DIVALCON ER
ABBOTT
divalproato
Anticonvulsivante. Antienxaquecoso. Estabilizador de humor.
Apresentações.
DIVALCON ER (divalproato de sódio) comprimido revestido de liberação prolongada de 250 mg: embalagem com 6, 30 e 60 comprimidos revestidos.
DIVALCON ER (divalproato de sódio) comprimido revestido de liberação prolongada de 500 mg: embalagem com 6, 30 e 60 comprimidos revestidos.
VIA ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 10 ANOS
Composição.
Cada comprimido revestido de DIVALCON ER 250 mg contém:
divalproato de sódio 269,10 mg
(equivalente a 250 mg de ácido valproico)
Excipientes: hipromelose, celulose microcristalina, dióxido de silício, sorbato de potássio, cobertura Opadry e Opadry II.
Cada comprimido revestido de DIVALCON ER 500 mg contém:
divalproato de sódio 538,10 mg
(equivalente a 500 mg de ácido valproico)
Excipientes: hipromelose, celulose microcristalina, lactose monoidratada, dióxido de silício, sorbato de potássio, cobertura Opadry e Opadry II.
Informações técnicas.
1. INDICAÇÕES
Mania: DIVALCON ER é indicado para o tratamento de episódios agudos de mania ou episódios mistos associados com transtornos afetivos bipolares, com ou sem características psicóticas.
Um episódio de mania é um período distinto de humor anormalmente e persistentemente elevado, expansivo ou irritável. Os sintomas típicos de mania incluem taquilalia, hiperatividade motora, redução da necessidade de dormir, fuga de ideias, grandiosidade, prejuízo da crítica, agressividade e possível hostilidade.
Um episódio misto é caracterizado pela presença simultânea de critérios diagnósticos para um episódio de mania e para um episódio depressivo (humor deprimido, perda do interesse ou prazer em quase todas as atividades).
A eficácia do divalproato de sódio de liberação prolongada foi baseada parcialmente em estudos de divalproato de sódio de liberação lenta para essa indicação e foi estabelecida em estudos de três semanas com pacientes que se enquadravam nos critérios da DSM-IV TR para transtorno afetivo bipolar I, tipo mania ou tipo misto, que foram hospitalizados com diagnóstico de mania aguda.
A eficácia de divalproato de sódio de liberação prolongada durante uso prolongado em mania, isto é, mais do que três semanas, não foram sistematicamente avaliadas nos estudos clínicos controlados. Portanto, os médicos que optam pelo uso de divalproato de sódio por períodos extensos deverão reavaliar continuamente a utilidade a longo prazo do medicamento para cada paciente.
Epilepsia: DIVALCON ER é indicado como monoterápico ou como terapia adjuvante ao tratamento de pacientes adultos e crianças acima de 10 anos com crises parciais complexas, que ocorrem tanto de forma isolada ou em associação com outros tipos de crises.
DIVALCON ER também é indicado como monoterápico ou como terapia adjuvante no tratamento de quadros de ausência simples e complexa.
Ausência simples é definida como breve obscurecimento sensorial ou perda de consciência, acompanhada de um certo número de descargas epilépticas generalizadas, sem outros sinais clínicos detectáveis. A ausência complexa é a expressão utilizada quando outros sinais também estão presentes.
Profilaxia da Migrânea (Enxaqueca): DIVALCON ER é indicado na profilaxia de enxaquecas em adultos. Não existe evidência de que o divalproato de sódio de liberação prolongada seja útil no tratamento agudo da enxaqueca. Uma vez que o ácido valproico pode ser perigoso para o feto, divalproato de sódio de liberação prolongada não deve ser considerado para mulheres em idade fértil a não ser que o medicamento seja essencial para o gerenciamento da sua condição médica.
DIVALCON ER é contraindicado para o uso na profilaxia de enxaquecas por mulheres grávidas.
2. RESULTADOS DE EFICÁCIA
Mania
A eficácia de divalproato de sódio de liberação prolongada para o tratamento de mania aguda foi demonstrada em um estudo multicêntrico, randomizado, duplo-cego, controlado por placebo e por grupo paralelo de 3 semanas. O estudo foi elaborado para avaliar a segurança e eficácia de divalproato de sódio no tratamento do transtorno bipolar I, tipo mania ou tipo misto, em adultos.
O estudo incluiu pacientes (homens e mulheres) adultos que preencheram os critérios do DSM-IV-TR para transtorno bipolar I, tipo mania ou tipo misto e que foram hospitalizados por mania aguda. O divalproato de sódio de liberação prolongada foi iniciado com doses de 25mg/kg/dia, administrados uma vez ao dia, aumentado para 500 mg/dia no dia 3, e então ajustado para atingir concentrações séricas de valproato em um intervalo de 85 a 125 mcg/mL. As doses médias de divalproato de sódio para os que completaram este estudo foram 2362 mg (variação: 500-4000), 2874 mg (variação: 1500-4500), 2993 mg
(variação: 1500-4500), 3181 mg (variação: 1500-5000), and 3353 mg (variação: 1500-5500) nos dias 1,
5, 10, 15 e 21, respectivamente. A média da concentração de valproato foi de 96,5 mg/mL, 102,1 mg/mL, 98,5 mg/mL, 89,5 mg/mL nos dias 5, 10, 15 e 21, respectivamente. Os pacientes foram avaliados pela escala de Mania (Mania Rating Scale - MRS; pontuação varia de 0-52).
O divalproato de sódio de liberação prolongada foi estatisticamente superior ao placebo na redução da pontuação total de MRS.
Epilepsia
Crises Parciais Complexas (CPC)
A eficácia do divalproato de sódio na redução da incidência de crises parciais complexas (CPC) que ocorrem de forma isolada ou em associação com outros tipos de crises foi estabelecida em dois ensaios controlados usando divalproato de sódio comprimidos revestidos.
Em um estudo multiclínico, placebo-controlado, empregado como terapia adjuvante, 144 pacientes que apresentaram oito ou mais CPCs durante oito semanas, por um período de oito semanas de monoterapia com doses de fenitoína ou carbamazepina suficientes para assegurar as concentrações plasmáticas no "intervalo terapêutico", foram randomizados para receber, em adição às suas medicações antiepiléticas originais, divalproato de sódio ou placebo.
Pacientes foram escolhidos ao acaso para prosseguir os estudos para um total de 16 semanas. A Tabela 1 descreve os achados.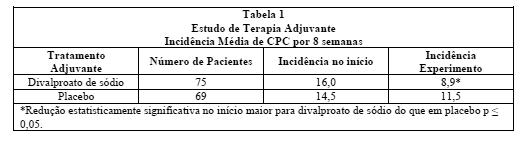
A Figura 1 apresenta a proporção de pacientes (eixo X) cuja porcentagem de redução das taxas de crises parciais complexas no início foi pelo menos tão elevada quanto a indicada no eixo Y no estudo de tratamento adjuvante. Uma redução percentual positiva indica uma melhora (ou seja, redução na frequência das crises), enquanto que uma redução percentual negativa indica uma piora. Deste modo, em uma exposição deste tipo, a curva que demonstra um tratamento efetivo é deslocada para a esquerda da curva do placebo. O resultado demonstrou que a proporção de pacientes que atingiram um determinado nível de melhoria com divalproato de sódio foi consistentemente maior do que os pacientes que usaram placebo. Por exemplo, 45% dos pacientes tratados com divalproato de sódio tiveram uma redução na taxa de CPCs maior ou igual a 50%, comparado a 23% de melhoria para os pacientes que usaram placebo.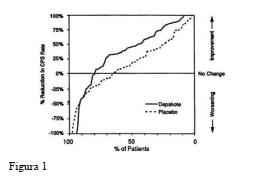
O segundo estudo avaliou a capacidade do divalproato de sódio em reduzir a incidência de CPCs como monoterapia antiepilética. O estudo comparou a incidência de CPCs entre os pacientes randomizados para receber altas ou baixas doses de tratamento. Os pacientes foram selecionados para participarem dos estudos somente se:
1) apresentassem duas ou mais CPCs por quarto semanas, durante um período de oito a doze semanas de monoterapia com doses adequadas de antiepiléticos (fenitoína, carbamazepina, fenobarbital, primidona); e
2) pacientes que passaram por uma transição de duas semanas bem sucedida para divalproato de sódio.
Os pacientes foram então submetidos à ingestão das doses determinadas, com diminuição gradual da medicação antiepilética concomitante, por um período de 22 semanas. Porém, menos de 50% dos pacientes finalizaram os estudos. Nos pacientes convertidos à monoterapia com divalproato de sódio, a média total das concentrações de valproato durante a monoterapia foram de 71 e 123 mcg/mL para a dose baixa e dose alta, respectivamente. A Tabela 2 apresenta os achados para todos os pacientes randomizados que passaram por pelo menos uma avaliação pós-randomização.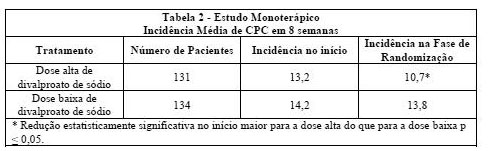
A Figura 2 apresenta a proporção de pacientes (eixo X) cuja porcentagem de redução nas taxas de crises parciais complexas no início foi pelo menos tão elevada quanto a indicada no eixo Y do estudo monoterápico. Uma redução percentual positiva indica uma melhora (ou seja, redução na frequência das crises), enquanto que uma redução percentual negativa indica uma piora. Deste modo, em uma exposição deste tipo, a curva que demonstra um tratamento mais efetivo é deslocada para a esquerda da curva que demonstra um tratamento menos efetivo.
Os resultados mostraram que a redução na incidência de CPCs foi significantemente maior quando administrada altas doses de divalproato de sódio. Por exemplo, quando da alteração da monoterapia de carbamazepina, fenitoína, fenobarbital ou primidona para administração de doses elevadas de divalproato de sódio como monoterapia, 63% dos pacientes sofreram nenhuma alteração ou uma redução de taxas de epilepsia parcial complexa, em comparação com 54% dos pacientes que receberam doses mais baixas de divalproato de sódio.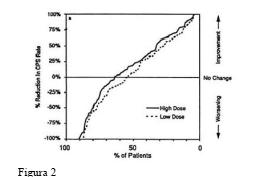
Profilaxia da Migrânea
Os resultados de um estudo clínico multicêntrico, randomizado, duplo-cego e placebo-controlado, demonstraram a eficácia do divalproato de sódio comprimidos revestidos na profilaxia da enxaqueca. Deste estudo, participaram pacientes com histórico de crises de enxaqueca, com ou sem aura que ocorreram duas vezes ou mais por mês pelos últimos três meses. Pacientes com cefaleia em crises ou dor de cabeça crônica diária foram excluídos dos estudos. Mulheres em idade fértil foram incluídas no estudo, desde que comprovassem o uso de um método contraceptivo eficaz.
Os pacientes que sofreram duas ou mais enxaquecas no período basal de quatro semanas foram escolhidos ao acaso para a administração de divalproato de sódio ou de placebo, na proporção 1:1 e tratados pelo período de doze semanas. Os pacientes iniciaram o tratamento com uma dose de 500 mg uma vez ao dia durante uma semana, que depois foi aumentada para 1000 mg uma vez ao dia, com a opção de diminuir a dose definitivamente para 500 mg uma vez ao dia durante a segunda semana de tratamento, caso ocorresse intolerância à medicação. Noventa e oito dos 114 pacientes tratados com divalproato (86%) e 100 dos 110 pacientes tratados com placebo (91%), por pelo menos duas semanas, mantiveram a dose de 1000 mg uma vez ao dia durante o período do tratamento. O resultado do tratamento foi avaliado com base na redução da enxaqueca após quatro semanas de tratamento, em comparação com o período inicial.
Pacientes (50 H e 187 M) com idade variando de 16 a 69 foram tratados com divalproato sódio de liberação prolongada (n = 122) ou placebo (n = 115). Quatro pacientes estavam abaixo dos 18 anos e três acima dos 65 anos de idade. Duzentos e dois pacientes (101 de cada grupo de tratamento) completaram o período de tratamento. A taxa de redução média de enxaqueca, em quatro semanas de tratamento, foi de 1,2 a partir de uma base média de 4,4 com uso de divalproato de sódio de liberação prolongada, versus 0,6 a partir de uma base média de 4,2 com uso de placebo. A diferença entre a eficácia dos tratamentos foi estatisticamente significante (veja a figura 3).
Referências Bibliográficas
Caso haja interesse em conhecer as referências bibliográficas e/ou estudos clínicos disponíveis para este medicamento, por favor, entre em contato com nosso Serviço de Atendimento ao Consumidor - Abbott Center através do telefone 0800 7031050.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Descrição
O divalproato de sódio é um composto de coordenação estável de valproato de sódio e ácido valproico em uma relação molar de 1:1, formado durante a neutralização parcial do ácido valproico com 0,5 equivalente de hidróxido de sódio.
Farmacodinâmica
O divalproato de sódio é dissociado em íon valproato no trato gastrintestinal. O mecanismo pelo qual o valproato exerce seu efeito terapêutico não está bem estabelecido. Foi sugerido que sua atividade na epilepsia está relacionada ao aumento das concentrações cerebrais de ácido gama-aminobutírico (GABA).
Farmacocinética
Absorção e Biodisponibilidade: a biodisponibilidade absoluta dos comprimidos de liberação prolongada de DIVALCON ER administrados em dose única após uma refeição foi de aproximadamente 90% em relação à infusão intravenosa. Quando administrados em doses equivalentes, a biodisponibilidade de divalproato de sódio ER é menor que a de divalproato de sódio comprimidos de liberação entérica. Em cinco estudos de doses múltiplas, realizados com indivíduos saudáveis (n = 82) e pacientes com epilepsia (n = 86), o comprimido de liberação prolongada, administrado uma vez ao dia em jejum ou imediatamente após refeições menores, teve uma biodisponibilidade média de 89% em relação aos comprimidos de liberação entérica administrados duas, três ou quatro vezes ao dia.
Nestes estudos, foram encontradas concentrações plasmáticas máximas (Cmáx) de valproato em média 4 a 17 horas após a ingestão de comprimidos de divalproato de sódio de liberação prolongada. Após múltiplas doses de divalproato de sódio de liberação prolongada, a concentração plasmática de valproato era 10 a 20% menor que a resultante da administração de divalproato de sódio administrado duas, três ou quatro vezes ao dia.
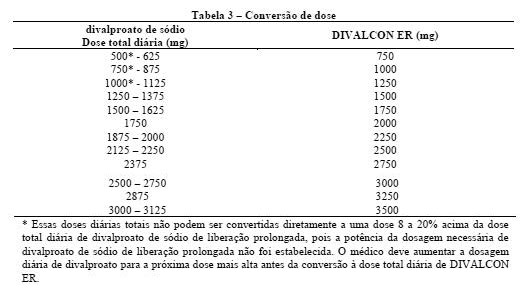
Conversão do divalproato de sódio para o divalproato de sódio de liberação prolongada: quando o divalproato de sódio de liberação prolongada é administrado em doses de 8 a 20% maior do que a dose total diária de divalproato de sódio, as duas formulações são bioequivalentes. Em dois estudos randomizados e cruzados, doses múltiplas diárias de divalproato de sódio foram de 8 a 20% maior do que a dose única diária do divalproato de sódio de liberação prolongada. Nestes estudos, o divalproato de sódio de liberação prolongada e o divalproato de sódio foram equivalentes com respeito à área sob a curva (ASC, uma medida de extensão da biodisponibilidade). Adicionalmente, a Cmáx foi menor, e a Cmin ou foi maior ou indiferente, para o divalproato de sódio de liberação prolongada, em relação ao divalproato de sódio.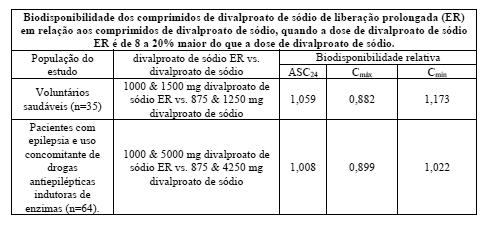
Foram avaliadas drogas antiepilépticas concomitantes, que induzem o sistema das isoenzimas do citocromo P450 (topiramato, fenobarbital, carbamazepina, fenitoína e lamotrigina), e não foram demonstradas alterações significativas na biodisponibilidade do valproato tanto para divalproato de sódio quanto para divalproato de sódio ER.
Distribuição:
Ligação às proteínas: a ligação do valproato a proteínas plasmáticas é dependente da concentração e a fração livre aumenta de aproximadamente 10% com concentração de 40 mcg/mL para 18,5% com concentração de 130 mcg/mL. A ligação protéica do valproato é reduzida em idosos, em pacientes com doenças hepáticas crônicas, em pacientes com insuficiência renal e na presença de outros fármacos (ex: ácido acetilsalicílico). Por outro lado, o valproato pode deslocar certas ligações protéicas de fármacos (ex: fenitoína, carbamazepina, varfarina e tolbutamida).
Distribuição no SNC: as concentrações de valproato no líquor aproximam-se das concentrações de valproato não ligado a proteínas no plasma (aproximadamente 10% da concentração total).
Metabolismo: o valproato é metabolizado quase que totalmente pelo fígado. Em pacientes adultos sob o regime de monoterapia, 30% a 50% de uma dose administrada aparece na urina como conjugado glicuronídeo. A beta-oxidação mitocondrial é outra via metabólica importante, metabolizando mais de 40% da dose. Geralmente, menos de 15 a 20% da dose administrada é eliminada por outros mecanismos oxidativos. Menos de 3% da dose administrada é excretada de forma inalterada pela urina. A relação entre dose e concentração total de valproato não é linear; a concentração não aumenta proporcionalmente à dose, mas numa extensão menor, devido à saturação da ligação protéica plasmática. A cinética do fármaco não ligado a proteínas plasmáticas é linear.
Eliminação: a média de depuração plasmática e o volume de distribuição do valproato total são de 0,56 L/h/1,73m2 e 11 L/1,73m2, respectivamente. A depuração plasmática média e o volume de distribuição do valproato livre são de 4,6 L/h/1,73 m2 e 92 L/1,73 m2, respectivamente. A meia-vida terminal média para a monoterapia com valproato varia de 9 a 16 horas após a administração oral de 250 a 1000 mg. As estimativas citadas aplicam-se principalmente a pacientes que não estão recebendo medicamentos que afetam os sistemas de metabolização de enzimas hepáticas. Por exemplo, pacientes que tomam medicamento anticonvulsivante que seja indutor enzimático (carbamazepina, fenitoína e fenobarbital) irão depurar valproato mais rapidamente. Devido a estas alterações na depuração do valproato, a monitorização das concentrações dos anticonvulsivantes deve ser intensificada, tanto na introdução quanto na descontinuação do tratamento.
Populações especiais
Neonatos: (Nota: A segurança e a eficácia do divalproato de sódio de liberação prolongada na profilaxia de enxaqueca em crianças não foram estabelecidas. Dessa forma, a informação abaixo é aplicável em pacientes pediátricos somente nos casos de epilepsia).
Crianças nos dois primeiros meses de vida tem capacidade de eliminação de valproato bastante reduzida quando comparadas com crianças mais velhas e adultos. Isto é resultado tanto da redução da depuração (talvez em virtude da demora no desenvolvimento da glicuronosil-transferase e outros sistemas enzimáticos envolvidos na eliminação do valproato), quanto do aumento do volume de distribuição (devido, em parte, à ligação protéica plasmática diminuída). Por exemplo, em um estudo, a meia-vida em crianças com menos de 10 dias variou de 10 a 67 horas e em crianças com mais de dois meses de vida, de 7 a 13 horas.
Crianças: (Nota: A segurança e a eficácia do divalproato de sódio de liberação prolongada na profilaxia de enxaqueca em crianças não foram estabelecidas. Dessa forma, a informação abaixo é aplicável em pacientes pediátricos somente nos casos de epilepsia).
Pacientes pediátricos (entre 3 meses e 10 anos) tem depuração 50% mais alta do que os adultos, em relação ao peso (mL/min/kg). Acima dos 10 anos de idade, as crianças tem parâmetros farmacocinéticos que se aproximam dos adultos.
O perfil farmacocinético do valproato após a administração de divalproato de sódio de liberação prolongada foi analisado em um estudo multicêntrico aberto de doses múltiplas com crianças e adolescentes.
A dosagem de DIVALCON ER variou de 250 mg a 1750 mg, uma vez ao dia.
Os parâmetros de concentração-tempo produzidos com a administração de DIVALCON ER em pacientes pediátricos (10-17 anos) foram semelhantes aos observados em adultos.
Idosos: pacientes idosos (entre 68 e 89 anos) tem uma capacidade diminuída de eliminação de valproato, quando comparados a adultos jovens (entre 22 e 26 anos). A depuração intrínseca está diminuída em 39% e a fração livre de valproato está aumentada em 44%; portanto, a dosagem inicial deverá ser reduzida em idosos.
Gênero: não há diferenças no clearance da droga não ligada quando se ajusta a área de superfície corporal ente homens e mulheres (4,8± 0,17 e 4,7 ± 0,07 L/h/1,73m2, respectivamente).
Etnia: os efeitos da etnia sobre a cinética do valproato não foram estudados.
Doenças hepáticas: Doenças hepáticas diminuem a capacidade de eliminação de valproato (ver Contraindicações e Precauções e Advertências - Hepatotoxicidade). Em um estudo, a depuração de valproato livre estava diminuída em 50% em sete pacientes com cirrose e em 16% em quatro pacientes com hepatite aguda, em relação a 6 indivíduos saudáveis. Neste estudo, a meia-vida do valproato foi aumentada de 12 para 18 horas. Doença hepática também está associada com decréscimo das concentrações de albumina e com frações mais altas de valproato não ligado (aumento de 2 a 2,6 vezes). Consequentemente, a monitoração de concentrações totais pode ser enganosa, uma vez que as concentrações livres podem estar substancialmente elevadas em pacientes com doença hepática, enquanto as concentrações totais podem parecer normais.
Doenças renais: uma pequena redução (27%) na depuração de valproato não ligado foi relatada em pacientes com insuficiência renal (clearance de creatinina < 10 mL/minuto); no entanto, a hemodiálise tipicamente reduz as concentrações de valproato em torno de 20%. Portanto, ajustes de doses parecem não ser necessários em pacientes com insuficiência renal. A ligação protéica nestes pacientes está substancialmente reduzida, assim, a monitoração das concentrações totais pode ser enganosa.
Níveis plasmáticos e efeitos clínicos: a relação entre concentração plasmática e resposta clínica não está totalmente esclarecida. Um fator contribuinte é a ligação protéica não linear e concentração-dependente do valproato, que afeta a depuração da substância. Então, o monitoramento do valproato sérico total não pode fornecer um índice confiável das espécies bioativas de valproato.
Por exemplo, tendo em vista que a ligação protéica plasmática do valproato é dependente da concentração, a fração livre aumenta cerca de 10% em 40 mcg/mL para 18,5% em 130 mcg/mL. Ocorrem frações livres maiores do que o esperado em idosos, pacientes hiperlipidêmicos e pacientes com doença hepática ou renal.
Mania: em um estudo clínico placebo-controlado de mania aguda, pacientes receberam doses obtendo resposta clínica com concentrações plasmáticas entre 85 e 125 mg/mL.
Epilepsia: a faixa terapêutica para epilepsia é geralmente considerada entre 50 e 100 mcg/mL de valproato total, embora alguns pacientes possam ser controlados com concentrações plasmáticas menores ou até maiores.
4. CONTRAINDICAÇÕES
DIVALCON ER é contraindicado para menores de 10 anos de idade.
DIVALCON ER é contraindicado para uso por pacientes com:
• Conhecida hipersensibilidade ao divalproato de sódio ou aos demais componentes da fórmula do produto;
• Doença ou disfunção no fígado significativas;
• Conhecida desordem na mitocôndria causada por mutação na DNA polimerase mitocondrial c (POLG; ou seja, Síndrome de Alpers-Huttenlocher) e crianças com menos de 2 anos com suspeita de possuir desordem relacionada à POLG;
• Distúrbio do ciclo da ureia (DCU);
• Doença do sangue conhecida como porfiria.
DIVALCON ER é contraindicado na profilaxia da migrânea em mulheres grávidas e mulheres em idade fértil que não utilizam métodos contraceptivos eficazes durante o tratamento, pois o risco de utilização em mulheres grávidas supera claramente qualquer possível benefício da droga.
Categoria de risco: X
Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
A possibilidade de gravidez deve ser excluída antes de iniciar o tratamento com DIVALCON ER.
5. ADVERTÊNCIAS E PRECAUÇÕES
Gerais: pelo fato de terem sido relatados casos de alterações na fase secundária da agregação plaquetária, trombocitopenia e anormalidade nos parâmetros da coagulação (ex. fibrinogênio baixo), recomenda-se a contagem de plaquetas e realização de testes de coagulação antes de iniciar o tratamento e depois, periodicamente. Os pacientes com indicação de cirurgias eletivas que estão recebendo divalproato de sódio devem ser monitorados com relação à contagem de plaquetas e testes de coagulação antes da cirurgia.
Em um estudo clínico de divalproato de sódio como monoterapia em pacientes com epilepsia, 34/126 pacientes (27%) recebendo aproximadamente 50mg/Kg/dia em média, tiveram pelo menos um valor menor que 75 x 109 plaquetas por litro.
Aproximadamente metade dos pacientes tiveram o tratamento descontinuado e o retorno na contagem normal das plaquetas. Nos pacientes restantes, a contagem de plaquetas normalizou-se com a continuidade do tratamento. Neste estudo, a probabilidade de trombocitopenia aparece aumentar significativamente em concentrações de valproato ≥110 mcg/mL em mulheres ou ≥135 mcg/mL em homens. Evidências de hemorragia, manchas roxas ou desordem na hemostasia/coagulação é um indicativo para a redução da dose ou interrupção da terapia.
Uma vez que o divalproato de sódio pode interagir com medicamentos administrados concomitantemente capazes de induzir enzimas, determinações periódicas da concentração plasmática de valproato e medicamentos concomitantes são recomendadas durante a terapia inicial. O valproato é eliminado parcialmente pela urina, como metabólito cetônico, o que pode prejudicar a interpretação dos resultados do teste de corpos cetônicos na urina.
Foram relatadas alterações nos testes da função da tireoide associadas ao uso de valproato. Desconhece-se o significado clínico desse fato.
Há estudos "in vitro" que sugerem que o valproato estimula a replicação dos vírus HIV e CMV em certas condições experimentais. A consequência clínica, se houver, não é conhecida. Adicionalmente, a relevância dessas descobertas "in vitro" é incerta para pacientes recebendo terapia antirretroviral supressiva máxima. Entretanto, estes dados devem ser levados em consideração ao se interpretar os resultados da monitorização regular da carga viral em pacientes infectados pelo HIV recebendo valproato ou no acompanhamento clínico de pacientes infectados por CMV.
Os pacientes com deficiência subjacente de carnitina-palmitoil transferase (CPT) tipo II devem ser advertidos do alto risco de rabdomiólise durante o tratamento com valproato.
Houve raros relatos de medicação residual nas fezes, alguns dos quais ocorreram em pacientes com distúrbios gastrointestinais anatômicos (incluindo ileostomia ou colostomia) ou distúrbios gastrointestinais funcionais com períodos de trânsito gastrointestinal diminuído. Alguns relatos de medicação residual ocorreram no contexto da diarreia. Em pacientes que apresentaram medicação residual nas fezes, recomenda-se avaliação do nível plasmático de valproato e a monitorização das condições clínicas do paciente. Se clinicamente indicado, tratamento alternativo pode ser considerado.
Hepatotoxicidade: casos de insuficiência hepática resultando em fatalidade ocorreram em pacientes recebendo ácido valproico. Estes incidentes usualmente ocorreram durante os primeiros seis meses de tratamento. Hepatotoxicidade grave ou fatal pode ser precedida por sintomas não específicos, como mal- estar, fraqueza, letargia, edema facial, anorexia e vômito. Em pacientes com epilepsia, a perda de controle de crises também pode ocorrer. Os pacientes devem ser cuidadosamente monitorizados quanto ao aparecimento desses sintomas. Testes de função hepática deverão ser realizados antes do início do tratamento e em intervalos frequentes após iniciado, especialmente durante os primeiros seis meses. No entanto, os médicos não devem confiar totalmente na bioquímica sérica, uma vez que estes exames nem sempre apresentam alterações, sendo, portanto, fundamental a obtenção de história clínica e realização de exames físicos cuidadosos. Deve-se ter muito cuidado quando divalproato de sódio for administrado em pacientes com história anterior de doença hepática. Pacientes em uso de múltiplos anticonvulsivantes, crianças, pacientes com doenças metabólicas congênitas, com doença convulsiva grave associada a retardo mental e pacientes com doença cerebral orgânica, podem ter um risco particular. A experiência tem demonstrado que crianças abaixo de dois anos de idade apresentam um risco consideravelmente maior de desenvolver hepatotoxicidade fatal, especialmente aquelas com condições anteriormente mencionadas. Quando o divalproato de sódio for usado neste grupo de pacientes, deverá ser administrado com extremo cuidado e como agente único. Os benefícios da terapia devem ser avaliados em relação aos riscos. A experiência em epilepsia tem indicado que a incidência de hepatotoxicidade fatal decresce consideravelmente, de forma progressiva, em pacientes mais velhos. O medicamento deve ser descontinuado imediatamente na presença de disfunção hepática significativa, suspeita ou aparente. Em alguns casos, a disfunção hepática progrediu apesar da descontinuação do medicamento.
O divalproato de sódio é contraindicado em pacientes com conhecida desordem da mitocôndria causada por mutação na DNA polimerase c (POLG, ou seja, Síndrome de Alpers-Huttenlocher) e crianças com menos de 2 anos com suspeita de possuir desordem relacionada a POLG.
Insuficiência hepática aguda induzida por valproato e mortes relacionadas à doença hepática têm sido reportadas em pacientes com síndrome neurometabólica hereditária causada por mutação no gene da DNA polimerase c (POLG, ou seja, Síndrome de Alpers-Huttenlocher) em uma taxa maior do que aqueles sem esta síndrome.
Deve-se suspeitar de desordens relacionadas à POLG em pacientes com histórico familiar ou sintomas sugestivos de uma desordem relacionada à POLG, incluindo, mas não limitado encefalopatia inexplicável, epilepsia refrataria (focal, mioclônica), estado de mal epilético na apresentação, atrasos no desenvolvimento, regressão psicomotora, neuropatia sensomotora axonal, miopatia, ataxia cerebelar, oftalmoplegia, ou migrânea complicada com aura occipital. O teste para mutação de POLG deve ser realizado de acordo com a prática clínica atual para avaliação diagnóstica dessa desordem.
Em pacientes maiores de 2 anos com suspeita clínica de desordem mitocondrial hereditária o divalproato de sódio deve ser usado apenas após tentativa e falha de outro anticonvulsivante. Este grupo mais velho de pacientes deve ser monitorado durante o tratamento com divalproato de sódio para desenvolvimento de lesão hepática aguda com avaliação clínica regular e monitoramento dos testes de função hepática.
Pancreatite: casos de pancreatite envolvendo risco de morte foram relatados tanto em crianças como em adultos que receberam valproato. Alguns desses casos foram descritos como hemorrágicos com rápida progressão dos sintomas iniciais a óbito. Alguns casos ocorreram logo após o início do uso, mas também após vários anos de uso. O índice baseado nos casos relatados excede o esperado na população em geral e houve casos nos quais a pancreatite recorreu após nova tentativa com valproato. Em estudos clínicos, ocorreram 2 casos de pancreatite em 2416 pacientes sem etiologia alternativa, representando 1044 pacientes ao ano do experimento. Pacientes e responsáveis devem ser advertidos que dor abdominal, náusea, vômito e/ou anorexia, podem ser sintomas de pancreatite, requerendo avaliação médica imediata. Se for diagnosticada pancreatite, o valproato deverá ser descontinuado. O tratamento alternativo para a condição médica subjacente deve ser iniciado conforme clinicamente indicado.
Distúrbios do ciclo da ureia (DCU): foi relatada encefalopatia hiperamonêmica, algumas vezes fatal, após o início do tratamento com valproato em pacientes com distúrbios do ciclo da ureia, um grupo de anormalidades genéticas incomuns, particularmente deficiência de ornitina-transcarbamilase. Antes de iniciar o tratamento com valproato, a avaliação com relação à presença de DCU deve ser considerada nos seguintes pacientes:
1) aqueles com história de encefalopatia inexplicável ou coma, encefalopatia associada a sobrecarga proteica, encefalopatia relacionada com a gestação ou pós-parto, retardo mental inexplicável, ou história de amônia ou glutamina plasmáticas elevadas;
2) aqueles com vômitos cíclicos e letargia, episódios de irritabilidade extrema, ataxia, baixos níveis de nitrogênio de ureia sanguínea, evacuação proteica;
3) aqueles com história familiar de DCU ou história familiar de óbitos infantis inexplicáveis (particularmente meninos);
4) aqueles com outros sinais ou sintomas de DCU. Pacientes que desenvolverem sinais ou sintomas de encefalopatia hiperamonêmica inexplicável durante o tratamento com valproato devem ser tratados imediatamente (incluindo a interrupção do tratamento com valproato) e ser avaliados com relação à presença de um distúrbio do ciclo da ureia subjacente.
Comportamento e ideação suicida: tem sido relatado um aumento no risco de pensamentos e comportamentos suicidas em pacientes que utilizam medicamentos antiepilépticos para qualquer indicação. O risco aumentado de comportamento ou pensamentos suicidas com medicamentos antiepilépticos foi observado logo uma semana após o início do tratamento medicamentoso com os antiepilépticos e persistiu durante todo o período em que o tratamento foi avaliado. O risco relativo de comportamento ou pensamentos suicidas foi maior em estudos clínicos para epilepsia do que em estudos para condições psiquiátricas ou outras, porém as diferenças com relação ao risco absoluto tanto para epilepsia quanto para indicações psiquiátricas foram similares. Pacientes tratados com um antiepiléptico para qualquer indicação devem ser monitorados para o aparecimento ou piora da depressão, comportamento ou pensamentos suicidas, e/ou qualquer mudança incomum de humor ou comportamento. Qualquer um que leve em consideração a prescrição do divalproato de sódio ou qualquer outro antiepiléptico deve levar em conta, o risco de comportamento ou pensamentos suicidas com o risco da doença não tratada. Epilepsia e muitas outras doenças para as quais os antiepilépticos são prescritos estão associadas com morbidade e um aumento no risco de comportamento e pensamentos suicidas. Caso o comportamento e os pensamentos suicidas surjam durante o tratamento, o prescritor deve considerar se o aparecimento destes sintomas em qualquer paciente pode estar relacionado à doença que está sendo tratada. Pacientes, seus responsáveis e familiares devem ser informados que os antiepilépticos aumentam o risco de comportamento e pensamentos suicidas e aconselhados sobre a necessidade de estarem alerta para surgimento ou piora dos sinais e sintomas de depressão, qualquer mudança incomum de humor ou comportamento, ou o surgimento de comportamento e pensamentos suicidas ou pensamentos sobre automutilação. Comportamentos suspeitos devem ser informados imediatamente aos profissionais de saúde.
Interação com antibióticos carbapenêmicos: antibióticos carbapenêmicos (ex. ertapenem, imipenem e meropenem) podem reduzir as concentrações séricas valproato a níveis subterapêuticos, resultando em perda de controle das crises. Os níveis séricos de valproato devem ser monitorados frequentemente após o início da terapia com carbapenêmicos. Tratamento alternativo com antibacterianos e anticonvulsivantes deve ser considerado se os níveis séricos de valproato tiverem queda significativa ou deterioração do controle das crises.
Trombocitopenia: a frequência de efeitos adversos (particularmente enzimas hepáticas elevadas e trombocitopenia) pode estar relacionada à dose. Em um estudo clínico de divalproato de sódio como monoterapia em pacientes com epilepsia, 34/126 pacientes (27%) recebendo aproximadamente 50 mg/kg/dia, em média, apresentaram pelo menos um valor de plaquetas ≤ 75 x 109 /L. Aproximadamente metade desses pacientes tiveram o tratamento descontinuado, com retorno das contagens de plaquetas ao normal. Nos pacientes remanescentes, as contagens de plaquetas normalizaram-se mesmo com a continuação do tratamento. Neste estudo, a probabilidade de trombocitopenia pareceu aumentar significativamente em concentrações totais de valproato ≥ 110 mcg/mL (mulheres) ou ≥ 135 mcg/mL (homens). O benefício terapêutico que pode acompanhar as maiores doses deverá, portanto, ser considerado contra a possibilidade de maior incidência de eventos adversos.
Disfunção hepática: ver "4. CONTRAINDICAÇÕES" e "5. ADVERTÊNCIAS E PRECAUÇÕES - Hepatoxicidade".
Hiperamonemia: foi relatado hiperamonemia em associação com a terapia com valproato e pode estar presente apesar dos testes de função hepática normais. Em pacientes que desenvolvem letargia inexplicada e vômito ou mudanças no status mental, a encefalopatia hiperamonêmica deve ser considerada e o nível de amônia deve ser mensurado. Hiperamonemia também deve ser considerada em pacientes que apresentam hipotermia. Se a amônia estiver elevada, a terapia com valproato deve ser descontinuada. Intervenções apropriadas para o tratamento da hiperamonemia deve ser iniciada, e os pacientes devem ser submetidos a investigação para determinar as desordens do ciclo da ureia.
Elevações assintomáticas de amônia são mais comuns, e quando presentes, requerem monitoramento intensivo dos níveis de amônia no plasma. Se a elevação persistir a descontinuação da terapia com valproato deve ser considerada.
Hiperamonemia e encefalopatia associadas com o uso concomitante de topiramato: a administração concomitante de topiramato e de ácido valproico foi associada com hiperamonemia, com ou sem encefalopatia, nos pacientes que toleraram uma ou outra droga isoladamente. Os sintomas clínicos de encefalopatia por hiperamonemia incluem frequentemente alterações agudas no nível de consciência e/ou na função cognitiva, com letargia ou vômito. Hipotermia também pode ser uma manifestação de hiperamonemia. Em muitos casos, os sintomas e sinais diminuem com a descontinuação de uma das drogas. Este evento adverso não está relacionado com uma interação farmacocinética. Não se sabe se a monoterapia com topiramato está associada a hiperamonemia.
Pacientes com erros inatos do metabolismo ou atividade mitocondrial hepática reduzida podem apresentar risco aumentado para hiperamonemia, com ou sem encefalopatia. Embora não estudada, a interação de topiramato e ácido valproico pode exacerbar defeitos existentes ou revelar deficiências em pessoas suscetíveis.
Pacientes e responsáveis devem ser informados sobre os sinais e sintomas associados à encefalopatia hiperamonêmica, requerendo avaliação médica se esses sintomas ocorrerem.
Hipotermia: definida como uma queda da temperatura central do corpo para menos de 35°C tem sido relatada em associação com a terapia com valproato em conjunto e na ausência de hiperamonemia. Esta reação adversa também pode ocorrer em pacientes utilizando topiramato e valproato concomitantes após o início do tratamento com topiramato ou após o aumento da dose diária de topiramato. Deve ser considerada a interrupção do valproato em pacientes que desenvolverem hipotermia, a qual pode se manifestar por uma variedade de anormalidades clínicas incluindo letargia, confusão, coma e alterações significativas em outros sistemas importantes como o cardiovascular e o respiratório. Monitoração e avaliação clínica devem incluir análise dos níveis de amônia no sangue.
Atrofia Cerebral/Cerebelar: houve relatos pós-comercialização de atrofia (reversível e irreversível) cerebral e cerebelar, temporariamente associadas ao uso de produtos que se dissociam em íon valproato. Em alguns casos, porém, a recuperação foi acompanha