DOVATO
GLAXOSMITHKLINE
dolutegravir + lamivudina
Anti-retroviral.
Apresentações.
Dovato® comprimidos revestidos, contendo 50 mg de dolutegravir e 300 mg de lamivudina, é apresentado em uma embalagem com 30 comprimidos.
USO ORAL
USO ADULTO E PEDIÁTRICO (ACIMA DE 12 ANOS COM PESO MÍNIMO DE 40 KG)
Composição.
Cada comprimido revestido de Dovato® contém: dolutegravir 50 mg (equivalentes a 52,6 mg de dolutegravir sódico), lamivudina 300 mg excipientes* q.s.p. 1 comprimido revestido
*celulose microcristalina, amidoglicolato de sódio, estearato de magnésio, manitol, povidona, estearilfumarato de sódio e AquariusTM Branco BP18237 ou Opadry® Branco OY-S-28876 (hipromelose, dióxido de titânio e macrogol).
Informações técnicas.
1. INDICAÇÕES
Dovato® é uma combinação de dois medicamentos contendo dolutegravir (um inibidor de transferência da fita da integrase -INSTI-) e lamivudina (inibidor da transcriptase reversa análogo de nucleosídeo -ITRN-) é indicado como um regime completo para o tratamento da infecção pelo vírus da imunodeficiência humana tipo 1 (HIV-1) em adultos e adolescentes acima de 12 anos pesando pelo menos 40 kg sem histórico de tratamento antirretroviral prévio ou em substituição ao regime antirretroviral atual em pessoas com supressão virológica (RNA do HIV-1 inferior a 50 cópias por mL) em um regime antirretroviral estável, sem histórico de falha ao tratamento e nenhuma substituição conhecida associada à resistência aos componentes individuais de Dovato® .
2. RESULTADOS DE EFICÁCIA
Indivíduos virgens de tratamento
A eficácia da associação em dose fixa de DTG/3TC é corroborada por dados de 2 estudos idênticos, de 148 semanas, de Fase III, randomizados, duplo-cegos, multicêntricos, de grupos paralelos, de não inferioridade, controlados (GEMINI-1 [204861] e GEMINI-2 [205543]). Um total de 1433 indivíduos adultos vivendo com HIV-1 virgens ao tratamento antirretroviral receberam tratamento nos estudos. Foram incluídos indivíduos que possuíam, no momento da triagem, HIV-1 RNA no plasma entre 1000 c/mL a ≤500.000 cópias/mL. Os indivíduos foram randomizados para um regime de dois fármacos com dolutegravir mais lamivudina administrados uma vez ao dia ou dolutegravir mais a associação em dose fixa de tenofovir/entricitabina administrados uma vez ao dia. O desfecho primário da eficácia para cada estudo GEMINI foi a proporção de indivíduos com HIV-1 RNA no plasma < 50 cópias/mL na Semana 48 (algoritmo Snapshot para a população ITT-E). No período basal, na análise agrupada, a idade mediana dos indivíduos foi de 33 anos, 15% eram do sexo feminino, 69% eram brancos, 9% eram de Estágio 3 de acordo com o CDC (AIDS), 20% tinham HIV-1 RNA > 100.000 cópias/mL e 8% tinham contagem de células CD4+ de menos de 200 células por mm3; essas características foram semelhantes entre os estudos e braços de tratamento. Na análise primária da semana 48, dolutegravir mais lamivudina foi não inferior a dolutegravir mais a associação em dose fixa de tenofovir/entricitabina nos estudos GEMINI-1 e GEMINI-2. Isto foi corroborado pela análise agrupada, ver Tabela 1.
Na semana 96 dos estudos GEMINI-1 e GEMINI-2, o grupo dolutegravir mais lamivudina (86% com HIV-1 RNA plasmático < 50 cópias/mL [dados combinados]) permaneceu não inferior ao grupo de dolutegravir mais a associação em dose fixa de tenofovir/entricitabina (90% com HIV-1 RNA plasmático < 50 cópias / mL [dados combinados]). A diferença ajustada em proporções e IC 95% foi de -3,4% (-6,7; 0,0). Os resultados da análise agrupada estavam alinhados com os dos estudos individuais, para os quais o desfecho secundário (diferença na proporção de HIV-1 RNA plasmático na semana 96 < 50 cópias / mL, com base no algoritmo Snapshot para dolutegravir mais lamivudina versus dolutegravir mais a associação em dose fixa de tenofovir/entricitabina). As diferenças ajustadas de -4,9 (IC 95%: -9,8; 0,0) para o GEMINI-1 e -1,8 (IC 95%: -6,4; 2,7) para o GEMINI-2 estavam dentro da margem de não inferioridade pré-especificada de -10%. O aumento médio na contagem de células T CD4 + foi de 269 células/nm3 no braço DTG + 3TC e 259 células/nm3 no braço DTG + FTC/TDF, na semana 96.
Na semana 144 dos estudos GEMINI 1 e GEMINI 2, o grupo dolutegravir mais lamivudina (82% com RNA do HIV 1 plasmático < 50 cópias/mL [dados agrupados]) permaneceu não inferior ao grupo dolutegravir mais dose fixa de tenofovir/entricitabina entricitabina (84% com RNA do HIV 1 no plasma < 50 cópias/mL [dados agrupados]). Os resultados da análise agrupada estavam de acordo com os dos estudos individuais, para os quais o desfecho secundário (diferença na proporção < 50 cópias/mL de RNA HIV 1 plasmático na Semana 144 com base no algoritmo Snapshot para dolutegravir mais lamivudina versus dolutegravir mais dose fixa de tenofovir/entricitabina) foi atendida. A diferença ajustada nas proporções e IC de 95% para os dados agrupados foi de -1,8% (-5,8, 2,1). As diferenças ajustadas de 3,6 (IC 95%: -9,4, 2,1) para GEMINI-1 e 0,0 (IC 95%: -5,3, 5,3) para GEMINI 2 estavam dentro da margem de não inferioridade préespecificada de -10%.
O aumento médio na contagem de células T CD4+ foi de 302 células/mm3 no braço DTG+3TC e 300 células/mm3 no braço DTG+FTC/TDF, na Semana 144
Indivíduos virologicamente suprimidos
A eficácia da associação em dose fixa de DTG/3TC em indivíduos vivendo com HIV, experientes à terapia antirretroviral e virologicamente suprimidos é apoiada por dados de 200 semanas de um estudo de Fase III controlado, de não inferioridade, randomizado, aberto, multicêntrico, de grupo paralelo, sem controle, (TANGO [204862]). Um total de 741 indivíduos adultos vivendo com HIV-1 que estavam em um regime supressivo estável contendo tenofovir alafenamida (TBR) receberam tratamento nos estudos. Os indivíduos foram randomizados em uma proporção de 1: 1 para receber a associação em dose fixa de DTG/3TC uma vez ao dia ou continuar com TBR por até 148 semanas. Na Semana 148, os indivíduos randomizados para continuar com TBR foram trocados para dose fixa de DTG/3TC uma vez ao dia. Todos os indivíduos foram acompanhados até a Semana 200. A randomização foi estratificada pela classe do terceiro agente na terapia de base (Inibidor da Protease [IP], Inibidor da Integrase [IN] ou Inibidor Não Nucleosídeo da Transcriptase Reversa [ITRNN]). O desfecho primário de eficácia foi a proporção de indivíduos com HIV1 RNA plasmático ≥50 c/mL (não resposta virológica) conforme a categoria do Snapshot FDA na semana 48 (algoritmo de Snapshot que ajusta para o fator de estratificação da randomização: classe do terceiro agente de base [IN, IP, ITRNN]). Na linha de base, a idade média dos indivíduos era de 39 anos, 8% eram do sexo feminino e 21% não brancos, 5% eram da classe C do CDC (AIDS) e 98% tinham a contagem de CD4 + basal ≥200 células/mm3; características semelhantes entre os braços de tratamento. Os indivíduos estavam em TARV por uma mediana de 2,8 anos antes do Dia 1 para o braço da associação em dose fixa DTG/3TC e 2,9 anos antes do Dia 1 para o braço TBR. A maioria dos indivíduos usava TBR baseado em INSTI, 78% e 80% nos braços da associação em dose fixa DTG/3TC e TBR, respectivamente. Na análise primária da Semana 48a dose fixa de DTG/3TC é não inferior ao TBR, com < 1% dos indivíduos em ambos os braços experimentando falha virológica (HIV-1 RNA ≥50 c/mL), com base no algoritmo Snapshot (Tabela 2)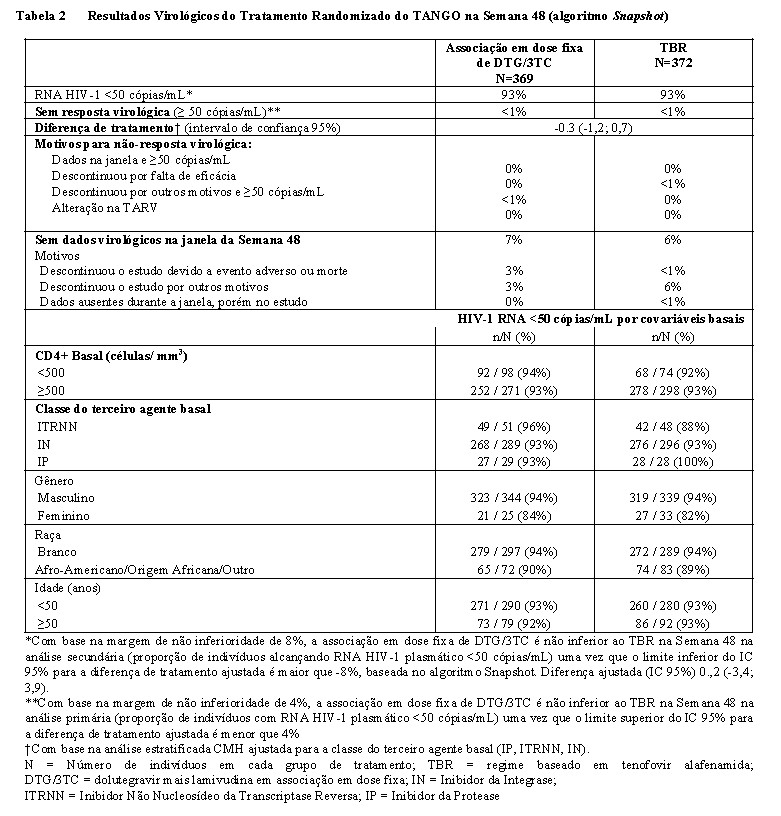
Na Semana 96 do estudo TANGO, a proporção de indivíduos com HIV-1 RNA ≥50 c/mL (Snapshot) foi de 0,3% e 1,1% nos grupos de dose fixa de DTG/3TC e TBR, respectivamente. Com base em uma margem de não inferioridade de 4%, o de dose fixa de DTG/3TC permaneceu não inferior ao TBR, pois o limite superior do IC de 95% para a diferença de tratamento ajustada (-2,0%, 0,4%) foi inferior a 4% para a população ITT E.
A alteração mediana da linha de base nas contagens de células T CD4+ na Semana 96 foi de 61 células/mm3 no braço de dose fixa de DTG+3TC e 45 células/mm3 no braço TBR.
Na Semana 144, a proporção de indivíduos com RNA do HIV-1 ≥50 c/mL (Snapshot) foi de 0,3% e 1,3% nos grupos com dose fixa de DTG/3TC e TBR, respectivamente. Com base em uma margem de não inferioridade de 4%, a dose fixa de DTG/3TC permaneceu não inferior ao TBR, pois o limite superior do IC de 95% para a diferença de tratamento ajustada (-2,4%, 0,2%) foi inferior a 4% para a população ITT E. A alteração mediana da linha de base nas contagens de células T CD4+ na Semana 144 foi de 36 células/mm3 no braço de dose fixa de DTG+3TC e 35 células/mm3 no braço TBR.
Antiretroviral Pregnancy Registry (APR)
O APR recebeu relatórios de mais de 600 exposições ao dolutegravir durante a gravidez, resultando em nascidos com vida, a partir de julho de 2019. Estes consistem em mais de 370 exposições durante o primeiro trimestre, mais de 230 exposições durante o segundo / terceiro trimestre e incluíram 12 e 9 defeitos congênitos, respectivamente. A prevalência (IC 95%) de defeitos entre bebês nascidos com vida e expostos ao dolutegravir no primeiro trimestre foi de 3,2% (1,7%, 5,5%) e no segundo / terceiro trimestre, 3,8% (1,7%, 7,0%). O APR recebeu relatórios de mais de 12.500 exposições à lamivudina durante a gravidez, resultando em nascidos com vida, em julho de 2019. Estes consistem em mais de 5.200 exposições durante o primeiro trimestre, mais de 7.400 exposições durante o segundo / terceiro trimestre e incluíram 161 e 216 defeitos de nascença, respectivamente. A prevalência (IC 95%) de defeitos entre bebês nascidos com vida expostos à lamivudina no primeiro trimestre foi de 3,1% (2,6, 3,6%) e no segundo / terceiro trimestre, 2,9% (2,5, 3,3%). Os dados disponíveis do APR não mostram aumento significativo no risco de defeitos congênitos importantes para dolutegravir ou lamivudina em comparação com as taxas de fundo nos dois sistemas de vigilância [Programa de Defeitos Congênitos da área Metropolitana de Atlanta (Metropolitan Atlanta Congenital Defects Program) com 2,72 defeitos por 100 nascidos com vida e o Registro de Defeitos Congênitos do Texas (Texas Birth Defects Registry) com 4,17 por 100 nascidos com vida].
Crianças Não existem dados de estudos clínicos com Dovato® na população pediátrica. Em um estudo de Fase I/II, de 48 semanas, multicêntrico, aberto (P1093/ING112578), os parâmetros farmacocinéticos, segurança, tolerabilidade e eficácia de dolutegravir foram avaliados em regimes de combinação em bebês, crianças e adolescentes vivendo com o HIV-1. Com 24 semanas, 16 de 23 (69%) adolescentes (com 12 a menos de 18 anos de idade) tratados com dolutegravir, uma vez ao dia (35 mg n=4, 50 mg n=19) mais terapia de base otimizada atingiram carga viral de menos de 50 cópias/mL.
Referências Bibliográficas:
Cahn P, Madero JS, Arribas J, Antinori, Ortiz R, Clarke A, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naïve adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet 2019; 393: 143-155.
VIANI, RM. et al. Safety, Pharmacokinetics and Efficacy of Dolutegravir in Treatment-experienced HIV-1 Infected Adolescents: Forty-eightweek Results from IMPAACT P1093. Pediatr Infect Dis J, 34(11): 1207-13, 2015.
Study 204861 (GEMINI-1) and Study 205543 (GEMINI-2), A Phase III, randomized, double-blind, multicentre, parallelgroup, non-inferiority study evaluating the efficacy, safety, and tolerability of dolutegravir plus lamivudine compared to dolutegravir plus tenofovir/emtricitabine in HIV-1-infected treatment-naïve adults - Week 96.
Study 204861 (GEMINI-1) and Study 205543 (GEMINI-2): A Phase III, randomized, double-blind, multicentre, parallelgroup, non-inferiority study evaluating the efficacy, safety, and tolerability of dolutegravir plus lamivudine compared to dolutegravir plus tenofovir/emtricitabine in HIV-1-infected treatment-naïve adults - Week 48-addendum 01.
Study 204861 (GEMINI-1) and Study 205543 (GEMINI-2), A Phase III, randomized, double-blind, multicentre, parallelgroup, non-inferiority study evaluating the efficacy, safety, and tolerability of dolutegravir plus lamivudine compared to dolutegravir plus tenofovir/emtricitabine in HIV-1-infected treatment-naïve adults - Week 96
Study 204862 (TANGO): A Phase III, randomized, multicenter, parallel-group, non-inferiority study evaluating the efficacy, safety, and tolerability of switching to dolutegravir plus lamivudine in HIV-1 infected adults who are virologically suppressed on a TAF-based regimen.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
Código ATC
Grupo farmacoterapêutico: Antiviral para uso sistêmico. Antivirais para tratamento de infecções pelo HIV, combinações. Código ATC: J05AR25
Mecanismo de Ação
O dolutegravir inibe a integrase do HIV ligando-se ao sítio ativo da integrase e bloqueando a etapa de transferência da fita da integração do ácido desoxirribonucleico (DNA) retroviral, que é essencial para o ciclo de replicação do HIV. Os ensaios bioquímicos de transferência da fita usando DNA da integrase do HIV 1 purificado e do substrato pré-processado resultaram em valores de IC50 de 2,7 nM e 12,6 nM. In vitro, dolutegravir se dissocia lentamente do sítio ativo do complexo integrase-DNA do tipo selvagem (t½ de 71 horas). A lamivudina é um inibidor nucleosídeo da transcriptase reversa (ITRN) e é um inibidor potente e seletivo do HIV-1 e HIV-2. A lamivudina é metabolizada sequencialmente por quinases intracelulares para o respectivo trifosfato (TP) que é o componente ativo com uma meia-vida intracelular prolongada, corroborando a administração uma vez ao dia (ver Eliminação, em Propriedades Farmacocinéticas abaixo). Lamivudina-TP é um substrato para e um inibidor competitivo da transcriptase reversa (TR) do HIV. No entanto, a sua principal atividade antiviral é por meio da incorporação da forma monofosfato na cadeia do DNA viral, resultando na terminação da cadeia. Lamivudina-TP mostra significativamente menos afinidade para as polimerases do DNA da célula hospedeira.
Efeitos Farmacodinâmicos
Em um estudo randomizado, de variação da dose, os indivíduos vivendo com HIV 1 tratados com a monoterapia de dolutegravir (ING111521) demonstraram atividade antiviral rápida e dose-dependente, com declínios médios desde o período basal até o dia 11 no HIV-1 RNA de 1,5, 2,0 e 2,5 log10 para dolutegravir 2 mg, 10 mg e 50 mg, uma vez ao dia, respectivamente. Esta resposta antiviral foi mantida por 3 a 4 dias após a última dose no grupo com 50 mg.
Atividade antiviral em cultura de células
O dolutegravir exibiu atividade antiviral contra cepas laboratoriais do HIV-1 do tipo selvagem com concentração média do fármaco necessária para efetuar a replicação viral em 50 por cento (EC50) em valores de 0,5 nM (0,21 ng por mL) a 2,1 nM (0,85 ng por mL) em células mononucleares do sangue periférico (PBMCs) e células MT-4. Em um ensaio de suscetibilidade da integrase viral usando a região de codificação da integrase de 13 isolados clinicamente diversos do subtipo B, dolutegravir demonstrou potência antiviral semelhante às cepas laboratoriais de referência, com uma EC50 média de 0,52 nM. Quando testado em ensaios com PBMC contra um painel consistindo de 24 isolados clínicos do HIV-1 [grupo M (subtipos A, B, C, D, E, F e G) e grupo O] e 3 isolados clínicos do HIV-2, a média geométrica da EC50 foi de 0,20 nM e os valores de EC50 variaram de 0,02 a 2,14 nM para HIV-1, enquanto a média geométrica da EC50 foi de 0,18 nM e os valores de EC50 variaram de 0,09 a 0,61 nM para os isolados do HIV-2. A atividade antiviral da lamivudina contra o HIV-1 foi avaliada em inúmeras linhas celulares incluindo monócitos e PBMCs usando ensaios de suscetibilidade padrão. Os valores de EC50 foram do intervalo de 0,003 a 0,17 mM. Os valores de EC50 da lamivudina contra subtipos diferentes do HIV-1 (A-G) variaram de 0,001 a 0,120 mM e contra isolados do HIV-2 de 0,003 a 0,120 mM em PBMCs.
Atividade antiviral em combinação com outros agentes antivirais
Nenhum fármaco com atividade anti-HIV inerente teve ação antagonista ao dolutegravir (avaliações in vitro foram realizadas em formato de checkerboard em combinação com estavudina, abacavir, efavirenz, nevirapina, lopinavir, amprenavir, enfuvirtida, maraviroque, adefovir e raltegravir). Além disso, os antivirais sem atividade anti-HIV inerente (ribavirina) não tiveram efeito aparente na atividade do dolutegravir. Não foram observados efeitos antagonistas in vitro com lamivudina e outros antirretrovirais (agentes testados: abacavir, didanosina, nevirapina, zalcitabina e zidovudina).
Efeito do Soro Humano e das Proteínas Séricas
Os estudos in vitro sugeriram um deslocamento de 75 vezes na EC50 de dolutegravir na presença de soro humano a 100% (pelo método de extrapolação) e a EC90 ajustada para proteína (PA-EC90) em PBMCs foi estimada em 64 ng/mL. A concentração mínima do dolutegravir para uma dose única de 50 mg em indivíduos virgens de tratamento com inibidor de integrase foi de 1,20 mg/mL, 19 vezes mais alta do que a PAEC90 estimada. A lamivudina apresenta farmacocinética linear ao longo do intervalo da dose terapêutica e apresenta baixa ligação à proteína no plasma (menos de 36%).
Resistência in vitro e in vivo (dolutegravir)
Vírus altamente resistentes ao dolutegravir não foram observados durante a passagem de 112 dias da cepa IIIB, com um fold change (FC) máximo de 4,1 vezes observado para as populações de vírus resistentes à passagem com substituições nas posições IN conservadas, S153Y e S153F. A passagem da cepa NL432 do HIV-1 do tipo selvagem na presença de dolutegravir selecionou substituições E92Q (FC do vírus da população de passagem=3,1) e G193E (FC do vírus da população de passagem=3,2) no Dia 56. A passagem adicional de vírus selvagens dos subtipos B, C e A/G na presença de dolutegravir selecionou G118R (FC do mutante direcionado ao sítio=10), S153T e R263K (FC do mutante direcionado ao sítio=1,5).
Indivíduos vivendo com o HIV-1 virgens de tratamento que receberam dolutegravir: Nenhuma mutação resistente ao INI ou aos ITRN de base foi isolada com dolutegravir 50 mg, uma vez ao dia, nos estudos com pacientes sem tratamento prévio em estudos randomizados de fase III.
Resistência in vitro e in vivo (lamivudina)
A resistência do HIV-1 à lamivudina envolve o desenvolvimento de uma alteração no aminoácido M184I ou M184V próxima ao sítio ativo da TR viral. Esta variante surge durante a seleção in vitro e em pacientes vivendo com HIV-1 tratados com terapia antirretroviral contendo lamivudina. Os mutantes M184V apresentam suscetibilidade consideravelmente reduzida à lamivudina e mostram redução na capacidade de replicação viral in vitro.
Resistência in vivo (dolutegravir mais lamivudina)
Nenhum indivíduo que tenha atendido aos critérios de retirada virológica confirmada (CVW) definida pelo protocolo nos estudos GEMINI-1 e GEMINI-2 agrupados até a Semana 144 ou no estudo TANGO até a semana 144 teve substituições de resistência a InSTI ou ITRN emergentes. Em pacientes com falha em terapias prévias, porém virgens de tratamento com a classe da integrase (estudo SAILING), as substituições do inibidor de integrase foram observadas em 4/354 pacientes (acompanhamento de 48 semanas) tratados com dolutegravir, que foi administrado em combinação com um regime de base selecionado pelo investigador. Desses quatro, dois indivíduos apresentaram uma substituição da integrase R263K exclusiva, com um fold change máximo de 1,93, um indivíduo teve uma substituição da integrase V151V/I polimórfica, com fold change de 0,92 e, um indivíduo tinha mutações na integrase pré-existentes e supõe-se que tenha tido experiência prévia a integrase ou sido infectado com vírus resistente à integrase por transmissão. Em pacientes virgens de tratamento com a classe da integrase e com falha no tratamento de primeira linha com ITRNN+ 2 ITRNs (estudo DAWNING) até a Semana 48, 2/314 indivíduos tratados com dolutegravir tiveram substituições da via G118R do inibidor de integrase conferindo fold changes do DTG de 15 e 30 com reduções respectivas na capacidade de replicação viral de 6,6 vezes e 18 vezes em comparação ao período basal. As mutações G118R e R263K também foram selecionadas in vitro (ver acima).
Resistência cruzada Vírus mutante a InSTI direcionado ao sítio: A atividade do dolutegravir foi determinada contra um painel de 60 vírus do HIV-1 mutante direcionado ao sítio resistente ao InSTI (28 com substituições únicas e 32 com 2 ou mais substituições). As únicas substituições de resistência ao InSTI T66K, I151L e S153Y conferiram uma redução de mais de 2 vezes na suscetibilidade ao dolutegravir (intervalo: 2,3 vezes a 3,6 vezes em relação à referência). As combinações de múltiplas substituições T66K/L74M, E92Q/N155H, G140C/Q148R, G140S/Q148H, R ou K, Q148R/N155H, T97A/G140S/Q148, e substituições em E138/G140/Q148 mostraram uma redução de mais de 2 vezes na suscetibilidade ao dolutegravir (intervalo: 2,5 vezes a 21 vezes em relação à referência).
Isolados clínicos recombinantes: A atividade do dolutegravir foi mensurada para 705 isolados recombinantes resistentes ao raltegravir da prática clínica; 93,9% (662/705) dos isolados tiveram um FC de dolutegravir ≤10 e 1,8% tiveram um FC de DTG > 25. Os mutantes com a via Y143 e N155 tiveram FCs médios de 1,2 e 1,5, respectivamente, enquanto que os FCs médios do mutante Q148 + 1 e mutantes Q148 + ≥2 foram de 4,8 e 6,0, respectivamente.
Resistência cruzada conferida pela transcriptase reversa M184V: A resistência cruzada é limitada dentro da classe do inibidor nucleosídeo dos agentes antirretrovirais. A zidovudina e a estavudina mantêm as suas atividades antirretrovirais contra HIV-1 resistente a lamivudina. O abacavir e o tenofovir mantêm a atividade antirretroviral contra o HIV-1 resistente a lamivudina portando apenas a mutação M184V.
Efeitos no Eletrocardiograma
Em um estudo randomizado, controlado por placebo, cruzado, 42 indivíduos sadios receberam administrações orais de dose única de placebo, dolutegravir 250 mg em suspensão (exposições aproximadamente 3 vezes da dose de 50 mg, uma vez ao dia em estado de equilíbrio) e moxifloxacino (400 mg, controle ativo) em sequência aleatória. O dolutegravir não prolongou o intervalo QTc por 24 horas após a dose. Após o ajuste para o período basal e placebo, a alteração média máxima no QTc com base no método de correção de Fridericia (QTcF) foi de 1,99 ms (IC 95% superior unilateral: 4,53 ms). Estudos semelhantes não foram realizados com lamivudina.
Efeitos na Função Renal
O efeito do dolutegravir no clearance de creatinina sérica (ClCr), taxa de filtração glomerular (TFG) usando ioexol como marcador e fluxo plasmático renal efetivo (FPRE) com uso de para-aminoipurato (PAH) como marcador foi avaliado em um estudo aberto, randomizado, de 3 braços, paralelo, controlado por placebo em 37 indivíduos sadios, que receberam dolutegravir 50 mg, uma vez ao dia (n=12), 50 mg duas vezes ao dia (n=13) ou placebo, uma vez ao dia (n=12) por 14 dias. Foi observada uma pequena redução no ClCr com dolutegravir na primeira semana de tratamento, compatível com o observado em estudos clínicos. O dolutegravir, nas duas doses, não teve efeito relevante sobre a TFG ou ERPF. Esses dados corroboram os estudos in vitro que sugerem que pequenos aumentos na creatinina observados em estudos clínicos são devido à inibição não patológica do transportador de cátions orgânicos tipo 2 (OCT2) nos túbulos renais proximais, o que medeia a secreção tubular da creatinina. Na análise agrupada dos estudos GEMINI-1 e GEMINI-2 em pacientes adultos virgens ao tratamento na análise da semana 144, dolutegravir mais lamivudina foi associado a impacto mais baixo nos parâmetros da segurança renal em comparação a dolutegravir mais a associação em dose fixa de tenofovir/entricitabina. O grupo dolutegravir mais lamivudina teve um aumento significativamente maior na TFG estimada usando a equação CKD-EPI ajustada para cistatina C, em comparação ao grupo dolutegravir mais associação em dose fixa de tenofovir/entricitabina (alteração média ajustada desde o período basal de 12,2 e 10,6 mL/min/1,73 m2, respectivamente; p < 0,008). A alteração desde a análise do período basal mostrou que as razões de albumina/creatinina urinária e proteína/creatinina foram mais baixas no grupo dolutegravir mais lamivudina em comparação ao grupo dolutegravir mais associação em dose fixa de tenofovir/entricitabina (razão de; a diferença foi estatisticamente significativa para a relação proteína/creatinina albumina/creatinina urinária da semana 144/período basal de 1,046 e 1,104, respectivamente; p = 0,261 e razão de proteína/creatinina da semana 144/período basal de 0,994 e 1,193, respectivamente; p < 0,001). As retiradas do estudo devido a eventos adversos relacionados à função renal ou por atender aos critérios pré-definidos de toxicidade renal (TFG < 50 mL/min/1,73m2) foram observadas com mais frequência em indivíduos no grupo dolutegravir mais associação em dose fixa de tenofovir/entricitabina em comparação ao grupo dolutegravir mais lamivudina.
Propriedades Farmacocinéticas
Quando administrado em jejum, a bioequivalência foi atingida para dolutegravir, ao comparar o comprimido da associação em dose fixa de DTG/3TC ao dolutegravir 50 mg coadministrado com lamivudina 300 mg, para AUC e Cmáx. Quando administrado em jejum, a bioequivalência foi atingida para AUC de lamivudina, quando comparado o comprimido da associação em dose fixa de DTG/3TC à lamivudina 300 mg coadministrado com dolutegravir 50 mg. A Cmáx de lamivudina para o comprimido da associação em dose fixa de DTG/3TC foi 32% mais alta do que lamivudina 300 mg coadministrada com dolutegravir 50 mg. Após a administração de múltiplas doses orais de Dovato® em indivíduos vivendo com HIV e experimentados ao tratamento no estudo de fase III TANGO, AUC e Cmáx de dolutegravir e lamivudina no estado de equilíbrio foram similares às exposições históricas.
Absorção
O dolutegravir e a lamivudina são rapidamente absorvidos após a administração oral. A biodisponibilidade absoluta de dolutegravir não foi estabelecida. A biodisponibilidade absoluta da lamivudina oral em adultos é de 80 a 85%. Para a associação em dose fixa de DTG/3TC, o tempo mediano até as concentrações máximas no plasma (Tmáx) é de 2,5 horas para dolutegravir e 1,0 hora para lamivudina, quando administrado em jejum. Após doses orais múltiplas de dolutegravir 50 mg, uma vez ao dia, a média geométrica das estimativas dos parâmetros farmacocinéticos no estado de equilíbrio é de 53,6 microgramas.h/mL para AUC24, 3,67 microgramas/mL para Cmáx, e 1,11 microgramas/mL para C24. Após a administração de doses orais múltiplas de lamivudina 300 mg, uma vez ao dia, por sete dias, a Cmáx média no estado de equilíbrio é de 2,04 microgramas/mL e a AUC24 média é de 8,87 microgramas.h/mL.
Efeito do Alimento
A administração do comprimido da associação em dose fixa de DTG/3TC com uma refeição com alto teor de gordura aumentou a AUC e Cmáx de dolutegravir em 33% e 21%, respectivamente e reduziu a Cmáx de lamivudina em 30% em comparação à condição de jejum. A AUC de lamivudina não foi afetada por uma refeição com alto teor de gordura. Essas alterações não são clinicamente significativas. A associação em dose fixa de DTG/3TC pode ser administrada com ou sem alimento.
Distribuição
O volume aparente de distribuição do dolutegravir (após administração oral da formulação em suspensão, Vd/F) é estimada em 12,5 L. Estudos com doses intravenosas de lamivudina mostraram que o volume aparente de distribuição médio é de 1,3 L/kg. O dolutegravir é altamente ligado (aproximadamente 99,3%) a proteínas no plasma humano com base nos dados in vitro. A ligação de dolutegravir a proteínas no plasma ocorreu independente da concentração. As razões de concentração da radioatividade relacionada ao medicamento no sangue total e no plasma foram, em média, entre 0,441 e 0,535, indicando associação mínima da radioatividade com componentes celulares do sangue. A fração livre de dolutegravir no plasma é estimada em aproximadamente 0,2 a 1,1% em indivíduos sadios, aproximadamente 0,4 a 0,5% em indivíduos com comprometimento hepático moderado, e de 0,8 a 1,0% em indivíduos com comprometimento renal grave e 0,5% em pacientes vivendo com HIV-1. A lamivudina apresenta farmacocinética linear ao longo do intervalo de dose terapêutico e apresenta baixa ligação à proteína no plasma (menos de 36%). O dolutegravir e a lamivudina estão presentes no líquido cefalorraquidiano (LCR). Em 12 indivíduos virgens ao tratamento que receberam um regime de dolutegravir mais abacavir/lamivudina por 16 semanas, a concentração de dolutegravir no LCR foi, em média, de 15,4 ng/mL na Semana 2 e 12,6 ng/mL na Semana 16, variando de 3,7 a 23,2 ng/mL (comparável à concentração não ligada no plasma). A razão da concentração no LCR: plasma de dolutegravir variou de 0,11 a 2,04%. As concentrações de dolutegravir no LCR excederam a IC50, corroborando a redução mediana desde o período basal no HIV-1 RNA no LCR de 2,2 log após 2 semanas e 3,4 log após 16 semanas de terapia (ver Propriedades Farmacodinâmicas, acima). A razão média das concentrações de lamivudina no LCR/soro 2 a 4 h após a administração oral foi aproximadamente de 12%. O verdadeiro grau de penetração no SNC de lamivudina e sua relação com qualquer eficácia clínica são desconhecidos. O dolutegravir está presente no trato genital feminino e masculino. A AUC no líquido cercovaginal, tecido vertebral e tecido vaginal foi de 6 a 10% aquela correspondente ao plasma no estado de equilíbrio. A AUC foi de 7% no sêmen e de 17% no tecido retal, daquela correspondente ao plasma no estado de equilíbrio.
Metabolismo
O dolutegravir é principalmente metabolizado por meio da UGT1A1 com um componente de menor importância da CYP3A (9,7% da dose total administrada em um estudo de balanço de massa no ser humano). O dolutegravir é o composto circulante predominante no plasma; a eliminação renal do medicamento inalterado é baixa ( < 1% da dose). Cinquenta e três por cento da dose oral total é excretada de modo inalterado nas fezes. Não se sabe se toda ou parte dela deve-se ao medicamento não absorvido ou à excreção biliar do conjugado glicuronídeo, que pode ser adicionalmente degradado para formar o composto original no lúmen intestinal. Trinta e um por cento da dose oral total é excretado na urina, representado por glicuronídeo de dolutegravir (18,9% da dose total), metabólito da N-desalquilação (3,6% da dose total) e um metabólito formado por oxidação no carbono benzílico (3,0% da dose total). O metabolismo da lamivudina é uma via de eliminação menor. A lamivudina é predominantemente eliminada inalterada por excreção renal. A probabilidade de interações metabólicas com lamivudina é baixa devido ao pequeno grau de metabolismo hepático (menos de 10%).
Eliminação
O dolutegravir possui uma meia-vida terminal de aproximadamente 14 horas e um clearance aparente (CL/F) de 0,56 L/h. A meia-vida de eliminação observada para lamivudina é de 18 a 19 horas. Para pacientes que receberam lamivudina 300 mg, uma vez ao dia, a meia-vida terminal intracelular de lamivudina-TP foi de 16 a 19 horas. O clearance sistêmico médio da lamivudina é de aproximadamente 0,32 L/h/kg, predominantemente por clearance renal (maior que 70%) por meio do sistema de transporte catiônico orgânico.
Populações Especiais de Pacientes
Crianças
A associação em dose fixa de DTG/3TC não foi estudada na população pediátrica. Em um estudo pediátrico incluindo 23 adolescentes de 12 a 18 anos de idade vivendo com HIV-1 com exposição prévia ao tratamento antirretroviral, a farmacocinética de dolutegravir foi avaliada em 10 adolescentes e mostrou que uma dose de dolutegravir 50 mg, uma vez ao dia, resultou em exposição de dolutegravir em indivíduos pediátricos comparável àquela observada em adultos que receberam dolutegravir 50 mg, uma vez ao dia (Tabela 3).
Dados limitados estão disponíveis sobre adolescentes que receberam uma dose diária de 300 mg de lamivudina. Os parâmetros farmacocinéticos são comparáveis àqueles relatados em adultos.
Idosos
A análise farmacocinética da população de dolutegravir usando dados em adultos vivendo com HIV-1 mostrou que não houve efeito clinicamente relevante da idade na exposição do dolutegravir. Os dados farmacocinéticos para dolutegravir e lamivudina em indivíduos com > 65 anos de idade são limitados.
Disfunção renal
Dados farmacocinéticos foram obtidos para dolutegravir e lamivudina isoladamente. A associação em dose fixa de DTG/3TC não deve ser usada em pacientes com clearance de creatinina de menos de 30 mL/min porque, embora não seja necessário ajuste posológico de dolutegravir em pacientes com comprometimento renal, é necessária a redução na dose para o componente lamivudina. Estudos com lamivudina mostram que as concentrações no plasma (AUC) estão aumentadas em pacientes com disfunção renal devido ao clearance reduzido. O clearance renal do medicamento não alterado é uma via de eliminação de menor importância para o dolutegravir. Foi realizado um estudo da farmacocinética do dolutegravir em indivíduos com comprometimento renal grave (ClCr < 30 mL/min). Não foram observadas diferenças farmacocinéticas clinicamente importantes entre indivíduos com comprometimento renal grave (ClCr < 30 mL/min) e indivíduos sadios pareados.
Disfunção hepática
Os dados farmacocinéticos foram obtidos para dolutegravir e lamivudina individualmente. Dados obtidos para lamivudina em pacientes com comprometimento hepático moderado a grave e para dolutegravir em pacientes com comprometimento hepático moderado mostram que a farmacocinética não é significativamente afetada pela disfunção hepática. O dolutegravir é principalmente metabolizado e eliminado pelo fígado. Em um estudo comparando 8 indivíduos com comprometimento hepático moderado (categoria B de acordo com Child-Pugh) a 8 controles adultos sadios pareados, a exposição à dose única de 50 mg de dolutegravir foi semelhante entre os dois grupos. O efeito do comprometimento hepático grave na farmacocinética de dolutegravir não foi estudado.
Polimorfismos em Enzimas Metabolizadoras do Medicamento
Não existe evidência de que polimorfismos comuns em enzimas metabolizadoras do fármaco alteram a farmacocinética do dolutegravir para um grau clinicamente significativo. Em uma metanálise usando amostras farmacogenômicas coletadas em estudos clínicos em indivíduos sadios, os indivíduos com genótipos UGT1A1 (n=7) conferindo baixo metabolismo do dolutegravir apresentaram um clearance 32% mais baixo de dolutegravir e AUC 46% mais alta em comparação aos indivíduos com genótipos associados ao metabolismo normal via UGT1A1 (n=41). Polimorfismos em CYP3A4, CYP3A5 e NR1I2 não foram associados a diferenças na farmacocinética do dolutegravir.
Gênero
As análises farmacocinéticas da população não revelaram efeito clinicamente relevante do gênero na exposição do dolutegravir. Não foram observadas diferenças clinicamente relevantes na farmacocinética de lamivudina entre homens e mulheres.
Raça
As análises farmacocinéticas da população usando dados farmacocinéticos agrupados dos estudos de Fase IIb e Fase III em adultos para dolutegravir não revelaram efeito clinicamente relevante da raça na exposição do dolutegravir. A farmacocinética do dolutegravir após administração de dose única oral a indivíduos japoneses parece ser semelhante aos parâmetros observados em ocidentais (EUA). Não existe evidência de que seria necessário um ajuste na dose de dolutegravir ou lamivudina com base nos efeitos da raça nos parâmetros farmacocinéticos.
Coinfecção com Hepatite B ou C
A análise farmacocinética da população indicou que a coinfecção com o vírus da hepatite C não teve efeito clinicamente relevante na exposição ao dolutegravir. Existem dados farmacocinéticos limitados sobre indivíduos com coinfecção com hepatite B (ver Advertências e Precauções).
Gravidez
A farmacocinética da lamivudina durante a gravidez é semelhante a aquela de adultos não-grávidos. Em seres humanos, consistente com a transmissão passiva de lamivudina pela placenta, as concentrações de lamivudina no soro do bebê no nascimento foram semelhantes a aquelas no soro materno e do cordão umbilical no parto. Não existem dados farmacocinéticos sobre o uso de dolutegravir na gravidez.
4. CONTRAINDICAÇÕES
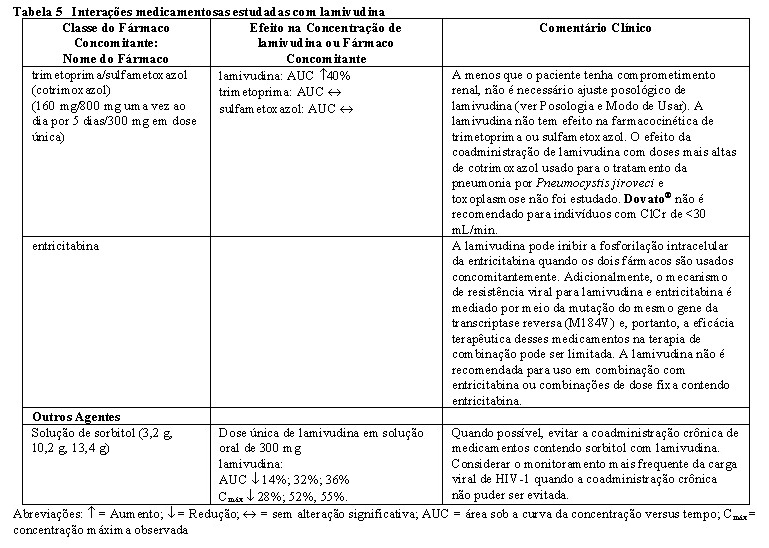
É contraindicada a administração de Dovato® a pacientes com hipersensibilidade conhecida ao dolutegravir, a lamivudina, ou a qualquer um dos excipientes. Dovato® não deve ser administrado em combinação com medicamentos que possuem janelas terapêuticas estreitas e que sejam substratos do transportador de cátions orgânicos 2 (OCT2), incluindo, mas não limitando-se a dofetilida, pilsicainida ou fampridina (também conhecida como dalfampridina) (ver Interações Medicamentosas).
Este medicamento é contraindicado para uso em combinação com dofetilida, pilsicainida ou fampridina.
5. ADVERTÊNCIAS E PRECAUÇÕES
As advertências e precauções especiais relevantes ao dolutegravir e lamivudina estão incluídas nesta seção. Não existem precauções e advertências adicionais relevantes a Dovato® .
Reações de hipersensibilidade
Reações de hipersensibilidade foram relatadas com inibidores de integrase, incluindo dolutegravir, foram caracterizadas por erupção cutânea, sintomas gerais inespecíficos e, às vezes, disfunção de órgãos, incluindo lesão hepática grave. Descontinuar imediatamente o uso de Dovato® e outros agentes suspeitos se ocorrer sinais ou sintomas de reações de hipersensibilidade (incluindo, entre outros, erupção cutânea intensa ou erupção cutânea acompanhada por febre, mal-estar geral, fadiga, dores musculares ou articulares, formação de vesículas, lesões orais, conjuntivite, edema facial, hepatite, eosinofilia, angioedema). Deve-se monitorar o estado clínico, incluindo as aminotransferases hepáticas e bilirrubina e a terapia apropriada deve ser iniciada. O atraso na interrupção do tratamento com Dovato® ou outros medicamentos suspeitos após o início da hipersensibilidade pode resultar em uma reação com risco à vida.
Acidose láctica/hepatomegalia grave com esteatose
A acidose láctica e hepatomegalia grave com esteatose, incluindo casos fatais, foram relatadas com o uso de antirretrovirais análogos de nucleosídeos isoladamente ou em combinação, incluindo lamivudina. A maioria desses casos ocorre