EBATZ
LIBBS
pitavastatina cálcica
Antilipêmico.
Apresentações.
Ebatz ® 2 mg: embalagem com 30 comprimidos revestidos.
Ebatz ® 4 mg: embalagem com 30 comprimidos revestidos.
EXCLUSIVAMENTE PARA ADMINISTRAÇÃO ORAL
USO ADULTO
Composição.
Cada comprimido revestido contém: Ebatz® 2 mg, 2,090 mg de pitavastatina cálcica, equivalente a 2 mg de pitavastatina em base livre. Ebatz® 4 mg, 4,180 mg de pitavastatina cálcica, equivalente a 4 mg de pitavastatina em base livre.
Excipientes: lactose monoidratada, hiprolose, hipromelose, silicato de alumínio e magnésio, estearato de magnésio, citrato de trietila, dióxido de silício e dióxido de titânio.
Informações técnicas.
1. INDICAÇÕES
Ebatz® é indicado como terapia adjunta à dieta para reduzir os níveis elevados de colesterol total (CT), lipoproteína de baixa densidade (LDL-colesterol), apolipoproteína B (Apo-B), triglicérides (TG) e para aumentar os níveis de lipoproteína de alta densidade (HDL-colesterol) em pacientes adultos com hiperlipidemia primária ou dislipidemia mista.
A terapia medicamentosa deve ser um componente da intervenção para múltiplos fatores de risco em indivíduos que requerem modificações no perfil lipídico. Agentes que alteram os lipídeos só devem ser usados em adição à dieta restrita de gorduras saturadas e colesterol quando a resposta à dieta e a outras medidas não farmacológicas forem inadequadas.
2. RESULTADOS DE EFICÁCIA
A pitavastatina cálcica não foi estudada especificamente em pacientes com dislipidemias de Fredrickson Tipo I, III e V.
Hiperlipidemia primária ou dislipidemia mista
Estudo de variação de dose: foi realizado um estudo multicêntrico, randomizado, duplo-cego, placebo-controlado, de variação de dose para avaliar a eficácia de pitavastatina cálcica comparada com placebo em 251 pacientes com hiperlipidemia primária. A pitavastatina cálcica administrada como uma dose única diária por 12 semanas reduziu significativamente o LDL-C, CT, TG e Apo-B plasmáticos comparado com placebo e foi associado com aumentos variáveis no HDL-C na variação de dose. 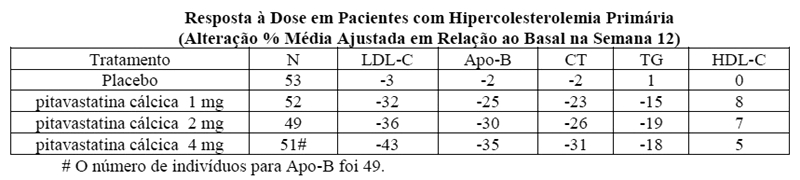
Estudos com comparadores ativos: todos os estudos de pitavastatina cálcica com comparadores ativos foram randomizados, multicêntricos, duplo-cegos, duplo-mascaramentos, com controle ativo, fase 3 de não inferioridade. Em todos os estudos, os pacientes participaram de um período introdutório de 6 a 8 semanas de eliminação/dieta e depois foram randomizados para uma dose uma vez ao dia de pitavastatina cálcica ou o comparador ativo por 12 semanas. A não-inferioridade de pitavastatina foi considerada demonstrada se o limite inferior do IC 95% para a diferença média de tratamento foi maior que -6% para a alteração percentual média no LDL-C.
Estudo com comparador ativo - atorvastatina (NK-104-301): pitavastatina cálcica foi comparado com o inibidor da HMG-CoA redutase, a atorvastatina, em um estudo com 817 pacientes com hiperlipidemia primária ou dislipidemia mista.
Os resultados lipídicos são mostrados na tabela abaixo. As comparações foram assim pareadas: pitavastatina cálcica 2 mg versus atorvastatina 10 mg e pitavastatina cálcica 4 mg versus atorvastatina 20 mg. As diferenças médias de tratamento (IC 95%) foram 0% (-3%, 3%) e 1% (-2%, 4%), respectivamente.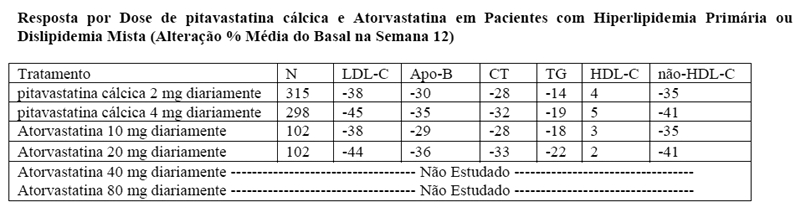
Estudo com comparador ativo - sinvastatina (NK-104-302): pitavastatina cálcica foi comparado com o inibidor da HMG-CoA redutase, a sinvastatina, em um estudo com 843 pacientes com hiperlipidemia primária ou dislipidemia mista. Os resultados lipídicos são mostrados na tabela abaixo. As comparações foram assim pareadas: pitavastatina cálcica 2 mg versus sinvastatina 20 mg e pitavastatina cálcica 4 mg versus sinvastatina 40 mg. As diferenças médias de tratamento (IC 95%) foram 4% (1%, 7%) e 1% (-2%, 4%), respectivamente.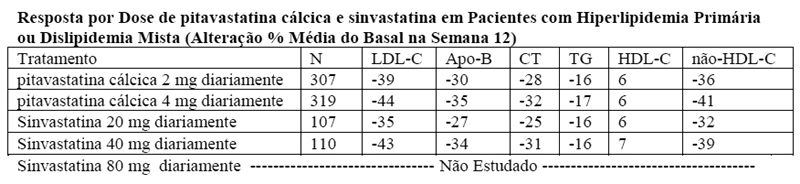
Estudo com comparador ativo, pravastatina, em idosos (NK-104-306): pitavastatina cálcica foi comparado com o inibidor da HMG-CoA redutase, a pravastatina, em um estudo com 942 pacientes idosos (≥ 65 anos) com hiperlipidemia primária ou dislipidemia mista.
Os resultados lipídicos são mostrados na tabela abaixo. A pitavastatina cálcica reduziu significativamente o LDL-C comparado à pravastatina como demonstrado pelas seguintes comparações pareadas da dose: pitavastatina cálcica 1 mg versus pravastatina 10 mg, pitavastatina cálcica 2 mg versus pravastatina 20 mg e pitavastatina cálcica 4 mg versus pravastatina 40 mg. As diferenças médias de tratamento (IC 95%) foram 9% (6%, 12%), 10% (7%, 13%) e 10% (7%, 13%), respectivamente.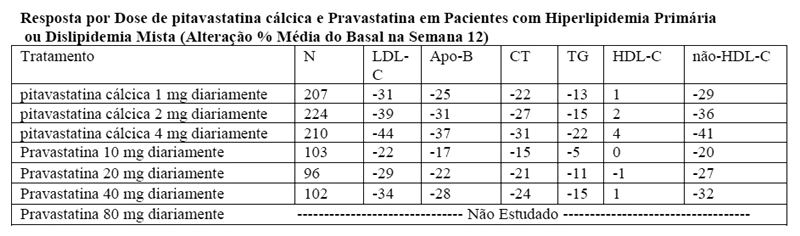
Estudo com comparador ativo, sinvastatina, em pacientes com ≥ 2 fatores de risco para doença coronariana cardíaca (NK-104-304): pitavastatina cálcica foi comparado com o inibidor da HMG-CoA redutase, a sinvastatina, em um estudo com 351 pacientes com hiperlipidemia primária ou dislipidemia mista com ≥ 2 fatores de risco para doença coronariana cardíaca.
Os resultados lipídicos são mostrados na tabela abaixo. A pitavastatina cálcica 4 mg foi não-inferior à sinvastatina 40 mg para a alteração percentual do basal até o desfecho no LDL-C. A diferença média de tratamento (IC 95%) foi 0% (-2%, 3%).
Estudo com comparador ativo, atorvastatina, em pacientes com diabetes mellitus tipo II (NK-104-305): pitavastatina cálcica foi comparado com o inibidor da HMG-CoA redutase, a atorvastatina, em um estudo com 410 indivíduos com diabetes mellitus tipo II e dislipidemia concomitantes.
Os resultados lipídicos são mostrados na tabela abaixo. A diferença de tratamento (IC 95%) para a alteração percentual no LDL-C em relação ao basal foi -2% (-6,2%, 1,5%). Os dois grupos de tratamento não foram estatisticamente diferentes para o LDL-C. Contudo, o limite inferior do IC foi -6,2%, excedendo levemente o limite de não-inferioridade de -6%, assim o objetivo de não-inferioridade não foi atingido.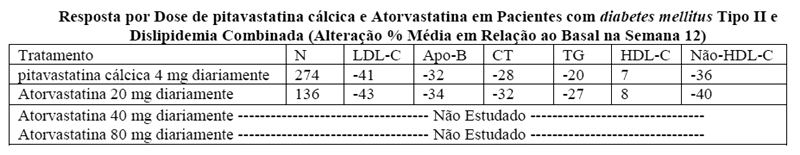
As diferenças de tratamento na eficácia na alteração do LDL-C em relação ao basal entre pitavastatina cálcica e os controles ativos nos estudos Fase 3 são resumidas na Figura abaixo.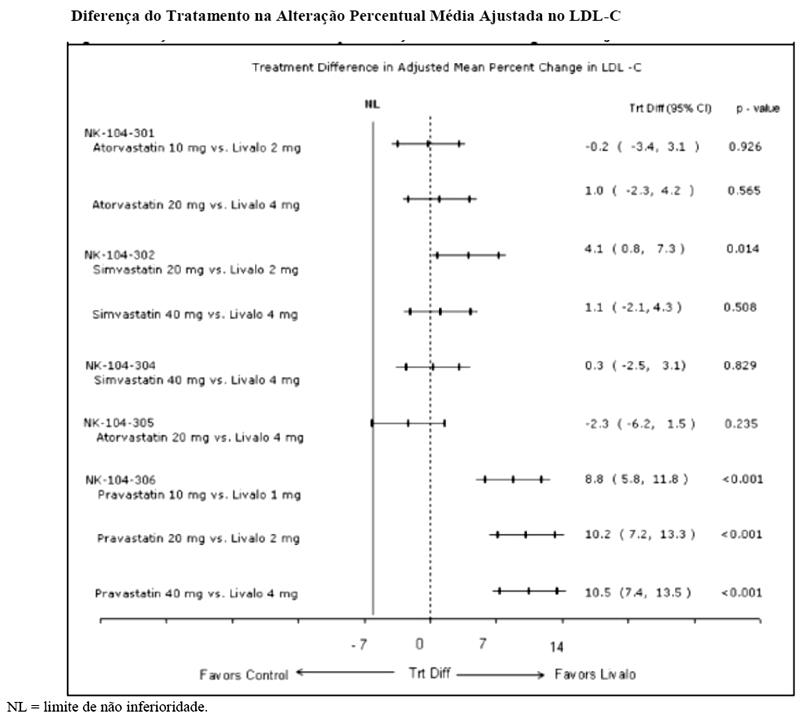
3. CARACTERÍSTICAS FARMACOLÓGICAS
Descrição: Ebatz®, um agente redutor de lipídeos sintético para administração oral, é um inibidor da HMG-CoA redutase. O nome químico da pitavastatina é (+) monocálcio bis{(3R, 5S, 6E)-7-[2-ciclopropil-4-(4-fluorofenil)-3-quinolil]-3,5-dihidroxi-6-heptenoato}.
A fórmula empírica da pitavastatina é C50H46CaF2N2O8 e o peso molecular é 880,98. A pitavastatina é inodora e apresentada como um pó branco a amarelo claro. É muito solúvel em piridina, clorofórmio, ácido clorídrico diluído e tetrahidrofurano, solúvel em etilenoglicol, moderadamente solúvel em octanol, levemente solúvel em metanol, muito pouco solúvel em água ou etanol e praticamente insolúvel em acetonitrila ou éter dietílico. A pitavastatina é higroscópica e levemente instável na luz.
Propriedades farmacodinâmicas: a pitavastatina inibe competitivamente a HMG-CoA redutase, que é uma enzima determinante da velocidade de reação envolvida na biossíntese do colesterol, numa forma de competição com o substrato, inibindo assim a síntese de colesterol no fígado. Como resultado, a expressão dos receptores de LDL seguida pela captação de LDL do sangue para o fígado é acelerada e depois o CT plasmático diminui. Além disso, a inibição contínua da síntese de colesterol no fígado diminui as concentrações das lipoproteínas de densidade muito baixa.
Em um estudo randomizado, duplo-cego, placebo-controlado, de 4 vias paralelas com comparador ativo com moxifloxacina em 174 participantes saudáveis, Ebatz® não foi associado com prolongamento clinicamente significativo do intervalo QTc ou da frequência cardíaca em doses diárias de até 16 mg (4 vezes a dose diária máxima recomendada).
Propriedades farmacocinéticas:
Absorção: as concentrações plasmáticas máximas de pitavastatina são atingidas cerca de 1 hora após a administração oral. Tanto a Cmáx quanto a AUC0-inf aumentaram de forma quase proporcional à dose com as doses únicas de pitavastatina de 1 a 24 mg uma vez ao dia. A biodisponibilidade absoluta da pitavastatina solução oral é de 51%. A administração de pitavastatina com uma refeição rica em gordura (50% de conteúdo de gordura) reduz a Cmáx de pitavastatina em 43% mas não reduz significativamente a AUC de pitavastatina. A Cmáx e a AUC da pitavastatina não diferiram após a administração da medicação à noite ou pela manhã. Em voluntários sadios recebendo 4 mg de pitavastatina, a alteração percentual do basal para o LDL-C após a administração à noite foi levemente maior do que após a administração pela manhã. A pitavastatina foi absorvida no intestino delgado, mas muito pouco no cólon.
Distribuição: a pitavastatina é ligada a mais de 99% das proteínas no plasma humano, principalmente à albumina e à glicoproteína alfa 1-ácida e o volume de distribuição médio é de aproximadamente 148 litros. A associação de pitavastatina e/ou seus metabólitos com as células sanguíneas é mínima.
Metabolismo: a pitavastatina é marginalmente metabolizada pelo CYP2C9 e em menor extensão pelo CYP2C8. O principal metabólito no plasma humano é a lactona, que é formada através de um conjugado glucuronida da pitavastatina tipo éster pela uridina 5'-difosfato (UDP) glucuronosiltransferase (UGT1A3 e UGT2B7).
Excreção: uma média de 15% da radioatividade da dose única administrada oralmente de 32 mg de pitavastatina 14C-marcado foi excretada na urina, enquanto uma média de 79% da dose foi excretada nas fezes dentro de 7 dias. A meia-vida de eliminação plasmática média é de aproximadamente 12 horas.
Farmacocinética em populações especiais:
Etnia: em estudos de farmacocinética, a Cmáx e AUC da pitavastatina foram 21% e 5% mais baixas, respectivamente nos negros ou afro-americanos voluntários sadios comparados aos voluntários sadios caucasianos. Na comparação farmacocinética entre voluntários caucasianos e japoneses, não houve nenhuma diferença significativa na Cmáx e AUC.
Sexo: em um estudo farmacocinético que comparou voluntários sadios homens e mulheres, a Cmáx e a AUC da pitavastatina foram 60% e 54% mais altas, respectivamente nas mulheres. Isso não teve nenhum efeito na eficácia ou segurança de pitavastatina em mulheres nos estudos clínicos.
Idosos: em um estudo farmacocinético que comparou voluntários jovens e idosos (≥ 65 anos) sadios, a Cmáx e a AUC da pitavastatina foram 10% e 30% mais altas, respectivamente, nos idosos. Isso não teve nenhum efeito na eficácia ou segurança de pitavastatina nos indivíduos idosos nos estudos clínicos.
Insuficiência renal: em pacientes com insuficiência renal moderada (taxa de filtração glomerular de 30 - 59 mL/min/1,73 m2) e doença renal em estágio terminal recebendo hemodiálise, a AUC0-inf da pitavastatina é 102% e 86% mais alta do que a de voluntários sadios, respectivamente, enquanto a Cmáx da pitavastatina é 60% e 40% mais alta do que a de voluntários sadios, respectivamente. Os pacientes receberam hemodiálise imediatamente antes da administração de pitavastatina e não fizeram hemodiálise durante o estudo farmacocinético. Os pacientes em hemodiálise tem aumentos de 33% e 36% na fração média não ligada de pitavastatina comparados com voluntários sadios e pacientes com insuficiência renal moderada, respectivamente.
Em outro estudo de farmacocinética, pacientes com insuficiência renal severa (taxa de filtração glomerular de 15 - 29 mL/min/1,73 m2) não recebendo hemodiálise, foi administrada dose única de pitavastatina 4 mg. A AUC0-inf e a Cmáx foram 36% e 18% mais alto, respectivamente, quando comparado com a dos voluntários sadios. Para ambos os pacientes, os com insuficiência renal severa e os voluntários sadios, a porcentagem média da fração não ligada de pitavastatina é de aproximadamente 0,6%.
O efeito da insuficiência renal leve na exposição da pitavastatina não foi estudado.
Insuficiência hepática: a disposição da pitavastatina foi comparada em voluntários sadios e pacientes com vários graus de insuficiência hepática. A razão da Cmáx da pitavastatina entre pacientes com insuficiência hepática moderada (Doença Child-Pugh B) e voluntários sadios foi de 2,7. A razão da AUCinf de pitavastatina entre pacientes com insuficiência hepática moderada e voluntários sadios foi de 3,8. A razão da Cmáx da pitavastatina entre pacientes com insuficiência hepática leve (Doença Child-Pugh A) e voluntários sadios foi de 1,3. A razão da AUCinf da pitavastatina entre pacientes com insuficiência hepática leve e voluntários sadios foi de 1,6. O tempo de meia-vida médio da pitavastatina para insuficiência hepática moderada, insuficiência hepática leve e indivíduos sadios foi de 15, 10 e 8 horas, respectivamente.
Interação droga-droga: a principal via de metabolismo da pitavastatina é a glucuronidação via UGTs hepáticas com formação subsequente de lactona de pitavastatina. Há apenas um metabolismo mínimo pelo sistema do citocromo P450.
varfarina: a farmacodinâmica em estado de equilíbrio [razão internacional normalizada (INR) e o tempo de protrombina (PT)] e a farmacocinética da varfarina em voluntários sadios não foram afetadas pela coadministração de pitavastatina 4 mg diariamente. Contudo, os pacientes recebendo varfarina devem ter seu PT ou INR monitorados quando a pitavastatina for adicionada ao seu tratamento.
atazanavir: com base nas informações atuais, não há nenhuma interação medicamentosa clinicamente significante entre a pitavastatina e o atazanavir.
enalapril e Diltiazem: também com base nas informações atuais, não há nenhuma interação medicamentosa clinicamente significante entre pitavastatina e enalapril ou pitavastatina e diltiazem.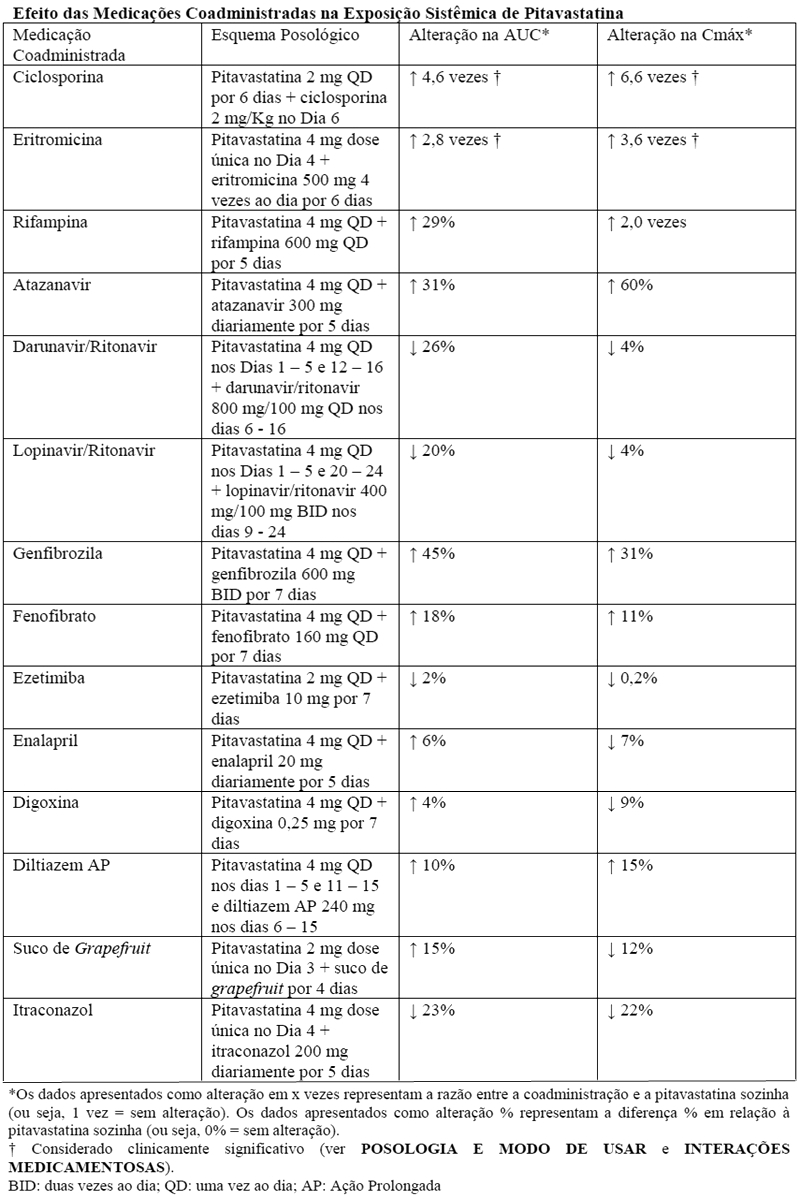
4. CONTRAINDICAÇÕES
Ebatz® é contraindicado nas seguintes condições:
• Pacientes com hipersensibilidade conhecida a qualquer componente desse produto. Foram relatadas reações de hipersensibilidade incluindo erupção da pele, prurido e urticária com pitavastatina (ver REAÇÕES ADVERSAS).
• Pacientes com doença hepática ativa que pode incluir elevações persistentes inexplicáveis das concentrações de transaminase hepática (ver ADVERTÊNCIAS E PRECAUÇÕES).
• Mulheres grávidas ou em idade fértil. Como os inibidores da HMG-CoA redutase reduzem a síntese de colesterol e possivelmente a síntese de outras substâncias biologicamente ativas derivadas do colesterol, Ebatz® pode causar dano fetal quando administrado a mulheres grávidas. Também, não há nenhum benefício aparente à terapia durante a gravidez e a segurança em mulheres grávidas não foi estabelecida. Se a paciente engravidar enquanto estiver tomando esse medicamento, a paciente deve ser informada sobre o risco potencial para o feto e a ausência de benefícios clínicos conhecidos com a continuação do uso durante a gravidez (ver ADVERTÊNCIAS E PRECAUÇÕES).
• Mulheres amamentando. Não se sabe se a pitavastatina é excretada no leite humano, contudo, foi demonstrado que uma outra droga dessa classe passa para o leite materno. Uma vez que os inibidores da HMG-CoA redutase tem o potencial de causar reações adversas sérias em bebês lactentes, mulheres que requerem tratamento com Ebatz® não devem amamentar seus bebês (ver ADVERTÊNCIAS E PRECAUÇÕES).
• Coadministração com ciclosporina (ver INTERAÇÕES MEDICAMENTOSAS).
• Coadministração com eritromicina (ver INTERAÇÕES MEDICAMENTOSAS).
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Efeitos no músculo esquelético: casos de miopatia e rabdomiólise com insuficiência renal aguda secundária à mioglobinúria foram relatados com inibidores da HMG-CoA redutase, incluindo pitavastatina. Esses riscos podem ocorrer com qualquer nível de dose, mas aumentam de forma dependente da dose.
Ebatz® deve ser prescrito com cautela para pacientes com fatores de predisposição para miopatia. Esses fatores incluem idade avançada (≥ 65 anos), insuficiência renal e hipotireoidismo tratado inadequadamente. O risco de miopatia também pode ser aumentado com a administração concomitante de fibratos ou doses de niacina modificadoras de lipídeos. Ebatz® deve ser administrado com cautela para pacientes com função renal comprometida, pacientes idosos ou quando usado concomitantemente com fibratos ou doses de niacina modificadoras de lipídeos (ver INTERAÇÕES MEDICAMENTOSAS).
Casos de miopatia, incluindo rabdomiólise, foram relatados com os inibidores da HMG-CoA redutase coadministrados com colchicina, portanto Ebatz® deve ser prescrito com cautela quando usado concomitantemente com colchicina (ver INTERAÇÕES MEDICAMENTOSAS).
Foram relatados casos raros de miopatia necrotizante imunomediada (MNI), uma miopatia autoimune, associada com o uso de estatinas. MNI é caracterizada por: fraqueza do músculo proximal e elevação da creatinaquinase sérica, que persiste mesmo após a descontinuação do tratamento com estatinas; biópsia muscular mostrando miopatia necrotizante sem inflamação significativa; melhoras com agentes imunosupressores.
O tratamento com Ebatz® deve ser descontinuado se ocorrerem concentrações marcantemente elevadas de creatinaquinase (CK),se for diagnosticada ou houver suspeita de miopatia. O tratamento com Ebatz® também deve ser temporariamente suspenso em todos os pacientes com uma condição séria aguda sugestiva de miopatia, ou predisposição para o desenvolvimento de insuficiência renal secundária à rabdomiólise (por exemplo: sepse, hipotensão, desidratação, cirurgia maior, trauma, distúrbios metabólicos, endócrinos e eletrolíticos severos ou convulsões não controladas). Todos os pacientes devem ser avisados para relatar prontamente dor, sensibilidade ou fraqueza muscular inexplicáveis, particularmente se acompanhadas por mal-estar, febre ou se os sinais e sintomas musculares persistirem depois de descontinuar o tratamento com Ebatz®.
Insuficiência hepática: foram relatados aumentos nas transaminases séricas [aspartato aminotransferase (AST)/transaminase glutâmica-oxaloacética sérica ou alanina aminotransferase (ALT)/transaminase glutâmica-pirúvica sérica] com inibidores da HMG-CoA redutase, incluindo Ebatz®. Na maior parte dos casos, as elevações foram transitórias e se resolveram ou melhoraram com a continuação do tratamento ou após uma breve interrupção do tratamento.
Nos estudos de Fase 2 placebo-controlados, a ALT > 3 vezes o limite superior de normalidade não foi observada nos grupos de placebo, pitavastatina 1 mg ou pitavastatina 2 mg. Um dos 202 pacientes (0,5%) que recebeu pitavastatina 4 mg teve ALT > 3 vezes o limite superior de normalidade.
É recomendado que os testes de enzimas hepáticas sejam realizados antes do início do Ebatz® e se aparecerem sinais e sintomas de insuficiência hepática.
Houve relatos raros, pós-comercialização, de casos de insuficiência hepática fatal e não fatal nos pacientes que tomam estatinas, incluindo pitavastatina. Se uma lesão hepática grave, com sintomas clínicos e/ou hiperbilirrubinemia, ou icterícia ocorrer durante o tratamento com Ebatz®, interromper o tratamento imediatamente. Se uma etiologia alternativa não for encontrada, não reiniciar Ebatz®.
Assim como outros inibidores da HMG-CoA redutase, Ebatz® deve ser usado com cautela em pacientes que consomem quantidades substanciais de álcool. Doença hepática ativa, que pode incluir elevações inexplicáveis persistentes da transaminase, é uma contraindicação para o uso de Ebatz® (ver CONTRAINDICAÇÕES).
Alterações das funções endócrinas: aumento nos níveis de HbA1c e glicemia em jejum foram relatados com os inibidores da HMG-CoA redutase, incluindo Ebatz®.
Insuficiência renal: a dose de pitavastatina deve ser individualizada em pacientes com insuficiência renal moderada e severa (taxa de filtração glomerular 30 - 59 mL/min/1,73 m2 e 15 - 29 mL/min/1,73 m2 não recebendo hemodiálise, respectivamente) bem como em doença renal em estágio terminal recebendo hemodiálise devem receber a dose inicial de Ebatz® de 1 mg uma vez ao dia e a dose máxima de Ebatz® de 2 mg uma vez ao dia (ver POSOLOGIA E MODO DE USAR).
Sistema cardiovascular: o efeito de Ebatz® na morbidade e mortalidade cardiovascular não foi determinado.
Gravidez categoria X - efeitos teratogênicos: Ebatz® é contraindicado em mulheres grávidas pois a segurança em mulheres grávidas não foi determinada e não há benefício aparente da terapia com Ebatz® durante a gravidez. Como os inibidores da HMG-CoA redutase diminuem a síntese de colesterol e, possivelmente a síntese de outras substâncias biologicamente ativas derivadas do colesterol, Ebatz® pode causar dano fetal quando administrado em mulheres grávidas. Ebatz® deve ser descontinuado se a paciente engravidar (ver CONTRAINDICAÇÕES). Os dados publicados sobre o uso de pitavastatina são limitados e insuficientes para determinar um risco associado à droga de malformações congênitas maiores ou aborto espontâneo. Em estudos de reprodução em animais, não foram observadas toxicidade embrio-fetal ou malformações congênitas quando ratas e coelhas grávidas foram submetidas à administração oral de pitavastatina durante a organogênese, sob exposições correspondentes, respectivamente, a 22 e 4 vezes a dose máxima humana recomendada (MRHD).
O risco estimado de maiores defeitos congênitos e aborto espontâneo para a população indicada é desconhecido. Efeitos adversos na gravidez ocorrem independentemente da saúde da mãe ou do uso de medicamentos.
Dados limitados publicados de pitavastatina não reportaram um risco associado à droga de malformações congênitas maiores ou aborto espontâneo. Raros relatos de anomalias congênitas foram recebidos após exposição intrauterina a inibidores da HMG-CoA redutase. Em uma revisão que acompanhou prospectivamente cerca de 100 gestações de mulheres expostas a outros inibidores da HMG-CoA redutase, as incidências de anomalias congênitas, abortos espontâneos e mortes fetais/natimortos não excederam a taxa esperada na população geral. O número de casos é adequado para excluir um aumento de anomalias congênitas maior ou igual do que 3 a 4 vezes a incidência histórica. Em 89% das gestações acompanhadas prospectivamente, o tratamento medicamentoso foi iniciado antes da gravidez e suspenso durante o primeiro trimestre quando a gravidez foi identificada.
Estudos de toxicidade reprodutiva mostraram que a pitavastatina atravessa a placenta em ratos e é detectada nos tecidos fetais a ≤ 36% das concentrações plasmáticas maternas, após uma dose única de 1 mg/Kg/dia durante a gestação.
Foram realizados estudos de desenvolvimento embriofetal em ratas prenhas tratadas com 3, 10 e 30 mg/Kg/dia de pitavastatina, por sonda oral, durante a organogênese. Não foram observados efeitos adversos a 3 mg/Kg/dia, exposições sistêmicas 22 vezes a exposição sistêmica humana a 4 mg/dia com base na AUC.
Estudos de desenvolvimento embriofetal foram realizados em coelhas prenhas tratadas com 0,1; 0,3 e 1 mg/Kg/dia de pitavastatina, por sonda oral, durante o período de organogênese fetal. Toxicidade materna, consistindo de peso corporal reduzido e aborto, foi observada em todas as doses testadas (4 vezes a exposição sistêmica humana a 4 mg/dia com base na AUC).
Em estudos perinatais/pós-natais em ratas prenhas recebendo doses por sonda oral de pitavastatina de 0,1; 0,3; 1; 3; 10 e 30 mg/Kg/dia da organogênese até o desmame, a toxicidade materna consistindo de mortalidade com ≥ 0,3 mg/Kg/dia e a lactação comprometida em todas as doses contribuiu para a redução na sobrevivência dos neonatos em todos os grupos de dose (0,1 mg/Kg/dia representa aproximadamente 1 vez a exposição sistêmica humana na dose de 4 mg/dia com base na AUC).
Lactação: Ebatz® é contraindicado durante a amamentação. Não há informação disponível sobre os efeitos do medicamento no bebê amamentado ou sobre os efeitos na produção do leite materno. Contudo, foi demonstrado que uma outra droga dessa classe passa para o leite materno.
Devido ao risco potencial de reações adversas sérias no bebê amamentando as pacientes devem ser aconselhadas de que a amamentação não é recomendada durante o tratamento com Ebatz® (ver CONTRAINDICAÇÕES).
Contracepção: Ebatz® pode causar dano fetal quando administrado a mulheres grávidas. Mulheres com potencial reprodutivo devem ser aconselhadas a utilizar um método contraceptivo eficaz durante o tratamento com Ebatz®.
Uso pediátrico: a segurança e a eficácia de Ebatz® em pacientes pediátricos não foram estabelecidas.
Uso geriátrico: dos 2.800 pacientes randomizados para Ebatz® 1 mg a 4 mg nos estudos clínicos controlados, 1.209 (43%) tinham 65 anos de idade ou mais. Não foram observadas diferenças significativas na eficácia ou segurança entre os pacientes idosos e os mais jovens. Contudo, a maior sensibilidade de alguns indivíduos mais velhos não pode ser excluída.
Carcinogênese, mutagênese e danos à fertilidade: em um estudo de carcinogenicidade de 92 semanas em camundongos recebendo pitavastatina, na dose máxima tolerada de 75 mg/Kg/dia, com exposições sistêmicas máximas (AUC) 26 vezes a exposição clínica máxima a 4 mg/dia, houve ausência de tumores relacionados com a droga.
Em um estudo de carcinogenicidade de 92 semanas em ratos recebendo pitavastatina a 1, 5 e 25 mg/Kg/dia por sonda oral, houve um aumento significativo na incidência de tumores da célula folicular da tireoide a 25 mg/Kg/dia, que representa 295 vezes as exposições sistêmicas humanas com base na AUC da dose máxima para humanos de 4 mg/dia.
Em um estudo de carcinogenicidade de 26 semanas em camundongos transgênicos (Tg rasH2) onde os animais receberam pitavastatina a 30, 75 e 150 mg/Kg/dia por sonda oral, não foram observados tumores clinicamente significativos.
A pitavastatina não foi mutagênica no teste de Ames com Salmonella typhimurium e Escherichia coli com e sem ativação metabólica, no teste de micronúcleo após a administração única em camundongos e administrações múltiplas em ratos, no teste de síntese de DNA não programada em ratos e no ensaio de Comet em camundongos. No teste de aberração cromossômica, foi observada clastogenicidade nas doses mais altas testadas que também induziram altos níveis de citotoxicidade.
A pitavastatina não teve efeitos adversos na fertilidade dos ratos machos e fêmeas nas doses orais de 10 e 30 mg/Kg/dia, respectivamente, em exposições sistêmicas 56 e 354 vezes a exposição clínica a 4 mg/dia com base na AUC.
O tratamento com pitavastatina em coelhos resultou em mortalidade em machos e fêmeas recebendo 1 mg/Kg/dia (30 vezes a exposição sistêmica clínica a 4 mg/dia com base na AUC) e recebendo uma dose mais alta durante um estudo de fertilidade. Apesar da causa da morte não ter sido determinada, os coelhos tinham sinais macroscópicos de toxicidade renal (rins esbranquiçados), indicativo de possível isquemia. Doses mais baixas (15 vezes a exposição sistêmica em humanos) não mostraram toxicidade significativa em adultos machos e fêmeas. Contudo, foi observada redução nas implantações, aumento nas reabsorções e redução na viabilidade dos fetos.
Toxicidade no sistema nervoso central (SNC): lesões vasculares no SNC, caracterizadas por hemorragias perivasculares, edema e infiltração celular mononuclear dos espaços perivasculares, foram observadas em cães tratados com vários outros membros dessa classe de droga. Uma droga quimicamente semelhante nessa classe produziu degeneração do nervo ótico dependente da dose (degeneração Walleriana das fibras retino-geniculadas) em cães, numa dose que produziu concentrações plasmáticas da droga cerca de 30 vezes mais altas do que a concentração média da droga em humanos tomando a dose mais alta recomendada. Não foi observada degeneração Walleriana com pitavastatina. Foi observada catarata e opacidade do cristalino nos cães tratados por 52 semanas ao nível de dose de 1 mg/Kg/dia (9 vezes a exposição clínica na dose máxima para humanos de 4 mg/dia com base nas comparações da AUC).
Efeito sobre a capacidade de dirigir e operar máquinas: não existe um padrão nos eventos adversos que sugira que os pacientes utilizando Ebatz® não apresentem nenhum comprometimento na capacidade de dirigir e utilizar máquinas perigosas, mas deve-se levar em consideração que há relatos de tontura e sonolência durante o tratamento com Ebatz®.
Este medicamento causa malformação ao bebê durante a gravidez.
Este medicamento contém LACTOSE. Portanto, deve ser usado com cautela em pacientes que apresentem intolerância à lactose.
6. INTERAÇÕES MEDICAMENTOSAS
Ciclosporina: a ciclosporina aumentou significantemente a exposição à pitavastatina. A coadministração de ciclosporina com Ebatz® é contraindicada (ver CONTRAINDICAÇÕES).
Eritromicina: a eritromicina aumentou significantemente a exposição à pitavastatina. A coadministração de eritromicina com Ebatz® é contraindicada (ver CONTRAINDICAÇÕES).
Rifampina: a rifampina aumentou significativamente a exposição à pitavastatina. Em pacientes tomando rifampina, a dose de Ebatz® 2 mg uma vez ao dia não deve ser excedida (ver POSOLOGIA E MODO DE USAR).
Genfibrozila: devido a um risco aumentado de miopatia/rabdomiólise quando os inibidores da HMG-CoA redutase são coadministrados com genfibrozila, a administração concomitante de Ebatz® com genfibrozila deve ser evitada.
Outros fibratos: como sabe-se que o risco de miopatia durante o tratamento com inibidores da HMG-CoA redutase é aumentado com a administração concomitante de outros fibratos, Ebatz® deve ser administrado com cautela quando usado concomitantemente com outros fibratos (ver ADVERTÊNCIAS E PRECAUÇÕES).
Niacina: o risco de efeitos no músculo esquelético pode aumentar quando Ebatz® é usado em combinação com niacina; deve-se levar em consideração a redução na dose de Ebatz® nesse grupo (ver ADVERTÊNCIAS E PRECAUÇÕES).
Colchicina: casos de miopatia, incluindo rabdomiólise, foram relatados com os inibidores da HMG-CoA redutase coadministrados com colchicina, portanto, Ebatz® deve ser prescrito com cautela quando usado concomitantemente com colchicina (ver ADVERTÊNCIAS E PRECAUÇÕES).
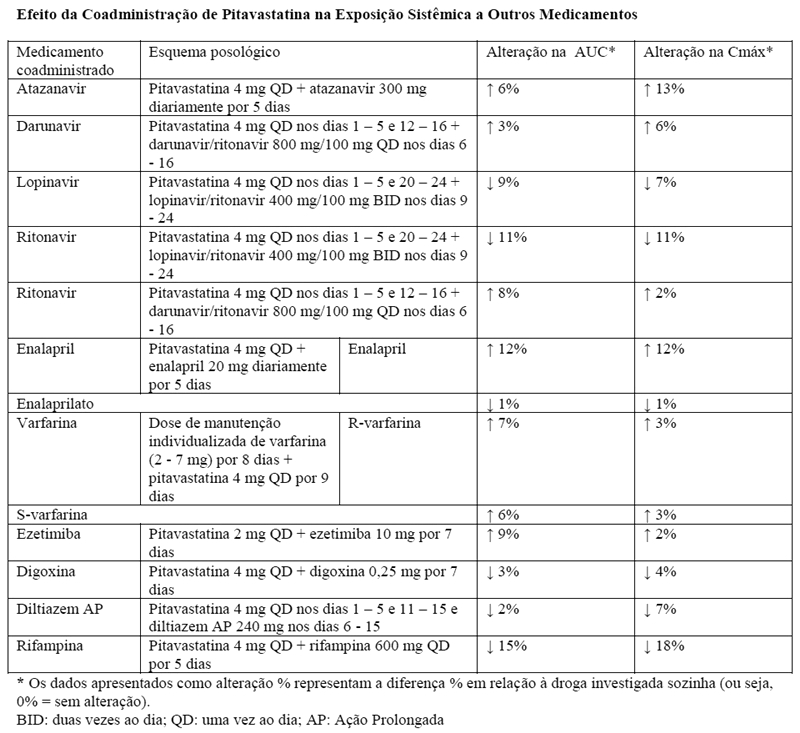
Varfarina: Ebatz® não teve nenhuma interação farmacocinética significativa com R- e S-varfarina. Pitavastatina não teve nenhum efeito significativo no tempo de protrombina (TP) e na razão normalizada internacional (INR) quando administrado em pacientes recebendo tratamento crônico com varfarina. Contudo, os pacientes recebendo varfarina devem ter seus TP e INR monitorados quando a pitavastatina é adicionada ao seu tratamento.
Atazanavir: com base nas informações atuais, não há nenhuma interação medicamentosa clinicamente significante entre a pitavastatina e o atazanavir.
Enalapril e diltiazem: com base nas informações atuais, não há nenhuma interação medicamentosa clinicamente significante entre a pitavastatina e enalapril ou pitavastatina e diltiazem.
Não foram realizados estudos para investigar a possível interação entre pitavastatina e plantas medicinais ou nicotina. Além disso, não há dados disponíveis da interação com testes laboratoriais. Assim como outros inibidores da HMG-CoA redutase, Ebatz® deve ser usado com cautela em pacientes que consomem quantidades substanciais de álcool.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Ebatz® deve ser conservado em temperatura ambiente (entre 15°C e 30°C), protegido da luz e umidade. O prazo de validade do produto nestas condições de armazenagem é de 30 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
2 mg: Ebatz® 2 mg apresenta-se na forma de comprimido revestido branco a quase branco, circular, biconvexo, com gravação "2" em uma das faces e liso na outra.
4 mg: Ebatz® 4 mg apresenta-se na forma de comprimido revestido branco a quase branco, circular, biconvexo, com gravação "4" em uma das faces e liso na outra.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
A variação de dose para Ebatz® é de 2 a 4 mg, por via oral, uma vez ao dia, a qualquer hora do dia, com ou sem alimento. A dose inicial recomendada é de 2 mg, e a dose máxima é de 4 mg. A dose inicial e as doses de manutenção de Ebatz® devem ser individualizadas, de acordo com as características do paciente, tal como o objetivo da terapia e da resposta.
Doses de Ebatz® acima de 4 mg uma vez ao dia foram associadas com aumento no risco de miopatia severa nos estudos clínicos realizados antes da comercialização. Não exceder a dose de 4 mg de Ebatz®, uma vez ao dia.
Após o início ou após a titulação de Ebatz®, as concentrações de lipídeos devem ser analisadas após 4 semanas, e a dose deve ser ajustada de acordo.
Em pacientes com insuficiência renal: a dose de pitavastatina deve ser individualizada em pacientes com insuficiência renal moderada e severa (taxa de filtração glomerular 30 - 59 mL/min/1,73 m2 e 15 - 29 mL/min/1,73 m2 não recebendo hemodiálise, respectivamente), bem como em pacientes com doença renal em estágio terminal recebendo hemodiálise, sendo a dose máxima de Ebatz® 2 mg, uma vez ao dia.
Uso com rifampina: em pacientes tomando rifampina, a dose de Ebatz® de 2 mg, uma vez ao dia, não deve ser excedida (ver INTERAÇÕES MEDICAMENTOSAS).
Este medicamento não deve ser partido, aberto ou mastigado.
9. REAÇÕES ADVERSAS
As seguintes reações adversas sérias são discutidas em maiores detalhes em outras seções da bula:
Reação muito rara < 1/10.000): rabdomiólise com mioglobinúria e insuficiência renal aguda e miopatia (incluindo miosite) (ver ADVERTÊNCIAS E PRECAUÇÕES) e anormalidades das enzimas hepáticas (ver ADVERTÊNCIAS E PRECAUÇÕES).
Dos 4.798 pacientes admitidos em 10 estudos clínicos controlados e 4 estudos com extensão aberta subsequente, 3.291 pacientes receberam pitavastatina 1 mg a 4 mg diariamente. A exposição contínua média de pitavastatina (1 mg a 4 mg) foi de 36,7 semanas (mediana 51,1 semanas). A idade média dos pacientes foi de 60,9 anos (variação: 18 anos - 89 anos) e a distribuição por sexo foi de 48% de homens e 52% de mulheres. Aproximadamente 93% dos pacientes eram caucasianos, 7% eram asiáticos/indianos, 0,2% eram afro-americanos e 0,3% eram hispânicos e de outras etnias.
Experiência nos estudos clínicos: como os estudos clínicos sobre pitavastatina são realizados em várias populações e vários desenhos de estudo, a frequência das reações adversas observadas nos estudos clínicos de pitavastatina não pode ser diretamente comparada com a dos estudos clínicos de outros inibidores da HMG-CoA redutase, e pode não refletir a frequência das reações adversas observadas na prática.
As reações adversas relatadas em ≥ 2% dos pacientes nos estudos clínicos controlados e com uma taxa maior ou igual à vista com placebo são mostradas abaixo. Esses estudos tiveram duração do tratamento de até 12 semanas.
Reação comum ( > 1/100 e < 1/10): dor nas costas, constipação, diarreia, mialgia, artralgia, cefaleia, gripe e nasofaringite.
Reação incomum ( > 1/1.000 e < 1/100): dor nas extremidades, elevação da creatina fosfoquinase e transaminases (ALT e AST).
Reação muito rara ( < 1/10.000): elevação da fosfatase alcalina, bilirrubina e glicose.
Nos estudos clínicos controlados e suas extensões abertas, 3,9% (1 mg), 3,3% (2 mg) e 3,7% (4 mg) dos pacientes tratados com pitavastatina foram retirados do estudo devido a reações adversas. As reações adversas mais comuns ( > 1/10) que levaram à descontinuação do tratamento foram: creatina fosfoquinase elevada (0,6% recebendo 4 mg) e mialgia (0,5% recebendo 4 mg).
Reações de hipersensibilidade incluindo erupções cutâneas, prurido e urticária foram relatadas com pitavastatina.
Relatos pós-comercialização:
As seguintes reações adversas foram identificadas durante a utilização de pitavastatina após aprovação. Por estas reações serem relatadas voluntariamente por uma população de dimensão incerta, nem sempre é possível estimar sua frequência ou estabelecer um relacionamento causal à exposição da droga.
As reações adversas relatadas associadas ao tratamento com pitavastatina, desde sua introdução no mercado, independentemente da avaliação de causalidade, incluem as seguintes:
Reação incomum ( > 1/1.000 e < 1/100): náusea, mal-estar, tontura, hipoestesia e espasmos musculares.
Reação rara ( > 1/10.000 e < 1/1.000): desconforto abdominal, dor abdominal, dispepsia, astenia, fadiga e insônia.
Reação muito rara ( < 1/10.000): hepatite, icterícia, insuficiência hepática fatal e não fatal, depressão, doença intersticial pulmonar e disfunção erétil.
Reação com frequência desconhecida: neuropatia periférica.
Houve relatos pós-comercialização de casos raros ( > 1/10.000 e < 1/1.000) de comprometimento cognitivo (por exemplo: perda de memória, esquecimento, amnésia, deterioração da memória e confusão) e miopatia necrotizante imunomediada (ver ADVERTÊNCIAS E PRECAUÇÕES) associados com o uso de estatina. Estes problemas cognitivos tem sido relatados para todas as estatinas. Os relatos são geralmente não graves e reversíveis com a descontinuação da estatina, com tempos variáveis para o início (1 dia a anos) e resolução dos sintomas (mediana de 3 semanas).
Em caso, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA, disponível em www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Não há um tratamento específico conhecido no caso de superdose de pitavastatina. No caso de superdose, o paciente deve ser tratado sintomaticamente e devem ser instituídas as medidas de suporte necessárias. É improvável que a hemodiálise seja benéfica devido à alta proporção de ligação proteica da pitavastatina.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS n°: 1.0033.0206
Venda sob prescrição médica.
Esta bula foi