DICOXIBE
WYETH PHARMA
celecoxibe
Antiinflamatório.
Apresentações.
DicoxibeTM 100 mg em embalagens contendo 20 cápsulas.
DicoxibeTM 200 mg em embalagens contendo 10, 15 ou 30 cápsulas.
VIA DE ADMINISTRAÇÃO: USO ORAL
USO ADULTO
Composição.
Cada cápsula de DicoxibeTM 100 mg ou 200 mg contém 100 mg ou 200 mg de celecoxibe, respectivamente.
Excipientes: lactose monoidratada, povidona, estearato de magnésio, croscarmelose sódica, laurilsulfato de sódio.
MEDICAMENTO SIMILAR EQUIVALENTE AO MEDICAMENTO DE REFERÊNCIA
Indicações.
DicoxibeTM (celecoxibe) está indicado para o tratamento dos sinais e sintomas da osteoartrite (OA) e da artrite reumatoide (AR); alívio dos sinais e sintomas da espondilite anquilosante (EA); alívio da dor aguda (principalmente no pós-operatório de cirurgia ortopédica ou dental e em afecções musculoesqueléticas), alívio dos sintomas da dismenorreia primária e da lombalgia (vide item 3. Características Farmacológicas - Informações Técnicas - Estudos Clínicos e item 5. Advertências e Precauções).
Resultados de eficácia.
Estudos Clínicos
Osteoartrite (OA)
O celecoxibe demonstrou uma redução significativa na dor articular em comparação com o placebo. O celecoxibe foi avaliado para o tratamento dos sinais e sintomas da osteoartrite do joelho e quadril em aproximadamente 4.200 pacientes de estudos clínicos controlados por placebo e por agente ativo com até 12 semanas de duração. Em pacientes com osteoartrite, o tratamento com celecoxibe 100 mg duas vezes ao dia ou 200 mg em dose única diária resultou em melhora do índice de osteoartrite de WOMAC (Western Ontario and McMaster Universities), um índice composto de dor, rigidez, e medidas funcionais em osteoartrite. Em três estudos de 12 semanas de duração em osteoartrite acompanhada de dor e vermelhidão, as doses de celecoxibe de 100 mg duas vezes ao dia ou 200 mg duas vezes ao dia proporcionaram redução significativa da dor dentro de 24-48 horas após o início da administração. Em doses de 100 mg duas vezes ao dia ou 200 mg duas vezes ao dia, a eficácia do celecoxibe mostrou ser semelhante à do naproxeno 500 mg duas vezes ao dia. Doses de 200 mg duas vezes ao dia não proporcionaram benefício adicional acima do observado com 100 mg duas vezes ao dia. Uma dose diária total de 200 mg mostrou ser igualmente eficaz quer seja administrada como 100 mg duas vezes ao dia ou como 200 mg em dose única diária.
Artrite Reumatoide (AR)
O celecoxibe demonstrou uma redução significativa na sensibilidade/dor articular e no inchaço articular em comparação com o placebo. O celecoxibe foi avaliado para o tratamento dos sinais e sintomas de artrite reumatoide em aproximadamente 2.100 pacientes em estudos clínicos controlados por placebo e por agente ativo com até 24 semanas de duração. O celecoxibe mostrou ser superior ao placebo nestes estudos, quando se utilizou o Índice de Resposta do American College of Rheumatology 20 (ACR20), um índice composto de medidas clínicas, laboratoriais e funcionais da artrite reumatoide. As doses de celecoxibe 100 mg duas vezes ao dia e 200 mg duas vezes ao dia apresentaram eficácia semelhante e ambas foram comparáveis à eficácia do naproxeno 500 mg duas vezes ao dia.
Embora o celecoxibe nas doses de 100 mg duas vezes ao dia e 200 mg duas vezes ao dia tenha proporcionado eficácia global semelhante, alguns pacientes obtiveram benefício adicional com a dose de 200 mg duas vezes ao dia. Doses de 400 mg duas vezes ao dia não proporcionaram benefício adicional acima do observado com 100 mg-200 mg duas vezes ao dia.
Analgesia em dor aguda, incluindo Dismenorreia Primária
Nos modelos de analgesia aguda de dor pós-cirúrgica oral, ortopédica e dismenorreia primária, o celecoxibe aliviou a dor classificada pelos pacientes como moderada a grave. Doses únicas de celecoxibe proporcionaram alívio da dor dentro de um período de 60 minutos (vide item 8. Posologia e Modo de Usar).
Espondilite Anquilosante (EA)
O celecoxibe foi avaliado em pacientes com espondilite anquilosante em dois estudos clínicos controlados por placebo e por agente ativo com 6 e 12 semanas de duração.
O celecoxibe nas doses de 100 mg duas vezes ao dia, 200 mg em dose única diária e 400 mg em dose única diária mostrou ser estatisticamente superior ao placebo nestes estudos para todas as três medidas de eficácia primárias que avaliam a intensidade de dor global (Escala Visual Analógica), atividade da doença global (Escala Visual Analógica) e comprometimento funcional (Índice Funcional de Espondilite Anquilosante de Bath). No estudo de 12 semanas, não houve diferença no nível de melhora entre as doses de 200 mg e 400 mg de celecoxibe em uma comparação de alteração média em relação ao basal, porém houve uma maior porcentagem de pacientes que responderam ao celecoxibe 400 mg (53%) do que ao celecoxibe 200 mg (44%), utilizando-se a Avaliação dos critérios de resposta de Espondilite Anquilosante (ASAS 20). A ASAS 20 define resposta de um paciente ao tratamento como melhora em relação ao basal de pelo menos 20% e melhora absoluta de pelo menos 10 mm, em uma escala de 0 a 100 mm, em pelo menos, três de quatro dos seguintes domínios: avaliação global do paciente, dor, Índice Funcional de Espondilite Anquilosante de Bath e inflamação. A análise de resposta também demonstrou ausência de alteração nas taxas de resposta em períodos superiores a 6 semanas.
Estudos em Dismenorreia
Dois estudos foram realizados para avaliar a eficácia do celecoxibe em dismenorreia, ambos randômicos, duplo-cegos, com 3 braços cruzados, que compararam celecoxibe (n=253) a naproxeno (n=251) e placebo (n=256). Nos dois estudos as pacientes receberam a dose inicial da medicação definida randomicamente (celecoxibe 400 mg, naproxeno 550 mg ou placebo) no primeiro dia do ciclo menstrual e, se necessário, doses das mesmas medicações (celecoxibe 200 mg, naproxeno 550 mg e placebo) eram repetidas a cada 12 horas por 3 dias.
Em todas as medidas de eficácia utilizadas (tempo até o alívio da dor, redução da intensidade da dor nas 8 e 12 horas após a dose inicial; manutenção da intensidade da analgesia durante os 3 dias de tratamento - quando necessário - e o uso de medicação analgésica de resgate) celecoxibe e naproxeno foram estatisticamente superiores ao placebo (p < 0,001). A avaliação do paciente em relação à eficácia do tratamento também foi superior (p < 0,01) nos braços em que as medicações ativas foram usadas.
Lombalgia
O celecoxibe foi utilizado para tratar pacientes que apresentavam lombalgia não neuropática preexistente de duração ≥12 semanas. Na tabela a seguir, os resultados de eficácia de 5 estudos clínicos são apresentados utilizando a Escala de Avaliação da Intensidade da Dor do Paciente (escala visual analógica de 100 mm), a partir da linha de base ao fim do tratamento: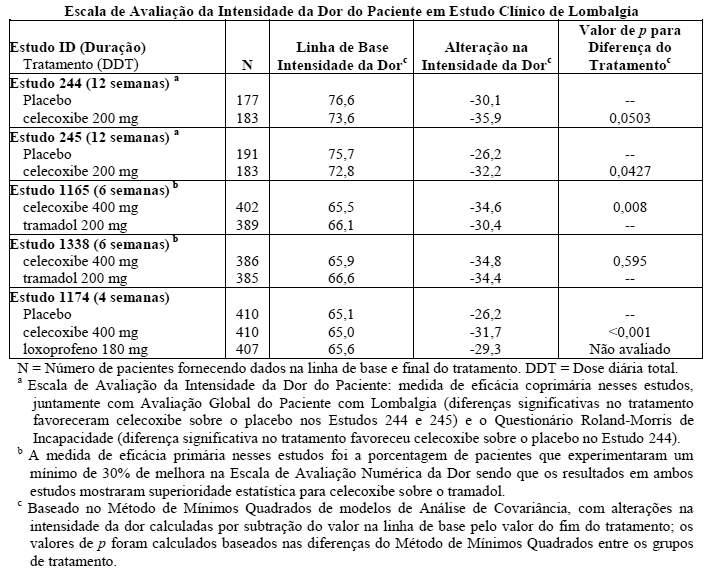
Informações Adicionais de Estudos Clínicos
Estudos Endoscópicos
As avaliações endoscópicas do trato gastrintestinal foram realizadas em mais de 4.500 pacientes com artrite que foram admitidos em 5 estudos randomizados e controlados de 12-24 semanas de duração que utilizaram agentes comparativos ativos, 2 dos quais também incluíram controles com placebo. Não houve relação consistente entre a incidência de úlceras gastroduodenais e a dose de celecoxibe dentro do intervalo estudado.
A Tabela 1 resume a incidência de úlceras endoscópicas em dois estudos de 12 semanas de duração que admitiram pacientes cujas endoscopias basais revelaram inexistência de úlceras.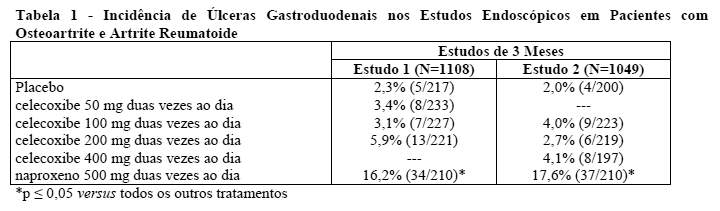
A Tabela 2 resume os dados de dois estudos de 12 semanas que incluiu pacientes cujas endoscopias basais revelaram ausência de úlceras. Os pacientes foram submetidos a intervalos entre as endoscopias a cada 4 semanas para fornecer informações sobre o risco de úlcera em função do tempo.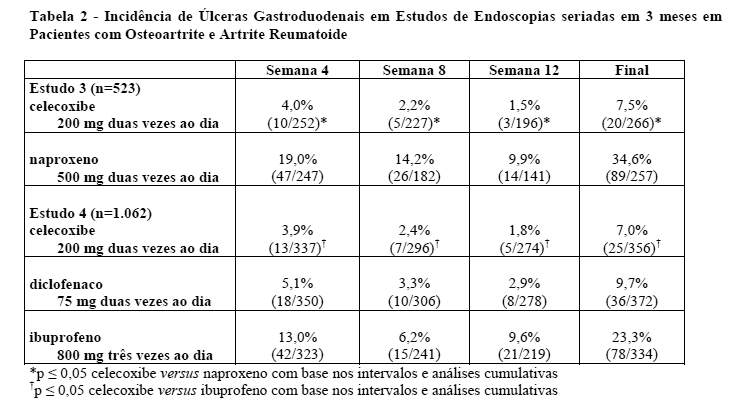
Foi conduzido um estudo randomizado e duplo-cego de 6 meses de duração em 430 pacientes com artrite reumatoide, no qual um exame endoscópico foi realizado no 6° mês.
A incidência de úlceras endoscópicas em pacientes recebendo celecoxibe 200 mg duas vezes ao dia foi de 4% versus 15% para pacientes recebendo diclofenaco SR (liberação prolongada) 75 mg duas vezes ao dia (p < 0,001).
Em 4 dos 5 estudos endoscópicos, aproximadamente 11% dos pacientes (440/4.000) estavam tomando ácido acetilsalicílico (≤ 325 mg/dia). Nos grupos celecoxibe, a taxa de úlcera endoscópica pareceu ser maior nos usuários de ácido acetilsalicílico do que nos não usuários. No entanto, a taxa aumentada de úlceras nestes usuários de ácido acetilsalicílico foi menor que a taxa de úlceras endoscópicas observada nos grupos com agentes comparativos ativos, com ou sem ácido acetilsalicílico.
A correlação entre os achados dos estudos endoscópicos e a incidência relativa de eventos sérios clinicamente significativos no trato gastrintestinal superior não foi estabelecida. Sangramento sério clinicamente significativo no trato gastrintestinal superior foi observado, embora infrequentemente, em pacientes recebendo celecoxibe em estudos controlados e abertos (vide item 5. Advertências e Precauções - Efeitos Gastrintestinais (GI)).
Meta-Análises em Segurança Gastrintestinal de Estudos em Osteoartrite e Artrite Reumatoide
Uma análise de 31 estudos clínicos controlados randomizados em osteoartrite (OA) e artrite reumatoide (AR), envolvendo 39.605 pacientes com osteoartrite (OA) (n=25.903), artrite reumatoide (AR) (n=3.232) ou pacientes com outras condições (n=10.470), comparou a incidência de eventos adversos gastrintestinais em pacientes tratados com celecoxibe à incidência em pacientes recebendo placebo ou AINEs (incluindo naproxeno, diclofenaco e ibuprofeno). A incidência clínica de úlcera e sangramento da úlcera com celecoxibe na dose diária total de 200 mg-400 mg foi de 0,2%, comparada à incidência de 0,6% com AINEs (RR=0,35; 95% IC 0,22-0,56).
Estudo de Segurança Prolongada do celecoxibe em Artrite (CLASS) incluindo o uso concomitante de ácido acetilsalicílico
Em um estudo prospectivo prolongado de resultados da segurança conduzido na fase pós-comercialização em aproximadamente 5.800 pacientes com osteoartrite e 2.200 pacientes com artrite reumatoide, os pacientes receberam celecoxibe 400 mg duas vezes ao dia (4 vezes e 2 vezes as doses recomendadas para osteoartrite e artrite reumatoide, respectivamente, ibuprofeno 800 mg 3 vezes/dia ou diclofenaco 75 mg duas vezes ao dia (doses terapêuticas usuais). As exposições medianas para o celecoxibe (n=3.987) e o diclofenaco (n=1.996) foram de 9 meses enquanto com o ibuprofeno (n=1.985) foi de 6 meses.
As taxas cumulativas de Kaplan-Meier em 9 meses são fornecidas para todas as análises. O endpoint primário deste estudo foi a incidência de úlceras complicadas (sangramento gastrintestinal, perfuração ou obstrução). Os pacientes podiam tomar ácido acetilsalicílico (AAS) em baixa dose concomitante (≤325 mg/dia) como profilático cardiovascular (subgrupos de AAS: celecoxibe, n=882; diclofenaco, n=445; ibuprofeno, n=412). As diferenças de incidência de úlceras complicadas entre o celecoxibe e o grupo combinado de ibuprofeno e diclofenaco não foram estatisticamente significativas. Os pacientes recebendo celecoxibe e AAS em baixa dose concomitante apresentaram taxas 4 vezes maiores de úlceras complicadas em comparação com os que não receberam AAS (vide item 5. Advertências e Precauções - Efeitos Gastrintestinais (GI)). Os resultados para celecoxibe encontram-se na Tabela 3.
Função Plaquetária
Em voluntários sadios, o celecoxibe em doses terapêuticas e em doses múltiplas de 600 mg duas vezes ao dia (três vezes a dose mais alta recomendada) não apresentou efeito sobre a agregação plaquetária e tempo de sangramento em comparação com o placebo. Todos os controles ativos (Inibidores inespecíficos da COX) reduziram significativamente a agregação plaquetária e prolongaram o tempo de sangramento (vide Figura 1).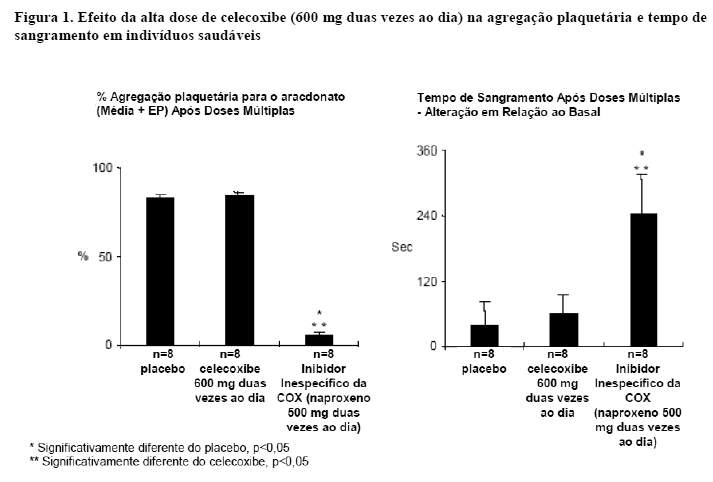
Estudo de celecoxibe versus omeprazol e diclofenaco em Pacientes sob Risco de Osteoartrite e Artrite Reumatoide (CONDOR)
Neste estudo prospectivo de 24 semanas em pacientes com idade ≥60 anos ou com histórico de úlcera gastroduodenal (excluindo usuários de ácido acetilsalicílico em baixa dose), a porcentagem de pacientes com eventos gastrintestinais clinicamente significativos (endpoint primário composto) foi menor em pacientes tratados com celecoxibe 200 mg duas vezes ao dia comparado aos pacientes tratados com diclofenaco SR (liberação prolongada) 75 mg duas vezes ao dia + omeprazol 20 mg uma vez ao dia. Este resultado é baseado na diminuição clinicamente significativa na hemoglobina (≥2g/dL) e/ou hematócrito (≥10%) de origem gastrintestinal definida ou suposta. Os resultados dos desfechos individuais desse endpoint composto foram os seguintes: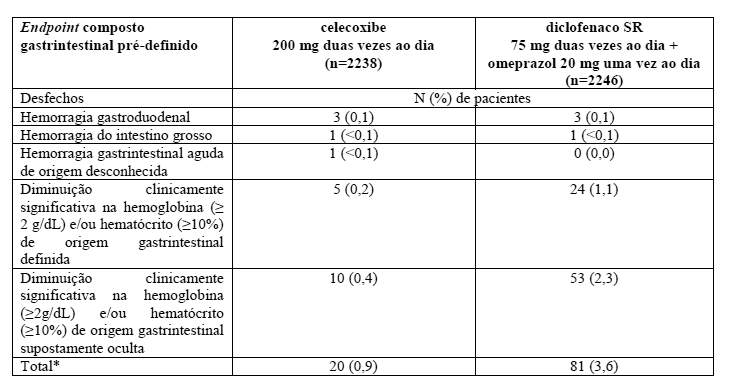
Para os seguintes componentes do endpoint composto gastrintestinal pré-definido, não houve eventos em ambos os grupos de tratamento: obstrução da saída gástrica; perfuração gastroduodenal, do intestino delgado ou do intestino grosso; hemorragia do intestino delgado. Todos os eventos compreendendo o endpoint composto gastrintestinal foram avaliados por um grupo de especialistas independente que não tinha conhecimento de qual grupo randomizado de tratamento o paciente fazia parte.
* Em uma análise de tempo para ocorrência de um desfecho, p < 0,0001 para a comparação entre o grupo de tratamento com celecoxibe e o grupo de tratamento com omeprazol + diclofenaco para este endpoint.
Segurança Cardiovascular - Estudos em Longo Prazo Envolvendo Pacientes com Pólipos Adenomatosos Esporádicos
Foram conduzidos dois estudos com celecoxibe envolvendo pacientes com pólipos adenomatosos esporádicos por ex., estudo APC (Adenoma Prevention with Celecoxib) e o estudo PreSAP (Prevention of Spontaneous Adenomatous Polyps). No estudo APC, houve um aumento relacionada à dose no endpoint composto de morte cardiovascular, infarto do miocárdio e acidente vascular encefálico (julgado) com celecoxibe comparado ao placebo por mais de 3 anos de tratamento. O estudo PreSAP não demonstrou um aumento de risco estatisticamente significativo para o mesmo endpoint.
No estudo APC, os riscos relativos comparados ao placebo para o endpoint composto de morte cardiovascular, infarto do miocárdio ou acidente vascular encefálico (julgado) foram 3,4 (95% IC 1,4-8,5) com celecoxibe 400 mg duas vezes ao dia e 2,8 (95% IC 1,1-7,2) com celecoxibe 200 mg duas vezes ao dia. As taxas cumulativas para o endpoint composto por mais de 3 anos de estudo foram de 3,0% (20/671) e 2,5% (17/685) para grupos de tratamento com celecoxibe 200 mg e 400 mg duas vezes ao dia, respectivamente, comparadas a 0,9% (6/679) para o grupo placebo. Os aumentos para ambos os grupos de doses de celecoxibe versus placebo foram devidos principalmente ao infarto do miocárdio.
No estudo PreSAP, o risco relativo comparado ao placebo para o mesmo endpoint composto foi de 1,2 (95% IC 0,6-2,4) com celecoxibe 400 mg uma vez ao dia. As taxas cumulativas para o endpoint composto por mais de 3 anos foram de 2,3% (21/933) comparadas a 1,9% (12/628 indivíduos) para o grupo placebo.
Segurança Cardiovascular -Estudo de Longa Duração e Prevenção da Doença de Alzheimer com uso de Anti-inflamatórios(ADAPT) Dados do estudo ADAPT (Prevenção da Doença de Alzheimer com uso de Anti-inflamatórios) não apresentou um aumento significativo do risco cardiovascular com o celecoxibe 200 mg duas vezes por dia em comparação com placebo. O risco relativo em comparação ao placebo para um endpoint semelhante (morte por alteração cardiovascular, infarto do miocárdio ou acidente vascular cerebral- AVC) foi de 1,14 (95% CI 0,61-2,12) com celecoxibe 200 mg duas vezes por dia.
Segurança Cardiovascular - Meta-Análise de Estudos com Uso Crônico
Não foi conduzido qualquer estudo clínico de longo prazo controlado e delineado especificamente para avaliar a segurança cardiovascular na administração crônica de celecoxibe com qualquer duração. Entretanto, foi conduzida uma meta-análise dos dados de segurança (eventos adversos considerados sérios pelo investigador) de 39 estudos clínicos completos com celecoxibe, de até 65 semanas de duração, representando 41.077 pacientes (23.030 (56,1%) pacientes expostos ao celecoxibe dose diária total de 200 mg-800 mg, 13.990 (34,1%) pacientes expostos aos AINEs não seletivos e 4.057 (9,9%) pacientes expostos ao placebo).
Nesta análise, a taxa de eventos considerados para o endpoint composto de morte cardiovascular, infarto do miocárdio não fatal e acidente vascular encefálico não fatal foi similar entre celecoxibe (n=19.773; 0,96 eventos/100 pacientes-ano) e o tratamento com AINEs não seletivos (n=13.990; 1,12 eventos/100 pacientes-ano) (RR=0,90; 95% IC 0,60-1,33). Este padrão de efeito foi mantido com ou sem o uso do ácido acetilsalicílico (≤325 mg). Houve uma incidência maior de infarto do miocárdio não fatal (RR=1,76; 95% IC 0,93-3,35). Entretanto, houve uma tendência de acidente vascular encefálico não fatal menor (RR=0,51; 95% IC 0,23-1,10) e a incidência de morte cardiovascular foi similar (RR=0,57; 95% IC 0,28-1,14) para celecoxibe comparado aos AINEs não seletivos combinados.
Nesta análise, a taxa de eventos considerados do endpoint composto de morte cardiovascular, infarto do miocárdio não fatal e acidente vascular encefálico não fatal foram de 1,42/100 paciente-ano para o tratamento com celecoxibe (n=7.462) e 1,20/100 pacientes-ano para placebo (n=4.057) (RR=1,11; 95% IC 0,47-2,67). Este padrão de efeito foi mantido com ou sem o uso de ácido acetilsalicílico (≤325 mg). Houve uma tendência maior de incidência de infarto do miocárdio não fatal (RR=1,56; 95% IC 0,21-11,90) e de morte cardiovascular (RR=1,26; 95% IC 0,33-4,77), e a de acidente vascular encefálico não fatal foi similar (RR=0,80; 95% IC 0,19-3,31) para celecoxibe comparada ao placebo.
Segurança Cardiovascular
Os resultados de segurança cardiovascular foram avaliados no estudo CLASS. As taxas cumulativas Kaplan-Meier para os eventos adversos tromboembólicos cardiovasculares sérios relatados pelo investigador (incluindo infarto do miocárdio, embolia pulmonar, trombose venosa profunda, angina instável, ataque isquêmico transitório e acidente cerebrovascular isquêmico) não demonstraram diferenças entre os grupos de tratamento com celecoxibe, diclofenaco ou ibuprofeno. As taxas cumulativas em todos os pacientes no nono mês para celecoxibe, diclofenaco e ibuprofeno foram 1,2%, 1,4% e 1,1% respectivamente. As taxas cumulativas em pacientes que não estavam utilizando o ácido acetilsalicílico no nono mês em cada um dos 3 grupos de tratamento foram menores que 1%. As taxas cumulativas para infarto do miocárdio em pacientes não usuários de ácido acetilsalicílico no nono mês em cada um dos 3 grupos de tratamento foram menores que 0,2%. Não havia grupo placebo no estudo CLASS, o que limita a possibilidade de determinar se os 3 fármacos testados não tinham aumento de risco de eventos cardiovasculares ou se eles todos tiveram o risco aumentado em um grau similar.
REFERENCIAS BIBLIOGRÁFICAS
1. Dawood MY. Dysmenorrhea. J Reprod Med 1985;30:154-167.
2. Benedetto C. Eicosanoids in primary dysmenorrhea, endometriosis and menstrual migraine. Gynecol Endocrinol 1989;3:71-94.
3. Burton AL, Boortz-Marx RL, Cabrera JA. Pre-emptive use of celecoxib in the treatment of primary dysmenorrhea. Reg Anesth Pain Med 2001;26(2):108.
4. Moore A, Derry S, Makinson G and McQuay H: Tolerability and adverse events in clinical trials of celecoxib in osteoarthritis and rheumatoid arthritis: systematic review and meta-analysis of information from company clinical trial reports. Arthritis Research & Therapy 2005, 7(3):R644-R665.
5. Arber N, Eagle CJ, Spicak J et al. Celecoxib for the Prevention of Colorectal Adenomatous Polyps. N Engl J Med 2006;355:885-95.
6. Solomon SD, McMurray JJ, Pfeffer MA et al. Effect of Celecoxib on Cardiovascular Events and Blood Pressure in Two Trials for the Prevention of Colorectal Adenomas. Circulation. 2006;114:1028-1035.
7. White WB, West CR, Borer JS, et al. Risk of Cardiovascular Events in Patients Receiving Celecoxib: A Meta-Analysis of Randomized Clinical Trials. Am J Cardiol (Jan 1, 2007);99:91-98.
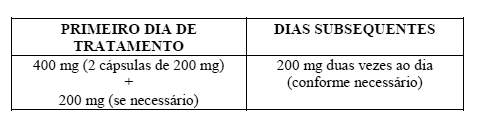
8. JB O'Donnell et al. The effectiveness of a weak opioid medication versus a Cyclo-oxygenase-2 (COX-2) selective non-steroidal anti-inflamatory drug in treating flare-up of chronic low back pain: Results from two randomized, double-blind, 6 week studies. The Journal of international medical research. 2009; 37: 1789-1802.
Caract. farmacológicas.
Propriedades Farmacodinâmicas
O mecanismo de ação do celecoxibe é via inibição da síntese das prostaglandinas, principalmente pela inibição da enzima ciclooxigenase 2 (COX-2). Em concentrações terapêuticas em humanos, celecoxibe não inibe a ciclooxigenase 1 (COX-1). A COX-2 é induzida em resposta a estímulos inflamatórios. Isto leva à síntese e ao acúmulo de prostanoides inflamatórios, em particular a prostaglandina E2, causando inflamação, edema e dor. O celecoxibe age como um agente anti-inflamatório, analgésico e antipirético em modelos animais pelo bloqueio da produção de prostanoides inflamatórios via inibição da COX-2. Em modelos animais de tumores de colo, celecoxibe reduziu a incidência e a multiplicidade dos tumores.
Estudos in vivo e ex vivo mostram que celecoxibe tem afinidade muito baixa pela enzima ciclooxigenase 1 (COX-1) de expressão constitutiva. Consequentemente, em doses terapêuticas, celecoxibe não tem efeito sobre prostanoides sintetizados pela ativação da COX-1, não interferindo, portanto, nos processos fisiológicos relacionados à COX-1 nos tecidos, particularmente no estômago, intestino e plaquetas.
Propriedades Farmacocinéticas
Absorção
A farmacocinética do celecoxibe foi avaliada em aproximadamente 1.500 indivíduos. Quando administrado em condições de jejum, o celecoxibe é bem absorvido atingindo concentrações plasmáticas máximas após aproximadamente 2-3 horas. A biodisponibilidade oral das cápsulas é de cerca de 99% em relação à administração em suspensão (forma farmacêutica oral de disponibilidade ideal). Em condições de jejum, tanto os níveis plasmáticos máximos (Cmáx) como as áreas sob a curva (AUC) são quase proporcionais à dose de até 200 mg duas vezes ao dia; em doses mais altas, ocorrem aumentos menos proporcionais na Cmáx e AUC.
Distribuição
A taxa de ligação às proteínas plasmáticas, que é independente da concentração, é de cerca de 97% em concentrações plasmáticas terapêuticas e o celecoxibe não se liga preferencialmente aos eritrócitos no sangue.
Metabolismo
O metabolismo de celecoxibe é mediado principalmente pela via citocromo P450 2C9. Foram identificados 3 metabólitos, inativos como os inibidores da COX-1 e COX-2, no plasma humano: álcool primário, o ácido carboxílico correspondente e seu glicuronídeo conjugado.
A atividade do citocromo P450 2C9 é reduzida em indivíduos com polimorfismos genéticos que levam à atividade reduzida da enzima, tais como aquelas homozigóticas para o polimorfismo CYP2C9*3.
Em um estudo farmacocinético de celecoxibe 200 mg administrado uma vez ao dia em voluntários sadios, genotipados como CYP2C9*1/*1, CYP2C9*1/*3 ou CYP2C9*3/*3, a média de Cmáx e AUC0-24 de celecoxibe no 7° dia foi de aproximadamente 4 vezes e 7 vezes, respectivamente, em indivíduos genotipados como CYP2C9*3/*3 comparados aos outros genótipos. Em três estudos diferentes de dose única, envolvendo um total de 5 indivíduos genotipados como CYP2C9*3/*3, AUC0-24 aumentada com dose única em aproximadamente 3 vezes comparado aos metabolizadores normais. É estimado que a frequência do genótipo homozigoto *3/*3 é 0,3-1,0% entre os diferentes grupos étnicos.
O celecoxibe deve ser administrado com cautela a pacientes com deficiência ou suspeita de deficiência de metabolizadores CYP2C9 baseados no histórico prévio/experiência com outros substratos CYP2C9. Considerar o início de tratamento com a metade da menor dose recomendada (vide item 8. Posologia e Modo de Usar e item 6. Interações Medicamentosas).
Excreção
O celecoxibe é eliminado predominantemente por metabolismo hepático, com menos de 1% da dose excretada inalterada na urina. Após múltiplas doses, a meia-vida de eliminação é de 8 a 12 horas e o clearance é de aproximadamente 500 mL/min. Com administrações múltiplas, as condições do estado de equilíbrio são atingidas até o 5° dia. A variação dos parâmetros farmacocinéticos (AUC, Cmáx e meia-vida) entre indivíduos é da ordem de 30%. O volume médio de distribuição é de aproximadamente 500 litros por 70 kg em indivíduos jovens adultos saudáveis, indicando extensa distribuição em todos os tecidos. Estudos pré-clínicos indicam que o celecoxibe atravessa a barreira hematoencefálica.
Efeitos dos alimentos
A administração com alimentos (refeição rica em gorduras) retarda a absorção do celecoxibe resultando em um Tmáx de cerca de 4 horas e aumenta a biodisponibilidade em cerca de 20% (vide item 8. Posologia e Modo de Usar).
Em voluntários adultos saudáveis, a exposição sistêmica global (AUC) de celecoxibe foi equivalente quando o celecoxibe foi administrado como cápsulas intactas ou cápsulas abertas cujo conteúdo foi misturado ao molho de maçã. Não houve alterações significantes no Cmáx, Tmáx ou T1/2 após a administração do conteúdo das cápsulas abertas misturadas ao molho de maçã.
Populações Especiais
Idosos: na população com idade > 65 anos, ocorre um aumento de 1,5 a 2 vezes a média de Cmáx e de AUC para o celecoxibe. Esta é uma alteração predominantemente relacionada ao peso em vez de ser relacionada à idade, os níveis de celecoxibe ficando mais altos em indivíduos de menor peso e, consequentemente, mais altos na população idosa, que geralmente apresenta peso médio inferior ao peso médio da população mais jovem. Portanto, as mulheres idosas tendem a apresentar concentrações plasmáticas do fármaco mais altas do que os homens idosos. Geralmente não é necessário ajuste de dose. No entanto, para pacientes idosos com menos de 50 kg, deve-se introduzir o tratamento com a menor dose recomendada.
Raça: uma meta-análise de estudos farmacocinéticos sugeriu que a AUC de celecoxibe é aproximadamente 40% maior em pacientes da raça negra quando comparada a pacientes da raça branca. A causa e o significado clínico desse achado não são conhecidos.
Insuficiência hepática: as concentrações plasmáticas de celecoxibe em pacientes com insuficiência hepática leve (classe A de Child-Pugh) não são significativamente diferentes dos controles pareados por sexo e idade. Em pacientes com insuficiência hepática moderada (classe B de Child-Pugh) a concentração plasmática de celecoxibe é cerca de 2 vezes a do grupo controle (vide item 8. Posologia e Modo de Usar).
Insuficiência renal: a farmacocinética do celecoxibe em indivíduos idosos com redução do ritmo de filtração glomerular (RFG) relacionada à idade (RFG médio > 65 mL/min/1,73 m2) e em pacientes com insuficiência renal crônica estável (RFG entre 35 e 60 mL/min/1,73 m2) foi comparável a de indivíduos com função renal normal. Não foi descoberta relação significante entre creatinina sérica (ou clearance de creatinina) e clearance de celecoxibe. Em insuficiência renal grave, não é esperada uma alteração do clearance de celecoxibe uma vez que a principal via de eliminação é hepática para metabólitos inativos.
Efeitos renais: os papéis das enzimas ciclooxigenase 1 e 2 na fisiologia renal ainda não são plenamente conhecidos. O celecoxibe reduz a excreção urinária de PGE2 e da 6-ceto-PGF1a (um metabólito da prostaciclina), mas não altera o nível sérico de tromboxano B2 (TXB2), e a excreção urinária de 11-deidro-TXB2, um metabólito do tromboxano inalterado (ambos resultantes da atividade da COX-1). Estudos específicos demonstraram que celecoxibe não produz diminuição do ritmo de filtração glomerular em idosos ou em pacientes com insuficiência renal crônica. Estes estudos também demonstraram reduções transitórias na excreção fracionada de sódio. Nos estudos conduzidos em pacientes com artrite, uma incidência comparável de edema periférico foi observada em relação à verificada com inibidores inespecíficos da COX (que também apresentam atividade inibitória da COX-2). Isto foi mais evidente em pacientes recebendo terapia diurética concomitante. No entanto, não foram observados aumentos das incidências de hipertensão e insuficiência cardíaca e o edema periférico foi leve e autolimitante.
Dados de Segurança Pré-clínica
Dados de segurança não clínicos revelaram a ausência de risco especial para humanos com base nos estudos convencionais de toxicidade de dose repetida, mutagenicidade ou carcinogenicidade.
O celecoxibe em doses orais ≥150 mg/kg / dia (aproximadamente 2 vezes a dose de exposição humana em 200 mg duas vezes ao dia, conforme medido por AUC0-24), causou um aumento da incidência de defeitos do septo ventricular, um evento raro, e alterações fetais, tais como costelas fundidas, esterno fundido e esterno disforme quando coelhos foram tratados durante toda a organogênese. Foi observado um aumento dose-dependente na hérnia diafragmática quando os ratos receberam celecoxibe em doses orais ≥30 mg / kg / dia (aproximadamente 6 vezes a dose de exposição humana com base na AUC0-24 em 200 mg duas vezes ao dia) durante toda a organogênese. Estes efeitos são esperados com a inibição da síntese de prostaglandinas. Em ratos, a exposição ao celecoxibe durante o desenvolvimento embrionário inicial resultou em perdas pré-implantação e pós-implantação, e reduziu a sobrevivência embrionária / fetal.
Toxicologia
Um aumento na incidência de achados experimentais de espermatocele com ou sem alterações secundárias, assim como hipoespermia epididimal mínima, assim como insignificante dilatação dos túbulos seminíferos tem sido encontrado em ratos jovens. Estes achados reprodutivos aparentemente relacionados ao tratamento, não aumentaram a incidência ou severidade com dose, e podem indicar uma exacerbação de uma condição espontânea. Achados reprodutivos similares não foram observados em estudos com cachorros jovens e adultos ou em ratos adultos tratados com celecoxibe. A significância clínica desta observação é desconhecida.
Contraindicações.
DicoxibeTM é contraindicado a pacientes com hipersensibilidade ao celecoxibe ou a qualquer componente da fórmula. DicoxibeTM é contraindicado, também, a pacientes com hipersensibilidade a sulfonamidas.
DicoxibeTM não deve ser administrado a pacientes que tenham apresentado asma, urticária ou reações alérgicas após uso de ácido acetilsalicílico (AAS) ou outros anti-inflamatórios não esteroides (AINEs), incluindo outros inibidores específicos da ciclooxigenase 2 (COX-2). Reações graves, raramente fatais, tipo anafiláticas a AINEs foram descritas em tais pacientes (vide item 5. Advertências e Precauções).
Não deve ser administrado a pacientes com doenças hepáticas (albumina sérica abaixo de 25 g/L) e com insuficiência renal grave (clearance de creatinina abaixo de 30 mL/min).
DicoxibeTM é contraindicado no tratamento da dor peri-operatória em pacientes submetidos à cirurgia de revascularização do miocárdio (vide item 5. Advertências e Precauções).
Advertências e precauções.
Eventos cardiovasculares trombóticos: DicoxibeTM pode causar um aumento no risco de eventos cardiovasculares (CV) trombóticos graves, infarto do miocárdio (IM) e acidente vascular encefálico, que pode ser fatal. Todos os anti-inflamatórios não esteroides podem ter um risco similar. Este risco pode aumentar com a dose, duração do tratamento e fator de risco cardiovascular basal. Pacientes com história médica conhecida de doença cardiovascular podem estar sob um risco maior. Para minimizar o risco potencial para um evento adverso cardiovascular em pacientes tratados com DicoxibeTM, deve-se usar a menor dose eficaz pelo menor período possível. Médicos e pacientes devem permanecer alertas para o desenvolvimento de tais eventos, mesmo na ausência de sintomas cardiovasculares prévios. Os pacientes devem ser informados sobre os sinais e sintomas de toxicidade cardiovascular grave e as medidas a serem tomadas se estes ocorrerem (vide item 3. Características Farmacológicas - Propriedades Farmacodinâmicas).
Foi observada incidência aumentada de infarto do miocárdio e acidente vascular encefálico em dois grandes estudos clínicos controlados com um anti-inflamatório não esteroide, seletivo para COX-2 diferente de DicoxibeTM, para o tratamento da dor nos primeiros 10-14 dias após cirurgia de revascularização do miocárdio (vide item 4. Contraindicações).
O celecoxibe não é um substituto do ácido acetilsalicílico na profilaxia de doença cardiovascular tromboembólica devido à falta de efeitos sobre a função plaquetária. Uma vez que o celecoxibe não inibe a agregação plaquetária, a terapia antiplaquetária (por ex., ácido acetilsalicílico) não deve ser descontinuada.
Efeitos gastrintestinais: perfurações, úlceras ou hemorragias gastrintestinais altas e baixas ocorreram em pacientes tratados com DicoxibeTM. Pacientes com maior risco para o desenvolvimento dessas complicações gastrintestinais com AINEs são os idosos, pacientes com doença cardiovascular, pacientes em uso concomitante de ácido acetilsalicílico, glicocorticoides ou outros AINEs, pacientes que fazem uso de álcool ou pacientes com história de doença gastrintestinal prévia ou doença ativa, tais como úlceras, hemorragia gastrintestinal ou condições inflamatórias. A maior parte dos relatos espontâneos de eventos gastrintestinais fatais aconteceu em idosos ou pacientes debilitados.
Embora se tenha demonstrado redução significativa do risco de desenvolvimento de complicações gastrintestinais comumente associadas ao uso de anti-inflamatórios, este risco não é completamente eliminado pelo uso de celecoxibe.
Para se reduzir o risco potencial de um efeito adverso GI, deve ser utilizada a menor dose eficaz durante o menor período de tempo possível.
Uso com outros AINEs: o uso concomitante de celecoxibe e um AINE, diferente do ácido acetilsalicílico, deve ser evitado.
Uso com anticoagulantes orais: o uso concomitante de AINEs com anticoagulantes orais aumenta o risco de hemorragia e deve ser administrado com cautela. Anticoagulantes orais incluem varfarina/cumarínicos e novos anticoagulantes orais (por exemplo apixabana, dabigatrana e rivaroxabana). Em pacientes em terapia concomitante com varfarina ou agentes similares, eventos hemorrágicos sérios, alguns deles fatais, foram relatados. Uma vez que aumento do tempo de protrombina (INR) foi relatado, a anticoagulação/INR deve ser monitorada em pacientes utilizando varfarina/anticoagulante cumarínico após o início do tratamento com DicoxibeTM ou após mudança de dose (vide item 6. Interações Medicamentosas).
Hipertensão: assim como ocorre com todos os AINEs, DicoxibeTM pode levar ao início de uma nova hipertensão ou piora da hipertensão preexistente, das quais podem contribuir para um aumento na incidência de eventos cardiovasculares. AINEs, incluindo DicoxibeTM, devem ser usados com cautela em pacientes com hipertensão. A pressão sanguínea deve ser cuidadosamente monitorada no início e durante a terapia com DicoxibeTM.
Retenção hídrica e edema: assim como ocorre com outros medicamentos inibidores da síntese de prostaglandinas, observou-se retenção hídrica e edema em pacientes recebendo DicoxibeTM. Portanto, pacientes com insuficiência cardíaca congestiva ou hipertensão preexistentes devem ser cuidadosamente monitorados. O DicoxibeTM deve ser usado com cautela em pacientes com função cardíaca comprometida, edema preexistente, ou outras condições que predisponham ou piorem a retenção hídrica, incluindo aqueles que fazem uso de diuréticos, ou sob risco de hipovolemia.
Efeitos renais: AINEs, incluindo DicoxibeTM, podem causar toxicidade renal. Estudos clínicos com DicoxibeTM mostraram efeitos renais similares àqueles observados com um AINEs comparativo. Pacientes sob um risco maior de toxicidade renal são aqueles com insuficiência renal, insuficiência cardíaca, disfunção hepática e os idosos. Tais pacientes devem ser cuidadosamente monitorados durante o tratamento com DicoxibeTM. Recomenda-se então, cuidado em pacientes com doença renal preexistente.
Deve-se ter cuidado ao iniciar o tratamento em pacientes com desidratação. É aconselhável reidratar o paciente antes de iniciar o tratamento com DicoxibeTM.
Doença renal avançada: a função renal deve ser cuidadosamente monitorada em pacientes com doença renal avançada em uso de DicoxibeTM (vide item 8. Posologia e Modo de Usar).
Efeitos hepáticos: pacientes com insuficiência hepática grave (classe C de Child-Pugh) não foram estudados. O uso de DicoxibeTM em pacientes com insuficiência hepática grave não é recomendado. DicoxibeTM deve ser utilizado com cuidado em pacientes com insuficiência hepática moderada (classe B de Child-Pugh), sendo iniciado com a menor dose recomendada (vide item 8. Posologia e Modo de Usar).
Raros casos de reações hepáticas severas, incluindo hepatite fulminante (algumas com consequência fatal), necrose do fígado e falência hepática (algumas com consequências fatais ou que requerem transplante de fígado) foram relatados com celecoxibe.
Um paciente com sinais e/ou sintomas de disfunção hepática, ou que tenha apresentado teste de função hepática anormal, deve ser monitorado cuidadosamente em relação à evidência de desenvolvimento de alteração hepática mais grave enquanto estiver em tratamento com DicoxibeTM. Deve-se interromper o uso de DicoxibeTM caso apareçam sinais e sintomas clínicos compatíveis com doença hepática, ou suas manifestações sistêmicas, (por ex., eosinofilia, erupção cutânea, etc.).
Reações anafilactoides: assim como ocorre com AINEs em geral, reações anafilactoides ocorreram em pacientes expostos ao celecoxibe (vide item 4. Contraindicações). Desde o início de sua comercialização, houve raros relatos de reações anafiláticas e angioedema em pacientes recebendo celecoxibe.
Geral: por reduzir a inflamação, DicoxibeTM pode reduzir a utilidade de sinais diagnósticos, como febre, na detecção de infecções.
Inibição do CYP2D6: o celecoxibe demonstrou ser um inibidor moderadamente potente do CYP2D6. Para os medicamentos que são metabolizados por CYP2D6, pode ser necessário uma redução da dose durante o início do tratamento com celecoxibe ou um aumento da dose após