptor HER1/EGFR. O receptor HER1/EGFR é expresso na superfície celular de células normais e de células cancerosas. Nos modelos não clínicos, a inibição da fosforilação do EGFR resulta em inibição da proliferação celular e/ou morte celular.
Farmacocinética
Após a dose oral de cloridrato de erlotinibe 150 mg, o tempo mediano para alcançar o estado de equilíbrio é de quatro horas, com a concentração plasmática mediana máxima em torno de 1,995 ng/mL. Antes da próxima dose em 24 horas, a concentração plasmática mediana mínima é de 1,238 ng/mL. A mediana da ASC observada durante os intervalos de dose no estado de equilíbrio é 41,300 < g*h/mL.
Absorção
Erlotinibe oral é bem absorvido e tem fase de absorção ampliada, com média de concentrações plasmáticas máximas ocorrendo quatro horas depois da administração oral. Um estudo em voluntários saudáveis normais
forneceu estimativa de biodisponibilidade de 59%. A exposição após uma dose oral pode ser aumentada ao ingerir alimentos.
Depois da absorção, erlotinibe é altamente ligado no sangue, com aproximadamente 95% ligado aos componentes do sangue, principalmente proteínas plasmáticas (por exemplo, albumina e alfa-1 glicoproteína ácida [AAG]), com uma fração livre de aproximadamente 5%.
Distribuição
Erlotinibe tem um volume médio aparente de distribuição de 232 litros e se distribui para o tecido tumoral em humanos. Em um estudo em quatro pacientes (três com CPNPC e um com câncer de laringe) com cloridrato de erlotinibe 150 mg, por via oral, uma vez ao dia, amostras de tumor de excisões cirúrgicas, no dia 9 de tratamento, revelaram concentrações de erlotinibe em média de 1.185 ng/g de tecido. Isso corresponde à média geral de 63% do estado de equilíbrio observado em concentrações plasmáticas máximas. Os principais metabólitos ativos estavam presentes no tumor em concentrações médias de 160 ng/g de tecido, o que correspondeu à média total de 113% das concentrações plasmáticas máximas em estado de equilíbrio. Os estudos de distribuição nos tecidos que usaram radiografia corporal total, após a administração oral de erlotinibe marcado com [14C] em camundongos atímicos, com enxertos de tumor HN5, mostraram distribuição tecidual rápida e extensa, com concentrações máximas de droga marcada radioativamente (aproximadamente 73% da concentração no plasma) observada em uma hora.
Metabolismo
Erlotinibe é metabolizado no homem pelas enzimas hepáticas do citocromo P450, principalmente CYP3A4 e em menor extensão por CYP1A2. O metabolismo extra-hepático pelo CYP3A4 no intestino, CYP1A1 no pulmão e CYP1B1 no tecido tumoral potencialmente contribui para a depuração metabólica de erlotinibe. Estudos in vitro indicam que aproximadamente 80 a 95% do metabolismo de erlotinibe é realizado por meio do CYP3A4. Existem três principais vias metabólicas identificadas: 1) O-desmetilação de uma cadeia lateral ou ambas, seguida por oxidação para ácidos carboxílicos; 2) oxidação da molécula acetileno seguida por hidrólise em ácido aril carboxílico; e 3) hidroxilação aromática da molécula fenil-acetileno. Os metabólitos principais de erlotinibe produzidos por O-desmetilação de cada cadeia lateral apresentam potência comparável à de erlotinibe em ensaios não clínicos in vitro e modelos tumorais in vivo. Eles estão presentes no plasma em concentrações < 10% de erlotinibe e apresentam farmacocinética semelhante à de erlotinibe.
Excreção
Os metabólitos e traços de erlotinibe são excretados predominantemente por meio das fezes ( > 90%), com a eliminação renal responsável por apenas uma pequena quantidade de uma dose oral.
Depuração
Uma análise farmacocinética populacional em 591 pacientes que receberam cloridrato de erlotinibe como monoterapia mostrou depuração aparente média de 4,47 L/h com meia-vida mediana de 36,2 horas. Portanto, é esperado que o tempo para atingir a concentração plasmática em estado de equilíbrio seja de, aproximadamente, sete ou oito dias. Não foram observadas relações significativas entre a depuração aparente prevista e idade, peso, sexo e etnia do paciente.
Fatores do paciente, que se correlacionam com a farmacocinética de erlotinibe, são bilirrubina total sérica, concentrações de AAG e tabagismo atual. Concentrações séricas aumentadas de bilirrubinas totais e concentrações de AAG foram associadas com a depuração mais lenta de erlotinibe. Fumantes apresentaram depuração mais rápida de erlotinibe (vide item "Interações medicamentosas").
Uma segunda análise farmacocinética populacional foi conduzida incorporando os dados de erlotinibe de 204 pacientes com câncer pancreático tratados com erlotinibe mais gencitabina. Essa análise demonstrou que as covariáveis que afetam a depuração de creatinina nos pacientes do estudo pancreático foram muito similares àqueles vistos anteriormente na análise farmacocinética do agente isolado. Não foram identificadas novas covariáveis. A coadministração de gencitabina não afetou a depuração plasmática de erlotinibe.
Farmacocinética em populações especiais
Não houve nenhum estudo específico em pacientes de faixa etária pediátrica ou em idosos.
Insuficiência hepática: erlotinibe é eliminado principalmente pelo fígado. A exposição a erlotinibe foi similar em pacientes com a função hepática moderadamente prejudicada (pontuação de Child-Pugh de 7 - 9) comparado com pacientes com a função hepática normal, incluindo pacientes com câncer de fígado primário ou metástases hepáticas.
Insuficiência renal: erlotinibe e seus metabólitos não são significativamente excretados pelos rins, porque menos de 9% de uma dose única é excretada na urina. Nenhum estudo clínico foi conduzido em pacientes com função renal comprometida.
Fumantes: um estudo de farmacocinética em indivíduos saudáveis não fumantes e fumantes de cigarros ativos mostrou que o tabagismo de cigarro leva à depuração aumentada de erlotinibe e diminui a exposição ao medicamento (vide item "Interações medicamentosas"). A ASC0-∞ em fumantes está em torno de 1/3 dos não fumantes (n = 16 em cada braço dos fumantes e não fumantes). Essa redução de exposição nos atualmente fumantes é presumidamente devido à indução do CYP1A1 no pulmão e CYP1A2 no fígado.
Em estudo pivotal de fase III em CPNPC, concluiu-se que o estado de equilíbrio dos fumantes ativos está em torno de 0,65 mg/mL (n = 16), aproximadamente duas vezes menor que nos ex-fumantes ou pessoas que nunca fumaram (1,28 mg/mL, n = 108). Esse efeito foi acompanhado pelo aumento de 24% da depuração plasmática do erlotinibe.
Em estudo fase I de escalonamento de dose em pacientes CPNPC fumantes ativos, as análises farmacocinéticas do estado de equilíbrio indicaram aumento proporcional da dose de exposição a erlotinibe, quando a dose de cloridrato de erlotinibe foi aumentada de 150 mg para 300 mg, que é a dose máxima tolerada. As concentrações plasmáticas do estado de equilíbrio da dose de 300 mg em fumantes ativos nesse estudo foi de 1,22 mg/mL (n = 17) (vide item "Posologia e modo de usar" e "Interações medicamentosas").
Segurança não clínica
Carcinogenicidade
Evidências de potencial carcinogênico não foram constatadas nos estudos não clínicos. Erlotinibe não se mostrou genotóxico nem clastogênico em estudos de toxicidade genética. Estudos de carcinogênese de dois anos, conduzidos com erlotinibe em ratos e camundongos, com exposições excedendo a exposição terapêutica humana foram negativos.
Genotoxicidade
Erlotinibe mostrou-se negativo na série padrão de ensaios de genotoxicidade.
Comprometimento da fertilidade
Não foi observado comprometimento da fertilidade em estudos com ratos machos e fêmeas em doses próximas aos níveis DMT.
Toxicidade reprodutiva
Dados de exames de toxicologia reprodutiva em ratos e coelhos indicam que, depois da exposição a erlotinibe em doses próximas à DMT e/ou doses tóxicas para a mãe, existe embriotoxicidade, mas não houve nenhuma evidência de comprometimento de fertilidade, teratogenicidade ou anormalidade física pré ou pós-natal ou de desenvolvimento comportamental. A toxicidade materna em ratos e coelhos nesses estudos ocorreu em níveis de exposição plasmática semelhantes às que ocorrem no homem depois de uma dose de 150 mg de erlotinibe.
Outros
Os efeitos da administração crônica observados em pelo menos uma espécie animal ou em pelo menos um estudo incluíram efeitos sobre córnea (atrofia, ulceração), pele (degeneração folicular e inflamação, vermelhidão e alopecia), ovário (atrofia), fígado (necrose hepática), rins (necrose papilar renal e dilatação tubular) e trato gastrintestinal (esvaziamento gástrico retardado e diarreia). Houve redução nos valores de eritrócitos, hematócrito e hemoglobina e aumento dos reticulócitos. Os leucócitos, principalmente os neutrófilos, ficaram aumentados. Houve elevações de alanina aminotransferase (ALT), aspartato aminotransferase (AST) e bilirrubinas relacionadas ao tratamento.
Estudos in vitro de erlotinibe mostraram inibição de canais hERG em concentrações pelo menos 20 vezes maiores que a concentração de droga livre no homem em doses terapêuticas. Estudos em cães não mostraram
prolongamento de QT. Uma revisão centralizada sistemática de dados de ECG de 152 indivíduos de sete estudos com voluntários saudáveis não mostrou nenhuma evidência de prolongamento de QT e estudos clínicos não encontraram nenhuma evidência de arritmias associadas a prolongamento de QT.
4. CONTRAINDICAÇÕES
O EGROTIB® está contraindicado a pacientes com hipersensibilidade severa a erlotinibe ou a qualquer componente da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Doença Pulmonar Intersticial: casos de doença pulmonar intersticial (DPI), incluindo óbitos, foram relatados de forma incomum em pacientes que receberam cloridrato de erlotinibe para o tratamento do CPNPC, câncer pancreático ou outros tumores sólidos avançados. No estudo de referência BR.21 em CPNPC, a incidência de eventos sérios tipo DPI foi de 0,8% em cada um dos braços de cloridrato de erlotinibe e de placebo. Em uma meta-análise de ensaios clínicos controlados randomizados com CPNPC, a incidência de eventos semelhantes a DPI foi de 0,9% em cloridrato de erlotinibe em comparação com 0,4% em pacientes nos braços de controle. Em estudo de câncer pancreático em combinação com gencitabina, a incidência de DPI-like foi de 2,5% no grupo de cloridrato de erlotinibe com gencitabina versus 0,4% no grupo tratado com placebo mais gencitabina. Alguns exemplos de diagnóstico descritos em pacientes com suspeita de eventos tipo DPI incluem pneumonite, pneumonite por radiação, pneumonite por hipersensibilidade, pneumonia intersticial, doença pulmonar intersticial, bronquiolite obliterante, fibrose pulmonar, Síndrome do Desconforto Respiratório Agudo, infiltrado pulmonar e alveolite. Esses eventos tipo DPI começaram poucos dias a vários meses após o início da terapia com cloridrato de erlotinibe. A maioria dos casos foi relacionada com fatores confundidores ou contribuintes, como quimioterapia concomitante ou prévia, radioterapia prévia, doença pulmonar parenquimatosa preexistente, doença pulmonar metastática ou infecções pulmonares.
Em pacientes que desenvolvem início agudo de sintomas pulmonares inexplicados, novos e/ou progressivos, como dispneia, tosse e febre, a terapia com cloridrato de erlotinibe deve ser interrompida e deve-se aguardar avaliação diagnóstica. Em caso de diagnóstico de DPI, EGROTIB® deve ser descontinuado e iniciado tratamento apropriado se necessário (vide item "Reações adversas").
Diarreia, desidratação, desequilíbrio eletrolítico e insuficiência renal: ocorreu diarreia em pacientes que receberam cloridrato de erlotinibe. Diarreias moderadas ou graves devem ser tratadas com loperamida. Em alguns casos pode ser necessária a redução da dose. Em caso de diarreia grave ou persistente, náusea, anorexia ou vômitos associados à desidratação, o tratamento com EGROTIB® deve ser interrompido, e as medidas apropriadas devem ser instituídas para tratar a desidratação (vide item "Reações adversas"). Houve raros relatos de hipocalemia e insuficiência renal secundária (incluindo óbitos). Alguns relatos de insuficiência renal foram secundários à desidratação grave causada pela diarreia, vômito e/ou anorexia, enquanto outros foram confundidos pelo uso de quimioterapia concomitante. Em casos de diarreia grave ou persistente, que leva à desidratação, particularmente em grupos de pacientes com fatores de risco agravantes (medicamentos concomitantes, sintomas ou outras condições predispostas, incluindo idade avançada), a terapia de EGROTIB® deve ser interrompida, e medidas apropriadas devem ser tomadas para hidratação intravenosa intensiva dos pacientes. Além do mais, função renal e exames laboratoriais, incluindo o de potássio, devem ser monitorados em pacientes com risco de desidratação.
O cloridrato de erlotinibe não foi testado em pacientes com metástases cerebrais sintomáticas, portanto, sua eficácia é desconhecida nesse grupo de pacientes.
Hepatite, insuficiência hepática: casos raros de insuficiência hepática (incluindo óbitos) foram relatados durante o uso de cloridrato de erlotinibe. Fatores confundidores estavam presentes, como doença hepática preexistente ou medicação hepatotóxica concomitante. Portanto testes periódicos para verificar a função do fígado devem ser considerados. A dosagem de cloridrato de erlotinibe deve ser interrompida se ocorrerem mudanças graves na função hepática (vide item "Reações adversas").
Perfurações gastrintestinais: pacientes tratados com cloridrato de erlotinibe possuem risco aumentado de apresentar perfurações gastrointestinais, as quais foram observadas incomumente (incluindo alguns casos fatais). Pacientes que recebem agentes antiangiogênicos concomitantemente, corticosteroides, agentes anti-inflamatórios não estereoidais (AINEs) e/ou quimioterapia baseada em taxano ou com histórico de ulceração peptídica ou doença diverticular possuem risco maior de desenvolvê-las. O cloridrato de erlotinibe deve ser permanentemente descontinuado em pacientes que desenvolverem perfuração gastrintestinal (vide item "Reações adversas").
Distúrbios bolhosos e esfoliativos da pele: foram relatadas condições bolhosas, vesiculares ou esfoliativas da pele, incluindo muito raramente casos sugestivos de Síndrome de Stevens-Johnson/ necrólise epidérmica tóxica, os quais, em alguns casos, foram fatais (vide item "Reações adversas"). O tratamento com EGROTIB® deve ser interrompido ou descontinuado se o paciente apresentar graves condições bolhosas, vesiculares ou esfoliativas.
Distúrbios na visão: casos muito raros de perfurações ou ulceração da córnea foram relatados durante o uso de cloridrato de erlotinibe. Outros distúrbios oculares, incluindo crescimento anormal dos cílios, ceratoconjuntivite sicca ou ceratite, foram observados no tratamento com cloridrato de erlotinibe, os quais também são fatores de risco para ulceração/ perfuração da córnea. O tratamento com EGROTIB® deve ser interrompido ou descontinuado em pacientes que apresentem distúrbios oftalmológicos graves ou agravamento de distúrbios oculares, tais como dor nos olhos (vide item "Reações adversas").
Interações medicamentosas: o EGROTIB® tem potencial para interações entre drogas clinicamente significativas (vide item "Interações medicamentosas").
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas: erlotinibe não possui influência ou possui influência insignificante na capacidade de conduzir e utilizar máquinas.
Uso em populações especiais
Potencial reprodutivo feminino e masculino
Contracepção:
Pacientes do sexo feminino: métodos contraceptivos adequados devem ser usados durante a terapia e durante, pelo menos, duas semanas depois de completar a terapia.
Gravidez: não existem estudos adequados ou bem controlados em gestantes que usaram cloridrato de erlotinibe. Estudos em animais mostraram alguma toxicidade reprodutiva (vide item "Segurança pré-clínica"). O potencial risco para humanos é desconhecido. Mulheres com possibilidade de engravidar devem ser alertadas para evitar a gravidez durante o tratamento com EGROTIB®. O tratamento somente deve ser mantido em gestantes se o potencial benefício para a mãe superar o risco para o feto.
Categoria de risco na gravidez: C
Os estudos em animais revelaram risco, mas não existem estudos adequados e bem controlados disponíveis realizados em mulheres grávidas.
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Lactação: não se sabe se erlotinibe é excretado no leite humano. Não foram realizados estudos para avaliar o impacto de cloridrato de erlotinibe a na produção de leite ou sua presença no leite materno. Como o potencial de perigo para o lactente é desconhecido, as mães devem ser orientadas a não amamentar enquanto recebem cloridrato de erlotinibe e pelo menos 2 semanas após a dose final.