DENBONLI
SANDOZ
denosumabe
Anticorpo monoclonal.
Apresentações.
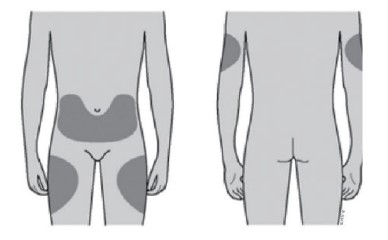
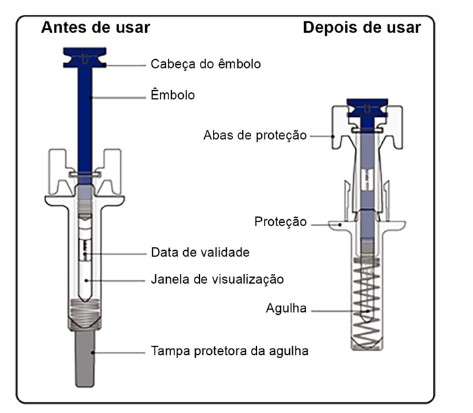
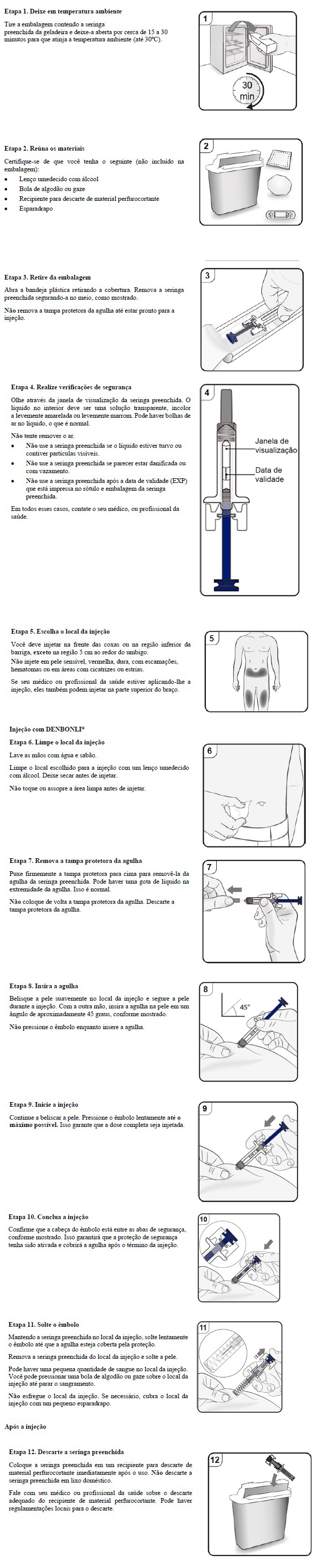
Cartucho com 1 seringa preenchida, com protetor de seringa, com 1 mL de solução injetável contendo 60 mg de denosumabe.
VIA SUBCUTÂNEA
USO ADULTO
Composição.
Cada seringa preenchida contém: denosumabe 60 mg, excipientes q.s.p. 1 mL (ácido acético, sorbitol, polissorbato 20, hidróxido de sódio, ácido clorídrico, nitrogênio e água para injetáveis)
Informações técnicas.
1. INDICAÇÕES
Osteoporose pós-menopáusica
DENBONLI® é indicado para o tratamento de osteoporose em mulheres na fase de pós-menopausa. Nessas mulheres, DENBONLI® aumenta a densidade mineral óssea (DMO) e reduz a incidência de fraturas de quadril, de fraturas vertebrais e não vertebrais.
Perda óssea em pacientes submetidos a ablação hormonal contra câncer
DENBONLI® é indicado para o tratamento de perda óssea em pacientes submetidos a ablação hormonal contra câncer de próstata ou de mama. Em pacientes com câncer de próstata, DENBONLI® reduz a incidência de fraturas vertebrais.
Osteoporose masculina
DENBONLI® é indicado para o tratamento de osteoporose em homens.
Osteoporose induzida por glicocorticoide
DENBONLI® é indicado para o tratamento de osteoporose associada à terapia sistêmica com glicocorticoides recém iniciada ou sustentada, tanto em homens quanto em mulheres sob risco aumentado de fratura.
2. RESULTADOS DE EFICÁCIA
DENBONLI® (denosumabe) é um medicamento biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre DENBONLI® e o medicamento comparador PROLIA (denosumabe).
*PROLIA é marca registrada da Amgen Biotecnologia do Brasil Ltda.
Resultados de eficácia do produto biológico comparador
Tratamento da osteoporose pós-menopáusica
A eficácia e a segurança de PROLIA administrado uma vez a cada 6 meses durante 3 anos foram investigadas em mulheres em pós-menopausa
(7.808 mulheres com 60 a 91 anos de idade, das quais 23,6% tinham principalmente fraturas vertebrais) com T-score basais de densidade mineral óssea na coluna lombar ou quadril total entre -2,5 e -4,0 e probabilidade média absoluta de fratura em 10 anos de 18,60% (decis: 7,9-32,4%) para fraturas osteoporóticas maiores e 7,22% (decis: 1,4-14,9%) para fratura de quadril. Mulheres com outras doenças ou recebendo terapias que podem afetar os ossos foram excluídas deste estudo. As mulheres receberam suplementos diários de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 400 UI).
As mulheres foram randomizadas para receber injeções subcutâneas de placebo (n = 3.906) ou denosumabe 60 mg (n = 3.902) uma vez a cada 6 meses. A variável de eficácia primária foi a incidência de novas fraturas vertebrais. As variáveis de eficácia secundárias incluíram a incidência de fraturas não vertebrais e de quadril, avaliadas após 3 anos.
PROLIA reduziu significativamente o risco de novas fraturas vertebrais, não vertebrais e de quadril, em comparação com placebo. Os 3 desfechos de eficácia em fraturas atingiram o nível de significância estatística (p < 0,05), com base no esquema predefinido de testes sequenciais.
Steven R. Cummings, M.D., Javier San Martin, M.D., Michael R. McClung, M.D., Ethel S. Siris, M.D., Richard Eastell, M.D., Ian R. Reid, M.D., et al. Denosumab for Prevention of Fractures in Postmenopausal Women with Osteoporosis. N Engl J Med, 2009, 361, 756-765.
Efeito sobre fraturas vertebrais
PROLIA reduziu significativamente o risco de novas fraturas vertebrais em 1, 2 e 3 anos (p < 0,0001) (vide tabela 1).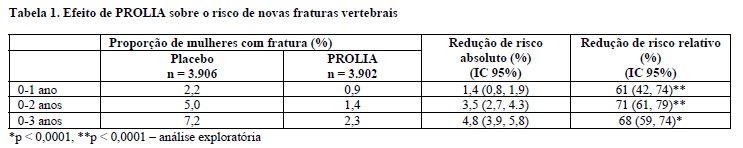
PROLIA também reduziu, em 3 anos, o risco de outras categorias de fratura predefinidas. São elas: novas fraturas vertebrais ou agravamento de fraturas vertebrais (redução do risco relativo de 67%, redução do risco absoluto não ajustada de 4,8%), novas fraturas vertebrais múltiplas (redução do risco relativo de 61%, redução do risco absoluto não ajustada de 1,0%) e fraturas vertebrais clínicas (redução do risco relativo de 69%, redução do risco absoluto não ajustada de 1,8%).
As reduções do risco de novas fraturas vertebrais pelo PROLIA durante 3 anos foram persistentes e significativas, independentemente do risco basal de fraturas osteoporóticas graves em 10 anos, avaliado pelo FRAX® (algoritmo de avaliação do risco de fraturas da OMS), e do fato de as mulheres terem fratura vertebral prevalente ou histórico de fratura não vertebral. A idade na avaliação basal, a DMO, a remodelação óssea e o uso prévio de produto medicinal para osteoporose também não influíram nessas reduções. Em mulheres na fase de pós-menopausa e com mais de 75 anos, o PROLIA reduziu a incidência de novas fraturas vertebrais (64%) e não vertebrais (16%).
Efeito sobre todas as fraturas clínicas
PROLIA reduziu significativamente as fraturas entre todos os tipos/grupos de fraturas (vide Tabela 2).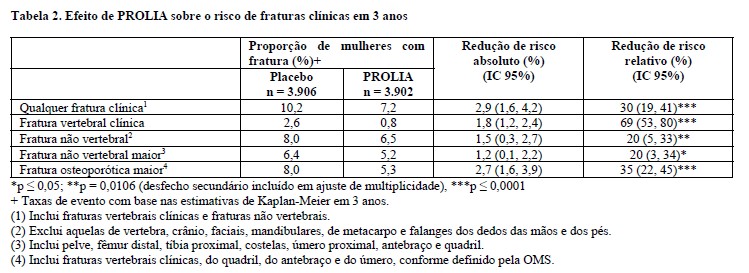
Em mulheres com DMO basal do colo do fêmur ≤-2,5, PROLIA reduziu o risco de fratura não vertebral (35% de redução do risco relativo, 4,1% de redução do risco absoluto, p < 0,001, análise exploratória).
A redução da incidência de novas fraturas vertebrais, fraturas de quadril e fraturas não vertebrais por PROLIA durante 3 anos foi consistente independentemente do risco de fratura basal em 10 anos.
Efeito sobre as fraturas de quadril
PROLIA demonstrou uma redução relativa de 40% (0,5% de redução do risco absoluto) no risco de fratura de quadril durante 3 anos (p < 0,05). A incidência de fratura de quadril foi de 1,2% no grupo de placebo em comparação com 0,7% no grupo de PROLIA em 3 anos. Tais reduções foram constantes e significativas e ocorreram independentemente de ter sido observada à entrada no estudo a probabilidade de ocorrência de fratura de quadril em 10 anos, de acordo com o FRAX®.
Em mulheres com alto risco de fraturas, conforme definido acima por idade basal, DMO e fratura vertebral prevalente, observou-se redução de 48% no risco relativo com PROLIA (redução do risco absoluto não ajustada de 1,1%).
Em análise post-hoc em mulheres na fase de pós-menopausa, com osteoporose e idade acima de 75 anos, PROLIA reduziu a incidência de fraturas de quadril (62%).
Efeito sobre a densidade mineral óssea (DMO)
PROLIA aumentou significativamente a DMO em todos os locais clínicos medidos, em relação ao tratamento com placebo, em 1, 2 e 3 anos. PROLIA aumentou a DMO em 9,2% na coluna lombar, 6,0% no quadril total, 4,8% no colo do fêmur, 7,9% no trocânter do fêmur, 3,5% no terço distal do rádio e 4,1% no corpo inteiro em 3 anos (todos p < 0,0001).
Em estudos clínicos que examinaram os efeitos da descontinuação de PROLIA, a DMO retornou aos níveis pré-tratamento aproximados e permaneceu superior ao placebo no período de 18 meses após a última dose. Estes dados indicam que o tratamento continuado com PROLIA é necessário para manter o efeito do medicamento. A reiniciação de PROLIA resultou em ganhos na DMO semelhantes àqueles observados durante a primeira administração de PROLIA.
Na DMO da coluna lombar, do quadril e do trocânter do fêmur, a alteração foi observada no período de um mês após a dose inicial. PROLIA aumentou a DMO da coluna lombar em relação à avaliação basal em 96% em mulheres pós-menopáusicas após 3 anos. Observaram-se efeitos consistentes sobre a DMO da coluna lombar, independentemente de idade basal, raça, peso/IMC, DMO e nível de remodelação óssea.
Bone HG, Bolognese MA, Yuen CK, Kendler DL, Wang H, Liu Y, et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women. J Clin Endocrinol Metab. 2008; 93(6):2149-57.
Engelke K, Libanati C, Liu Y, Wang H, Austin M, Fuerst T, et al. Quantitative computed tomography (QCT) of the forearm using general purpose spiral whole body CT scanners: accuracy, precision and comparison with dual-energy X-ray absorptiometry (DXA). Bone. 2009; 45(1):110-8.
Lewiecki EM, McClung MR, Cohen SB, et al. Two-year treatment with denosumab in a randomized phase 2 study of postmenopausal women with low bone mineral density. J Bone Miner Res. 2007; 22:1832-1841. McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006; 354:821-831.
Histologia óssea
As avaliações da histologia mostraram ossos com arquitetura e qualidade normais, bem como a redução esperada de remodelação óssea em relação ao tratamento com placebo. Não houve evidências de defeitos de mineralização, osso trançado (não lamelar) ou fibrose de medula.
Steven R. Cummings, M.D., Javier San Martin, M.D., Michael R. McClung, M.D., Ethel S. Siris, M.D., Richard Eastell, M.D., Ian R. Reid, M.D., et al. Denosumab for Prevention of Fractures in Postmenopausal Women with Osteoporosis. N Engl J Med, 2009, 361, 756-765.
Kendler DL, Roux C, Benhamou CL, Brown JP, Lillestol M, Siddhanti S, et al. Effects of Denosumab on Bone Mineral Density and Bone Turnover in Postmenopausal Women Transitioning from Alendronate Therapy. J Bone Miner Res. 2009; 25:72-81.
Estudo de extensão aberto no tratamento de osteoporose pós-menopáusica
Um total de 4.550 mulheres (2.343 PROLIA e 2.207 placebo), que perderam não mais que uma dose do medicamento investigacional no estudo principal descrito acima e que completaram a visita do mês 36 do estudo, concordaram em ser incluídas em um estudo de extensão multinacional, multicêntrico, aberto e de braço único, com 7 anos de duração, para avaliar a segurança e a eficácia em longo prazo de PROLIA. Todas as mulheres no estudo de extensão receberam PROLIA 60 mg a cada 6 meses, bem como cálcio (pelo menos 1 g) e vitamina D (pelo menos 400 UI) diariamente. Um total de 2.626 pacientes (58% das mulheres incluídas no estudo de extensão, ou seja, 34% das mulheres incluídas no estudo principal) concluíram o estudo de extensão.
Nos pacientes tratados com PROLIA por até 10 anos, a DMO aumentou desde a avaliação basal do estudo principal em 21,7% na coluna lombar, 9,2% no quadril total, 9,0% no colo do fêmur, 13,0% no trocânter e 2,8% no terço distal do rádio. A pontuação média da DMO da coluna lombar no final do estudo foi de 1,3 em pacientes tratados por 10 anos.
A incidência de fratura foi avaliada como um desfecho de segurança, mas a eficácia na prevenção de fraturas não pode ser estimada devido ao alto número de descontinuações na fase aberta. A incidência cumulativa de novas fraturas vertebrais e não vertebrais foi de aproximadamente 6,8% e 13,1%, respectivamente, em pacientes que permaneceram em tratamento com denosumabe por 10 anos (n = 1.278). Pacientes que não concluíram o estudo por qualquer motivo apresentaram maiores taxas de fraturas durante o tratamento. Nos anos 4 a 10, as taxas de novas fraturas vertebrais e não vertebrais não aumentaram com o tempo; as taxas anualizadas foram de aproximadamente 1,0% e 1,3%, respectivamente.
Treze casos adjudicados de osteonecrose de mandíbula (ONM) e dois casos adjudicados de fraturas femorais atípicas ocorreram durante o estudo de extensão.
Dados clínicos comparativos com os de alendronato em mulheres pós-menopáusicas com baixa massa óssea
Em 2 estudos randomizados, duplo-cegos e controlados com ativo, um em mulheres não submetidas a tratamento anterior e outro em mulheres previamente tratadas com alendronato, o denosumabe mostrou aumentos significativamente maiores da DMO e reduções dos marcadores de remodelação óssea (por exemplo, CTX sérico), em comparação com alendronato.
Observaram-se aumentos consistentemente maiores da DMO de coluna lombar, quadril, colo do fêmur, trocânter e terço distal do rádio em mulheres tratadas com o denosumabe, em comparação com as que continuaram a receber alendronato (para todos, p < 0,05).
Kendler DL, Roux C, Benhamou CL, Brown JP, Lillestol M, Siddhanti S, et al. Effects of Denosumab on Bone Mineral Density and Bone Turnover in Postmenopausal Women Transitioning from Alendronate Therapy. J Bone Miner Res. 2009; 25:72-81.
Brown JP, Prince RL, Deal C, Recker RR, Kiel DP, de Gregorio LH, et al. Comparison of the effect of denosumab and alendronate on BMD and biochemical markers of bone turnover in postmenopausal women with low bone mass: a randomized, blinded, phase 3 trial. J Bone Miner Res. 2009 Jan; 24(1):153-61.
Eficácia clínica do tratamento da perda óssea associada com ablação hormonal
Tratamento da perda óssea associada com privação androgênica
A eficácia e a segurança de PROLIA uma vez a cada 6 meses por 3 anos foram investigadas em homens com câncer de próstata não metastático, confirmado histologicamente, recebendo ADT (1.468 homens com 48 a 97 anos), que estavam sob risco aumentado de fratura (definido como > 70 anos ou < 70 anos com um T-score de DMO na coluna lombar, quadril total ou colo do fêmur < -1,0 ou história de uma fratura osteoporótica). Os pacientes receberam injeções subcutâneas de denosumabe 60 mg (n = 734) ou placebo (n = 734) uma vez a cada 6 meses, além de suplementos diários de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 400 UI).
PROLIA aumentou significativamente a DMO em todos os locais clínicos medidos, em relação ao tratamento com placebo, após 3 anos: 7,9% na coluna lombar, 5,7% no quadril total, 4,9% no colo do fêmur, 6,9% no trocânter do fêmur, 6,9% no terço distal do rádio e 4,7% no corpo inteiro (todos p < 0,0001). Em uma análise exploratória planejada prospectivamente, aumentos significativos na DMO foram observados na coluna lombar, quadril total, colo do fêmur e trocânter do fêmur 1 mês após a dose inicial.
Os efeitos do tratamento sobre a DMO da coluna lombar foram persistentes independentemente dos dados basais relativos a idade, raça, região geográfica, peso/IMC, DMO, nível de remodelação óssea, duração da privação androgênica e presença de fratura vertebral.
PROLIA demonstrou uma redução significativa no risco relativo de novas fraturas vertebrais: 85% (1,6% de redução do risco absoluto) em 1 ano, 69% (2,2% de redução do risco absoluto) em 2 anos e 62% (2,4% de redução do risco absoluto) em 3 anos (todos p < 0,01). Este medicamento reduziu ainda em 72% a incidência de mais de uma fratura osteoporótica em um mesmo paciente, em qualquer local, em 3 anos, com relação a placebo (taxa de 2,5% com placebo versus 0,7% com denosumabe; p = 0,0063).
Smith MR, Egerdie B, Hernández Toriz N, Feldman R, Tammela TL, Saad F, et al. Denosumab HALT Prostate Cancer Study Group. Denosumab in men receiving androgen deprivation therapy for prostate cancer. N Engl J Med. 2009 Aug 20; 361(8):745-55.
Tratamento da perda óssea em mulheres sob tratamento com inibidores da aromatase para câncer de mama
A eficácia e a segurança de PROLIA no tratamento da perda óssea decorrente da terapia adjuvante com inibidores da aromatase foram avaliadas em um estudo multinacional randomizado, duplo-cego, controlado com placebo, com duração de 2 anos, em 252 mulheres com câncer de mama não metastático e idade de 35 a 84 anos. As pacientes tinham pontuações T-score de DMO de -1,0 a -2,5 na coluna lombar, no quadril ou no colo do fêmur. Foram randomizadas para receber injeções subcutâneas de denosumabe 60 mg (n = 127) ou placebo (n = 125) uma vez a cada 6 meses. Receberam suplementos diários de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 400 UI).
A variável de eficácia primária foi a alteração percentual da DMO da coluna lombar; a eficácia de fratura não foi avaliada. PROLIA aumentou significativamente a DMO em todos os locais clínicos medidos, em relação ao tratamento com placebo, após 2 anos: 7,6% na coluna lombar, 4,7% no quadril total, 3,6% no colo do fêmur, 5,9% no trocânter do fêmur, 6,1% no terço distal do rádio e 4,2% no corpo inteiro (todos p < 0,0001).
Os aumentos da DMO da coluna lombar já foram significativos 1 mês após a dose inicial. Os efeitos do tratamento sobre a DMO da coluna lombar foram persistentes independentemente de idade inicial, duração da terapia com inibidores da aromatase, peso/IMC, quimioterapia prévia, uso prévio de moduladores dos receptores seletivos de estrogênio (SERM) e tempo desde a menopausa.
Ellis GK, Bone HG, Chlebowski R, Paul D, Spadafora S, et al. Randomized trial of denosumab in patients receiving adjuvant aromatase inhibitors for non-metastatic breast cancer. J Clin Oncol. 2008 Oct 20; 26(30):4875-82.
Ellis GK, Bone HG, Chlebowski R, Paul D, Spadafora S, Fan M, Kim D. Effect of denosumab on bone mineral density in women receiving adjuvant aromatase inhibitors for non-metastatic breast cancer: subgroup analyses of a phase 3 study. Breast Cancer Res Treat. 2009 Nov; 118(1):81-7.
Tratamento da osteoporose em homens
A eficácia e a segurança de PROLIA no tratamento de homens com osteoporose foram demonstradas em um estudo multinacional, controlado com placebo, duplo-cego, randomizado, com duração de 1 ano, em homens com baixa massa óssea, que apresentaram pontuação T-score basal de DMO entre -2,0 e -3,5 na coluna lombar ou no colo do fêmur. Homens com pontuação T-score de DMO entre -1,0 e -3,5 na coluna lombar ou no colo do fêmur e com histórico de fratura anterior por fragilidade também foram inscritos. Homens com outras doenças (tais como artrite reumatoide, osteogênese imperfeita e doença de Paget) ou em terapias capazes de afetar os ossos foram excluídos deste estudo.
Os 242 homens inscritos no estudo tinham idades na faixa de 31 a 84 anos e foram randomizados para receber injeções subcutâneas de placebo (n = 121) ou de denosumabe 60 mg (n = 121) uma vez a cada 6 meses. Os pacientes também receberam pelo menos 1.000 mg de cálcio e pelo menos 800 UI de suplemento de vitamina D diariamente.
A variável primária de eficácia foi o percentual de mudança na DMO da coluna lombar em 1 ano. As variáveis secundárias de eficácia incluíram
o percentual de mudança na DMO do quadril total, do trocânter do fêmur, do colo do fêmur e do terço do rádio distal em 1 ano e mudança no telopeptídeo C (CTX) no dia 15. PROLIA aumentou significativamente a DMO em todos os locais medidos, com relação ao tratamento com placebo, em 12 meses: 4,8% na coluna lombar, 2,0% no quadril total, 2,2% no colo do fêmur, 2,3% no trocânter do fêmur e 0,9% no terço distal do rádio (todos p < 0,05). PROLIA aumentou a DMO da coluna lombar a partir da avaliação basal em 94,7% dos homens em 1 ano. Aumentos significativos da DMO na coluna lombar, quadril total, colo femoral e trocânter do fêmur foram observados em 6 meses (p < 0,0001).
Foram observados efeitos consistentes na DMO da coluna lombar independentemente da idade, da raça, do peso/índice de massa corporal (IMC), da DMO e de remodelação óssea.
Histologia e histomorfometria do osso
A histologia óssea foi avaliada em 62 mulheres em pós-menopausa com osteoporose ou com massa óssea baixa, que nunca foram tratadas para osteoporose ou que transitaram de uma terapia prévia com alendronato, seguindo-se 1-3 anos de tratamento com PROLIA. Um total de 59 mulheres participaram do subestudo de biópsia óssea no mês 24 (n = 41) e/ou mês 84 (n = 22) do estudo de extensão em mulheres na pós menopausa com osteoporose. A histologia óssea também foi avaliada em 17 homens com osteoporose e em 6 pacientes com osteoporose associada à terapia com glicocorticoide após 1 ano de tratamento com PROLIA. Os resultados das biópsias ósseas mostraram ossos de arquitetura e qualidade normais, sem evidências de defeitos de mineralização, osso trançado ou fibrose de medula. Achados de histomorfometrial no estudo de extensão em mulheres na pós-menopausa com osteoporose mostraram que os efeitos antirreabsortivos de PROLIA, medido por frequência de ativação e taxas de formação óssea, foram mantidos ao longo do tempo.
Orwoll E, Teglbjærg CS, Langdahl BL, Chapurlat R, Czerwinski E, Kendler DL, Reginster JY, Kivitz A, Lewiecki EM, Miller PD, BologneseMA, McClung MR, Bone HG, Ljunggren Ö, Abrahamsen B, Gruntmanis U, Yang YC, Wagman RB, Siddhanti S, Grauer A, Hall JW, Boonen
S. A randomized, placebo-controlled study of the effects of denosumab for the treatment of men with low bone mineral density. J Clin Endocrinol Metab. 2012 Sep; 97(9):3161-9. doi: 10.1210/jc.2012-1569. Epub 2012 Jun 21.
Tratamento de osteoporose induzida por glicocorticoide
A eficácia e a segurança de PROLIA uma vez a cada 6 meses durante 2 anos foram investigadas em 795 pacientes (69,9% mulheres e 30,1% homens) com 20 a 94 anos de idade tratados com ≥ 7,5 mg de prednisona por via oral por dia (ou equivalente) e com planejamento para continuar
o tratamento por um total de pelo menos 6 meses.
Duas subpopulações foram estudadas: glicocorticoide continuado (≥ 7,5 mg de prednisona por dia ou o seu equivalente por ≥ 3 meses antes da inclusão no estudo; n = 505) e glicocorticoide inicial (≥ 7,5 mg de prednisona por dia ou o seu equivalente por < 3 meses antes da inclusão no estudo; n = 290). Dentro de cada subpopulação, a randomização foi estratificada por gênero e os pacientes foram randomizados (1:1) para receber PROLIA 60 mg por via subcutânea uma vez a cada 6 meses ou risedronato 5 mg por via oral uma vez por dia (controle ativo) por 2 anos. Os pacientes receberam suplemento diário de cálcio (pelo menos 1.000 mg) e vitamina D (pelo menos 800 UI).
Os pacientes incluídos com < 50 anos de idade tinham de ter uma história de fratura osteoporótica. Os pacientes incluídos com ≥ 50 anos de idade, que estavam na subpopulação sob glicocorticoide continuado, tinha de ter um T-score basal de DMO ≤ -2,0 na coluna lombar, quadril total ou colo do fêmur; ou um T-score de DMO ≤ -1,0 na coluna lombar, quadril total ou colo do fêmur e história de fratura osteoporótica. A maioria dos pacientes incluídos no estudo apresentavam alto risco de fratura.
O desfecho primário de eficácia foi a alteração percentual da linha de base na DMO da coluna lombar por DXA (absorciometria com raio X de dupla energia) aos 12 meses (não inferioridade). Os desfechos secundários de eficácia incluíram a alteração percentual da linha de base na DMO total da coluna lombar e do quadril aos 12 e 24 meses.
Efeito sobre a densidade mineral óssea
Na subpopulação sob glicocorticoide continuado, PROLIA demonstrou um aumento significativamente maior na DMO da coluna lombar em comparação com risedronato em 1 ano (PROLIA 4,4%, risedronato 2,3%) com uma diferença no tratamento de 2,2% (p < 0,001) e em 2 anos (PROLIA 6,4%, risedronato 3,2%) com uma diferença no tratamento de 3.2% (p < 0,001). Na subpopulação sob glicocorticoide inicial, PROLIA demonstrou um aumento significativamente maior na DMO da coluna lombar em comparação com risedronato em 1 ano (PROLIA 3,8%, risedronato 0,8%) com uma diferença no tratamento de 2,9% (p < 0,001) e em 2 anos (PROLIA 6,2%, risedronato 1,7%) com uma diferença de tratamento de 4,5% (p < 0,001).
Foram observados efeitos consistentes na DMO da coluna lombar em 1 ano independentemente do gênero; raça; estado de pós-menopausa; e idade basal e T-score de DMO da coluna lombar e dose de glicocorticoide dentro de cada subpopulação.
Além disso, PROLIA demonstrou um aumento significativamente maior na DMO a partir da avaliação basal em comparação com risedronato em 1 e 2 anos no quadril total, colo do fêmur e trocânter do fêmur em ambas as subpopulações, terço distal do rádio na subpopulação sob glicocorticoide continuado, e em 2 anos no terço distal do rádio na subpopulação de iniciação de glicocorticoide.
Comparabilidade farmacocinética de DENBONLI (GP2411) ao comparador PROLIA (denosumabe)
Farmacocinética
Um perfil farmacocinético comparável entre GP2411 (denosumabe biossimilar) e o medicamento comparador (PROLIA) foi demonstrado em um estudo randomizado, duplo-cego em mulheres pós-menopáusicas com osteoporose (Estudo CGP24112301). Neste estudo, foi demonstrada semelhança com relação aos parâmetros farmacocinéticos AUCinf e Cmáx após a primeira dose entre o GP2411 e o medicamento comparador.
Os resultados dos estudos de farmacocinética confirmaram a biossimilaridade entre GP2411 e o medicamento comparador.
Os resumos estatísticos dos parâmetros PK primários do estudo CGP24112301 são apresentados abaixo.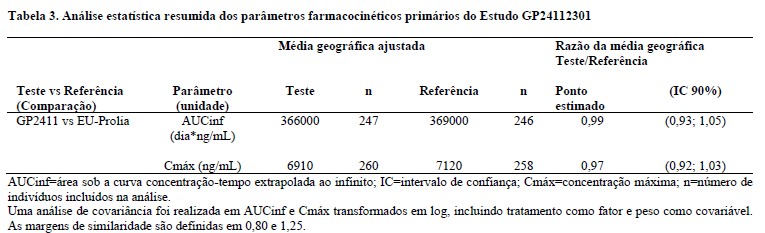
Comparabilidade farmacodinâmica de GP2411 com o medicamento comparador (PROLIA)
A equivalência farmacodinâmica de GP2411 e do medicamento comparador, em termos de inibição do marcador de renovação óssea sérica CTX (como alteração percentual em relação ao basal), foi demonstrada através da área sob a curva de efeito (AUEC) após a primeira dose em um estudo duplo-cego, randomizado, em mulheres pós-menopáusicas com osteoporose. Um sumário dos parâmetros farmacodinâmicos é apresentado abaixo.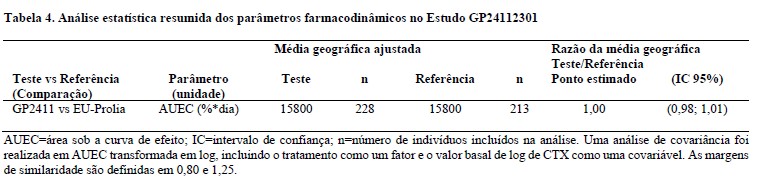
Comparabilidade de GP2411 com o medicamento comparador em termos de eficácia
Resultados do estudo CGP24112301 em mulheres na pós-menopausa com osteoporose
O estudo CGP24112301 demonstrou eficácia clínica semelhante ao GP2411 e EU-Prolia em mulheres com osteoporose pós-menopáusica (PMO) em termos de variação percentual do basal (%CfB) da densidade mineral óssea da coluna lombar (LS-BMD) na Semana 52. Melhoria em LSBMD foi semelhante em ambos os grupos de tratamento. Os resultados são apresentados na Tabela 5, abaixo.
Uma análise de medidas repetidas de modelo misto foi realizada para %CfB em LS-BMD, incluindo as seguintes variáveis: tratamento (GP2411, EU-Prolia), uso prévio de bisfosfonatos (sim, não), tipo de máquina de absorciometria de raios X de dupla energia (Lunar, Hologic), visita (Semana 26, Semana 52), interação entre visita e tratamento e valor basal LS-BMD como uma covariável contínua. Presume-se que os dados ausentes estejam ausentes aleatoriamente. As margens de equivalência são definidas em [-1,45, 1,45].
Comparabilidade de GP2411 com o medicamento comparador em termos de segurança
Os tipos, frequência e gravidade das reações adversas foram comparáveis entre GP2411 e o medicamento comparador.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Mecanismo de ação
O denosumabe é um anticorpo monoclonal humano (IgG2) que tem como alvo o RANKL, ao qual se liga com grande afinidade e especificidade, impedindo que o ligante ative seu único receptor, o RANK, na superfície dos osteoclastos e seus precursores, independentemente da superfície óssea. A prevenção da interação RANKL/RANK inibe a formação, a função e a sobrevivência de osteoclastos. O denosumabe, portanto, reduz a reabsorção óssea e aumenta a massa e a resistência dos ossos corticais e trabeculares.
Efeitos farmacodinâmicos
Em estudos clínicos, o tratamento com 60 mg de denosumabe resultou em rápida redução (de aproximadamente 70%) do marcador de reabsorção óssea tipo 1 no soro, o telopeptídeo C (CTX), no período de 6 horas após a administração subcutânea, atingindo-se cerca de 85% de redução em 3 dias. As reduções do CTX se mantiveram ao longo do intervalo de administração de 6 meses. Ao final de cada intervalo, eram parcialmente atenuadas, com máxima ≥ 87% e mínima ≥ 45% (faixa de 45% a 80%), o que reflete a reversibilidade dos efeitos do denosumabe sobre a remodelação óssea assim que os níveis séricos diminuem. Esses efeitos foram mantidos com a continuação do tratamento. De maneira condizente com o acoplamento fisiológico de formação e reabsorção óssea na remodelação esquelética, observaram se reduções dos marcadores de formação óssea (por exemplo, fosfatase alcalina específica dos ossos [BSAP] e propeptídeo N-terminal sérico do colágeno de tipo 1 [P1NP]), iniciadas 1 mês após a primeira dose do denosumabe.
De modo geral, os marcadores de remodelação óssea (marcadores de reabsorção e formação óssea) atingiram os níveis pré-tratamento no período de 9 meses após a última dose subcutânea de 60 mg. A cada retomada do tratamento, o grau de inibição de CTX obtido com o denosumabe foi similar ao observado no uso inicial desse medicamento.
Imunogenicidade
Anticorpos anti-denosumabe podem se desenvolver durante o tratamento com denosumabe. Nenhuma correlação aparente do desenvolvimento de anticorpos com farmacocinética, resposta clínica ou evento adverso foi observada.
Em um estudo clínico em mulheres pós-menopáusicas com baixa massa óssea (N = 504) que haviam recebido alendronato pelo período mediano de 3 anos, as pacientes que passaram a ser tratadas com denosumabe apresentaram reduções adicionais do CTX sérico, em comparação às que continuaram recebendo alendronato. Nesse estudo, as alterações dos níveis séricos de cálcio foram similares entre os 2 grupos.
Farmacocinética
Após a administração subcutânea, o denosumabe exibiu farmacocinética não linear com as doses em uma grande variedade delas, além de aumentos de exposição proporcionais à dose a partir de 60 mg (ou 1 mg/kg).
Absorção
Após dose subcutânea do denosumabe, a biodisponibilidade foi de 61% e as concentrações séricas máximas (Cmáx), de 6 mcg/mL (faixa de 1 a 17 mcg/mL), ocorreram em 10 dias (faixa de 2 a 28 dias). Em seguida à Cmáx os níveis séricos diminuíram, sendo a meia-vida de 26 dias (faixa de 6 a 52 dias) durante o período de 3 meses (faixa de 1,5 a 4,5 meses). Cinquenta e três por cento dos pacientes não apresentaram quantidades mensuráveis do denosumabe 6 meses pós-dose.
Distribuição
Nem acúmulo nem alteração da farmacocinética do denosumabe foram observados com o passar do tempo após doses múltiplas de 60 mg por via subcutânea 1 vez a cada 6 meses.
Metabolismo
O denosumabe é composto unicamente de aminoácidos e carboidratos, como imunoglobulina nativa. Com base em dados não clínicos, espera-se que o metabolismo do denosumabe siga as vias de eliminação da imunoglobulina, resultando em degradação para pequenos peptídeos e aminoácidos individualizados.
Eliminação
O denosumabe é composto unicamente de aminoácidos e carboidratos, como imunoglobulina nativa, e não se prevê que sejam eliminados por meio de mecanismos metabólicos hepáticos (como enzimas do citocromo P450, ou CYP). Tomando-se por base os dados não clínicos, prevê-se que a eliminação do denosumabe seguirá as vias de eliminação da imunoglobulina, resultando em degradação para pequenos peptídeos e aminoácidos individualizados.
Interações medicamentosas
Em um estudo com 17 mulheres na fase pós-menopausa com osteoporose, foi administrado midazolam (oral, 2 mg) duas semanas após uma dose única de denosumabe (subcutâneo, 60 mg), que corresponde ao tempo máximo de efeitos farmacodinâmicos de denosumabe. O denosumabe não afetou a farmacocinética de midazolam que é metabolizado pelo citocromo P450 3A4 (CYP3A4). Isto indica que denosumabe não altera a farmacocinética de medicamentos metabolizados pelo CYP3A4.
Linearidade/não linearidade
Em estudos de variação de dose, o denosumabe exibiu farmacocinética não linear e dependente da dose, com menor depuração em doses ou concentrações mais altas, mas aumentos aproximadamente proporcionais à dose em exposições para doses de 60 mg e maiores.
Populações especiais de pacientes
Idosos (a partir de 65 anos de idade)
A idade não foi considerada um fator significativo na farmacocinética do denosumabe em uma análise farmacocinética da população de pacientes de 28 a 87 anos.
Crianças e adolescentes (até 18 anos de idade)
DENBONLI® não deve ser usado em populações pediátricas. Em um estudo de fase 3 de pacientes pediátricos com osteogênese imperfeita (N = 153), as concentrações séricas máximas de denosumabe foram observadas no dia 10 em todas as faixas etárias. Para cada dosagem de 3 meses e 6 meses, as concentrações séricas médias de denosumabe foram observadas como sendo maiores em crianças de 11 a 17 anos de idade, enquanto crianças de 2 a 6 anos de idade apresentaram as menores concentrações médias.
Raça
A farmacocinética do denosumabe não foi afetada pelo fator raça em mulheres na fase de pós-menopausa nem em pacientes com câncer de mama sob tratamento de ablação hormonal.
Insuficiência renal
Em um estudo com 55 pacientes que apresentavam graus variados de função renal, incluindo-se os que se submetiam a diálise, o grau de insuficiência renal não teve nenhum efeito sobre a farmacocinética nem sobre a farmacodinâmica do denosumabe. Portanto, não é necessário ajuste de dose para pacientes com insuficiência renal.
Insuficiência hepática
Não foram conduzidos estudos clínicos para avaliar o efeito da insuficiência hepática sobre a farmacocinética do denosumabe. Em geral, os anticorpos monoclonais não são eliminados por mecanismos metabólicos hepáticos. Não se espera que a farmacocinética do denosumabe seja afetada pela insuficiência hepática.
4. CONTRAINDICAÇÕES
O uso deste medicamento é contraindicado para pacientes que apresentam hipocalcemia.
O uso deste medicamento é contraindicado para pacientes que apresentam hipersensibilidade clinicamente significativa à denosumabe ou qualquer componente de DENBONLI® .
5. ADVERTÊNCIAS E PRECAUÇÕES
Suplementação de cálcio e vitamina D
Uma ingestão adequada de cálcio e vitamina D é importante para todos os pacientes.
Precauções de utilização
Hipocalcemia
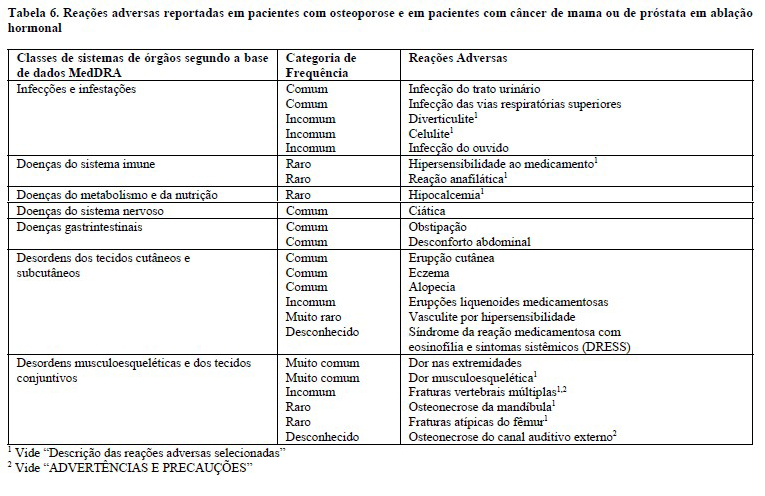
É importante identificar os pacientes em risco de desenvolver hipocalcemia. A hipocalcemia deve ser corrigida através de uma administração adequada de cálcio e de vitamina D antes de se iniciar o tratamento. Recomenda-se monitoramento clínico dos valores de cálcio antes de cada dose e nos pacientes com predisposição para a hipocalcemia (com insuficiência renal grave [depuração de creatinina < 30 mL/min], em diálise ou em tratamento com outros medicamentos que reduzem o cálcio) nas duas semanas, após a dose inicial. Se algum paciente apresentar quaisquer sintomas suspeitos de hipocalcemia durante o tratamento (vide "REAÇÕES ADVERSAS") os valores de cálcio devem ser medidos. Os pacientes devem ser encorajados a reportar sintomas indicadores de hipocalcemia.
Na fase de pós-comercialização, têm sido reportados casos de hipocalcemia sintomática grave (resultando em hospitalização, eventos de risco à vida e casos fatais), particularmente em pacientes com insuficiência renal grave, em diálise ou em tratamento com outros medicamentos que reduzem o cálcio. Embora a maioria dos casos tenha ocorrido nas primeiras semanas após o início do tratamento, também ocorreu mais tarde.
O tratamento concomitante com glicocorticoide é um fator de risco adicional para hipocalcemia.
Insuficiência renal
Pacientes com insuficiência renal grave (depuração da creatinina < 30 mL/min) ou em diálise apresentam um risco maior de desenvolver hipocalcemia. Os riscos de desenvolver hipocalcemia e concomitante elevação do hormônio da paratireoide aumentam com o aumento do nível de comprometimento renal. Casos graves e fatais foram relatados. A administração adequada de cálcio, vitamina D e a monitorização regular dos níveis de cálcio é especialmente importante nestes pacientes, ver acima.
Infecções na pele
Os pacientes recebendo DENBONLI® podem desenvolver infeções na pele (predominantemente celulite) levando à hospitalização (vide "REAÇÕES ADVERSAS"). Os pacientes devem ser aconselhados a procurar cuidados médicos imediatos se desenvolverem sinais ou sintomas de celulite.
Osteonecrose da mandíbula (ONM)
A ONM tem sido notificada raramente em pacientes recebendo denosumabe para a osteoporose (vide "REAÇÕES ADVERSAS").
O início do tratamento/novo ciclo de tratamento deve ser adiado em pacientes com feridas abertas e não cicatrizadas nos tecidos moles na boca.É recomendada uma avaliação dentária com odontologia preventiva apropriada e uma avaliação individual do benefício-risco antes do tratamento com denosumabe em pacientes com fatores de risco concomitantes.
Os seguintes fatores de risco devem ser considerados na avaliação de um paciente com risco de desenvolver ONM:
• potência do medicamento que inibe a reabsorção óssea (maior risco para compostos mais potentes), via de administração (maior risco para administração parenteral) e doses cumulativas de terapêutica de reabsorção óssea.
• câncer, comorbilidades (exemplo: anemia, coagulopatias, infecção), tabagismo.
• terapias concomitantes: corticosteroides, quimioterapia, inibidores da angiogênese, radioterapia da cabeça e pescoço.
• higiene oral deficiente, doença periodontal, próteses dentárias mal ajustadas, doença dentária preexistente, procedimentos orais invasivos, por exemplo: extrações dentárias.
Todos os pacientes devem ser encorajados a manter boas práticas de higiene oral, efetuar exames gerais dentários de rotina e reportar imediatamente qualquer sintoma oral como mobilidade dentária, dor ou edema ou não cicatrização de feridas ou supuração durante o tratamento com denosumabe. Durante o tratamento, procedimentos orais invasivos devem ser realizados apenas após consideração cuidadosa e ser evitados próximo da administração com DENBONLI® .
O plano de gestão de pacientes que desenvolvem ONM deve ser estabelecido em colaboração próxima entre o médico e um dentista ou um cirurgião-dentista com experiência em ONM. Interrupções temporárias do tratamento devem ser consideradas até a situação estar resolvida e os fatores de risco estarem mitigados sempre que possível.
Osteonecrose do canal auditivo externo
A osteonecrose do canal auditivo externo tem sido reportada associada à utilização de denosumabe. Potenciais fatores de risco para a osteonecrose do canal auditivo externo incluem a utilização de esteroides e quimioterapia e/ou fatores de risco locais como infeção ou trauma. A possibilidade de osteonecrose do canal auditivo externo deve ser considerada em pacientes em tratamento com denosumabe, que apresentem sintomas do ouvido, incluindo infeções crônicas do ouvido.
Fraturas atípicas do fêmur
Fraturas atípicas do fêmur têm sido reportadas em pacientes recebendo denosumabe (vide "REAÇÕES ADVERSAS"). As fraturas atípicas do fêmur podem ocorrer após um pequeno traumatismo ou sem traumatismo em regiões femorais subtrocantéricas e diafisárias. Estes acontecimentos são caracterizados por alterações radiográficas específicas. Fraturas atípicas do fêmur também têm sido notificadas em pacientes com certas comorbilidades (exemplos: deficiência em vitamina D, artrite reumatoide, hipofosfatasia) e com a utilização de certos agentes farmacêuticos (exemplos: bifosfonatos, glucocorticoides, inibidores da bomba de prótons). Estes acontecimentos também ocorreram sem terapia antirreabsortiva. Fraturas semelhantes reportadas em associação com bifosfonatos são frequentemente bilaterais; por isso o fêmur contralateral deve ser examinado nos pacientes tratados com denosumabe que têm uma fratura da diáfise do fêmur estável. A descontinuação do tratamento com DENBONLI® em pacientes com suspeita de terem uma fratura atípica do fêmur deve ser considerada após avaliação do paciente baseada numa avaliação individual do risco benefício. Durante o tratamen