DANYELZA
ADIUM
naxitamabe
Anticorpo monoclonal.
Apresentações.
Solução para diluição para infusão
DANYELZA® (naxitamabe) 4 mg/mL é fornecido em embalagens contendo 1 frasco-ampola de 10 mL.
USO INTRAVENOSO
USO ADULTO E PEDIÁTRICO DE 1 ANO DE IDADE OU MAIS
Composição.
Cada mL de DANYELZA® (naxitamabe) contém: naxitamabe 4 mg. Excipientes: ácido cítrico, poloxâmer, cloreto de sódio, citrato de sódio di-hidratado e água para injetáveis q.s para 1mL
ADVERTÊNCIA: REAÇÕES GRAVES RELACIONADAS À INFUSÃO E NEUROTOXICIDADE Reações graves relacionadas à infusão: DANYELZA® pode causar reações graves à infusão, incluindo parada cardíaca, anafilaxia, hipotensão, broncoespasmo e estridor. Reações à infusão de qualquer Grau ocorreram em 94-100% dos pacientes. Reações graves à infusão ocorreram em 32-68% e reações graves à infusão ocorreram em 4 - 18% dos pacientes em estudos clínicos DANYELZA® [ver Advertências e Precauções (5)]. Pré-medicar antes de cada infusão de DANYELZA® conforme recomendado e monitorar os pacientes por pelo menos 2 horas após o término de cada infusão. Reduzir a taxa, interromper a infusão ou permanentemente descontinuar DANYELZA® com base na gravidade [ver Posologia e Administração (8), Contraindicações (4) e Advertências e Precauções (5)]. Neurotoxicidade: DANYELZA® pode causar neurotoxicidade grave, incluindo dor neuropática grave, mielite e síndrome de leucoencefalopatia posterior reversível (SLPR). Dor de qualquer Grau ocorreu em 94-100% dos pacientes dos estudos clínicos de DANYELZA® [ver Advertências e Precauções (5)]. Pré-medicar para tratar a dor neuropática conforme recomendado. Descontinuar permanentemente DANYELZA® com base na reação adversa e gravidade [ver Posologia e Administração (8) e Advertências e Precauções (5)].
Informações técnicas.
1. INDICAÇÕES
DANYELZA® (naxitamabe) é indicado, em combinação com sargramostim, para o tratamento de pacientes pediátricos de 1 ano de idade ou mais e pacientes adultos que apresentam neuroblastoma de alto risco recidivado ou refratário nos ossos ou na medula óssea que apresentaram resposta parcial, resposta mínima ou doença estável à terapia anterior.
O medicamento sargramostim não foi avaliado pela ANVISA.
2. RESULTADOS DE EFICÁCIA
A eficácia de DANYELZA® (naxitamabe) em combinação com sargramostim foi avaliada em dois estudos clínicos abertos de braço único em pacientes com neuroblastoma de alto risco com doença refratária ou recidivada nos ossos ou na medula óssea, Estudo 201 e Estudo 12-230.
Estudo 201
A eficácia de DANYELZA® (naxitamabe) em combinação com sargramostim foi avaliada no Estudo 201, um estudo multicêntrico aberto, de braço único, em uma subpopulação de pacientes com neuroblastoma de alto risco recidivado ou refratário nos ossos ou na medula óssea e que apresentaram resposta parcial, resposta mínima ou doença estável à terapia anterior. Pacientes que apresentaram doença progressiva foram excluídos. Todos os pacientes receberam pelo menos uma terapia sistêmica para tratar a doença fora dos ossos ou da medula óssea antes da inclusão no estudo. Os pacientes receberam DANYELZA® (naxitamabe) 9 mg/kg/ciclo administrado como três infusões intravenosas de 3 mg/kg nos Dias 1, 3 e 5 de cada ciclo. Os pacientes receberam sargramostim via subcutânea na dose de 250 mg/m2/dia nos Dias -4 a 0 e na dose de 500 mg/m2/dia nos Dias 1 a 5. Radiação pré- planejada para o local primário foi permitida.
A principal medida de desfecho de eficácia foi a taxa de resposta global (TRG) de acordo com os Critérios Internacionais de Resposta do Neuroblastoma (International Neuroblastoma Response Criteria - INRC) revisados, conforme determinado por patologia independente e revisão de exames de imagem e confirmado por pelo menos uma avaliação subsequente. Uma medida de desfecho de eficácia adicional foi a duração da resposta.
Dos 52 pacientes incluídos na análise de eficácia data limite de 31 de dezembro de 2021, 50% apresentavam doença refratária e 50% apresentavam doença recidivada; a idade mediana foi 5.7 anos (intervalo de 2 a 18 anos), 60% eram do sexo masculino; 40% eram brancos, 56% eram asiáticos e 4% eram negros.
A amplificação do gene MYCN estava presente em 13% dos pacientes e 88% dos pacientes eram estágio 4 segundo o Sistema Internacional de Estadiamento de Neuroblastoma (International Neuroblastoma Staging System - INSS) no momento do diagnóstico. Quanto ao local doença, 56% eram apenas nos ossos, 4% apenas na medula óssea e 40% em ambos. As terapias anteriores incluíam cirurgia (88%), quimioterapia (100%), radiação (40%), transplante autólogo de células-tronco (TACT) 27%) e tratamento com anticorpo anti-GD2 (25%).
Os resultados de eficácia do Estudo 201 estão descritos na Tabela 1.
Em uma análise exploratória no subgrupo de pacientes previamente tratados com um anticorpo anti-GD2 (n=13), a ORR foi de 31% [95% CI: 9% a 61%] três pacientes demonstraram uma resposta completa confirmada e um paciente demonstrou uma resposta parcial confirmada.
Estudo 12-230
A eficácia de DANYELZA® (naxitamabe) em combinação com sargramostim foi avaliada no Estudo 12-230, um estudo de centro único, aberto, de braço único, em uma subpopulação de pacientes que apresentavam neuroblastoma de alto risco recidivado ou refratário nos ossos ou na medula óssea e que apresentaram resposta parcial, resposta mínima ou doença estável à terapia anterior. Pacientes que apresentaram doença progressiva foram excluídos. Todos os pacientes receberam pelo menos uma terapia sistêmica para tratar a doença fora dos ossos ou da medula óssea antes da inclusão. Os pacientes deveriam ter recebido pelo menos uma dose de DANYELZA® (naxitamabe) na dose de 3 mg/kg ou maior por infusão e apresentar doença avaliável no período basal de acordo com a revisão independente, conforme os INRC revisados.
Os pacientes receberam DANYELZA® (naxitamabe) 9 mg/kg/ciclo administrado como três infusões intravenosas de 3 mg/kg (nos Dias 1, 3 e 5) na primeira semana de cada ciclo. Os pacientes receberam sargramostim via subcutânea na dose de 250 mg/m2/dia nos Dias -4 a 0 e na dose de 500 mg/m2/dia nos Dias 1 a 5. Radiação para lesões ósseas não alvo e lesões de tecido mole foi permitida a critério do investigador; a avaliação de resposta excluiu os locais que receberam radiação. As principais medidas de desfecho de eficácia foram a TRG e a duração da resposta, conforme determinado por patologia independente e revisão de exames de imagem, de acordo com os INRC revisados e confirmado por pelo menos uma avaliação subsequente.
Dos 38 pacientes incluídos na análise de eficácia, 55% apresentavam neuroblastoma recidivado e 45% apresentavam doença refratária; 50% eram do sexo masculino, a idade mediana foi 5 anos (intervalo de 2 a 23 anos), 74% eram brancos, 8% asiáticos, 5% eram negros, 5% nativos americanos/índios americanos/nativos do Alasca, 3% de outras raças e 5% não estavam disponíveis. A amplificação do gene MYCN estava presente em 16% dos pacientes e a maioria dos pacientes era estágio 4 do INSS (95%). Cinquenta por cento (50%) dos pacientes apresentavam envolvimento da doença apenas nos ossos, 11% apenas na medula óssea e 39% em ambos. As terapias anteriores incluíram cirurgia (100%), quimioterapia (100%), radiação (47%), TACT (42%) e tratamento com anticorpo anti-GD2 (58%).
Os resultados de eficácia são fornecidos na Tabela 2.
Em uma análise exploratória no subgrupo de pacientes tratados anteriormente com um anticorpo anti-GD2 (n=22), a TRG foi 18% (IC de 95% 5%, 40%), sem nenhum dos pacientes apresentando uma resposta documentada de 6 meses ou mais.
Referências bibliográficas
1) Estudo clínico Estudo 201
2) Estudo clínico Estudo 12-230
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
Naxitamabe se liga ao glicolipídeo GD2. GD2 é um disialogangliosídeo que é superexpresso em células de neuroblastoma e outras células de origem neuroectodérmica, incluindo o sistema nervoso central e os nervos periféricos. In vitro, naxitamabe foi capaz de se ligar a GD2 da superfície celular e induzir citotoxicidade dependente de complemento (CDC) e citotoxicidade mediada por células dependente de anticorpo (CCDA).
Farmacodinâmica
A relação exposição-resposta e a evolução temporal da resposta farmacodinâmica para a segurança e a eficácia de naxitamabe não foram totalmente caracterizadas.
Farmacocinética
A média geométrica (CV%) da concentração plasmática máxima (Cmax) de naxitamabe foi 57,4 mg/mL (49%) após a infusão intravenosa de 3 mg/kg de DANYELZA durante 30 minutos.
Eliminação
A meia-vida terminal média de naxitamabe foi 8,2 dias.
Metabolismo
Espera-se que naxitamabe seja metabolizado em pequenos peptídeos por vias catabólicas.
Populações específicas
As análises de farmacocinética populacional sugerem que a idade (intervalo: 1 a 34 anos), o sexo e a raça não possuem efeito clinicamente importante sobre a depuração (clearance - CL) de naxitamabe. Não se espera que a exposição sistêmica (AUC) a naxitamabe na dose de 150 mg/dia (450 mg por ciclo) para pacientes que apresentam peso corporal acima de 50 kg seja clinicamente diferente das exposições a naxitamabe na dose de 3 mg/kg/dia (9 mg/kg por ciclo) para pacientes que apresentam peso corporal entre 30-50 kg.
4. CONTRAINDICAÇÕES
DANYELZA® (naxitamabe) é contraindicado em pacientes com uma história de reação de hipersensibilidade grave a naxitamabe. As reações incluíam anafilaxia [consulte 5. Advertências e Precauções].
5. ADVERTÊNCIAS E PRECAUÇÕES
Reações graves relacionadas à infusão
DANYELZA® (naxitamabe) pode causar reações à infusão graves que exigem intervenção urgente, incluindo ressuscitação com fluidos, administração de broncodilatadores e corticosteroides, hospitalização em unidade de terapia intensiva, redução da taxa de infusão ou interrupção da infusão de DANYELZA® (naxitamabe). As reações relacionadas à infusão incluem hipotensão, broncoespasmo, hipóxia e estridor (vide item 9. Reações Adversas). Reações relacionadas à infusão graves ocorreram em 4% dos pacientes no Estudo 201 e em 18% dos pacientes no Estudo 12-230. Reações relacionadas à infusão de qualquer grau ocorreram em 100% dos pacientes no Estudo 201 e em 94% dos pacientes no Estudo 12-230. Hipotensão de qualquer grau ocorreu em 100% dos pacientes no Estudo 201 e em 89% dos pacientes no Estudo 12-230.
No Estudo 201, 68% dos pacientes apresentaram reações à infusão Grau 3 ou 4 e, no Estudo 12-230, 32% dos pacientes apresentaram reações à infusão Grau 3 ou 4. Anafilaxia ocorreu em 12% dos pacientes e 2 pacientes (8%) descontinuaram permanentemente DANYELZA® (naxitamabe) devido à anafilaxia no Estudo 201. Um paciente no Estudo 12-230 (1,4%) apresentou uma parada cardíaca Grau 4, 1,5 hora após a conclusão da infusão de DANYELZA® (naxitamabe).
No Estudo 201, as reações à infusão geralmente ocorreram no período de 24 horas após a conclusão de uma infusão de DANYELZA® (naxitamabe), mais frequentemente no período de 30 minutos após o início da infusão. As reações à infusão foram mais frequentes durante a primeira infusão de DANYELZA® (naxitamabe) em cada ciclo. Oitenta por cento dos pacientes necessitaram de redução da taxa de infusão e 80% dos pacientes tiveram uma infusão interrompida por pelo menos uma reação relacionada à infusão.
Pré-medicar com um anti-histamínico, acetaminofeno, um antagonista de H2 e corticosteroide, conforme recomendado (vide item 8. Posologia e Modo de usar). Monitorar os pacientes atentamente quanto a sinais e a sintomas de reações à infusão durante e por pelo menos 2 horas após a conclusão de cada infusão de DANYELZA® (naxitamabe) em um cenário em que medicação e equipamento de ressuscitação cardiopulmonar estejam disponíveis.
Reduzir a taxa, interromper a infusão ou descontinuar permanentemente DANYELZA® (naxitamabe) com base na gravidade e instituir o tratamento médico apropriado conforme necessário (vide item 8. Posologia e modo de usar e 4. Contraindicações).
Neurotoxicidade
DANYELZA® (naxitamabe) pode causar neurotoxicidade grave, incluindo dor neuropática grave, mielite transversa e síndrome da leucoencefalopatia posterior reversível.
Dor
Dor, incluindo dor abdominal, dor óssea, cervicalgia e dor nas extremidades, ocorreu em 100% dos pacientes no Estudo 201 e em 94% dos pacientes no Estudo 12-230. Dor Grau 3 ocorreu em 72% dos pacientes no Estudo 201. Um paciente no Estudo 201 (4%) necessitou de interrupção de uma infusão devido à dor. A dor tipicamente começou durante a infusão de DANYELZA® (naxitamabe) e durou uma mediana de menos de um dia no Estudo 201 (intervalo de menos de um dia a até 62 dias) (vide item 9. Reações Adversas). Pré-medicar com medicamentos que tratam dor neuropática (por exemplo, gabapentina) e opioides orais. Administrar opioides intravenosos conforme necessário para dor disruptiva (vide item 8. Posologia e modo de usar). Descontinuar permanentemente DANYELZA® (naxitamabe) com base na gravidade (vide item 8. Posologia e modo de usar).
Mielite transversa
Mielite transversa ocorreu com DANYELZA® (naxitamabe). Descontinuar permanentemente DANYELZA® (naxitamabe) em pacientes que desenvolvem mielite transversa (vide item 8. Posologia e modo de usar).
Síndrome de leucoencefalopatia posterior reversível
Síndrome da leucoencefalopatia posterior reversível (SLPR) (também conhecida como síndrome da encefalopatia posterior reversível ou SEPR) ocorreu em 2 (2,8%) pacientes no Estudo 12-230. Os eventos ocorreram 2 e 7 dias após a conclusão do primeiro ciclo de DANYELZA® (naxitamabe). Monitorar a pressão arterial durante e após a infusão de DANYELZA® (naxitamabe) e avaliar os sintomas neurológicos (vide item 5. Advertências e Precauções). Descontinuar permanentemente DANYELZA® (naxitamabe) em caso de SLPR sintomática (vide itens 8. Posologia e modo de usar e 9. Reações Adversas].
Neuropatia periférica
Neuropatia periférica, incluindo neuropatia sensorial periférica, neuropatia motora periférica, parestesia e neuralgia, ocorreu em 32% dos pacientes no Estudo 201 e em 25% dos pacientes no Estudo 12-230. A maioria dos sinais e sintomas de neuropatia começou no dia da infusão e a neuropatia durou uma mediana de 5,5 dias (intervalo de 0 a 22 dias) no Estudo 201 e 0 dias (intervalo de 0 a 22 dias) no Estudo 12-230 [vide item 9. Reações Adversas].
Descontinuar permanentemente DANYELZA® (naxitamabe) com base na gravidade (vide item 8. Posologia e modo de usar).
Distúrbios neurológicos dos olhos
Distúrbios neurológicos dos olhos, incluindo pupilas desiguais, visão turva, distúrbio de acomodação, midríase, comprometimento visual e fotofobia, ocorreram em 24% dos pacientes no Estudo 201 e em 19% dos pacientes no Estudo 12-230.
Os distúrbios neurológicos dos olhos duraram uma mediana de 17 dias (intervalo de 0 a 84 dias) no Estudo 201, com dois pacientes (8%) apresentando um evento que não havia sido resolvido no momento do corte de dados e uma mediana de 1 dia (intervalo menos de um dia a 21 dias) no Estudo 12-230. Descontinuar permanentemente DANYELZA® (naxitamabe) com base na gravidade (vide itens 8. Posologia e modo de usar e 9. Reações Adversas).
Retenção urinária prolongada
Retenção urinária ocorreu em 1 (4%) paciente no Estudo 201 e em 3 pacientes (4%) no Estudo 12-230. Todos os eventos em ambos os estudos ocorreram no dia da infusão de DANYELZA® (naxitamabe) e duraram entre 0 e 24 dias. Descontinuar permanentemente DANYELZA® (naxitamabe) em pacientes que apresentam retenção urinária que não é resolvida após a descontinuação dos opioides (vide itens 8. Posologia e modo de usar e 9. Reações Adversas).
Hipertensão
Hipertensão ocorreu em 44% dos pacientes no Estudo 201 e em 28% dos pacientes no Estudo 12-230 que receberam DANYELZA® (naxitamabe). Hipertensão Grau 3 ou 4 ocorreu em 4% dos pacientes no Estudo 201 e em 7% dos pacientes no Estudo 12-230. Quatro pacientes (6%) no Estudo 12-230 descontinuaram permanentemente DANYELZA® (naxitamabe) devido à hipertensão. Em ambos os estudos, a maioria dos eventos ocorreu no dia da infusão de DANYELZA® (naxitamabe) e ocorreu até 9 dias após uma infusão de DANYELZA® (naxitamabe).
Não iniciar DANYELZA® (naxitamabe) em pacientes que apresentam hipertensão não controlada. Monitorar a pressão arterial durante a infusão e pelo menos uma vez ao dia nos Dias 1 a 8 de cada ciclo de DANYELZA® (naxitamabe) e avaliar quanto a complicações da hipertensão, incluindo SLPR (vide item 5. Advertências e Precauções). Interromper a infusão de DANYELZA® (naxitamabe) e retomar em uma taxa reduzida, ou descontinuar permanentemente DANYELZA® (naxitamabe) com base na gravidade (vide itens 8. Posologia e modo de usar e 9. Reações Adversas).
Toxicidade embriofetal
Com base em seu mecanismo de ação, DANYELZA® (naxitamabe) pode causar lesão fetal quando administrado a uma mulher grávida.
Aconselhar as mulheres com potencial reprodutivo, incluindo mulheres grávidas, sobre o potencial risco para o feto. Aconselhar as mulheres com potencial reprodutivo a utilizar contracepção efetiva durante o tratamento com DANYELZA® (naxitamabe) e por dois meses após a dose final. (vide item Uso em populações especiais).
Uso em populações especiais Populações Especiais
Verifique as seções "8. Posologia e modo de usar" e "Características farmacológicas".
Gravidez e lactação
Gravidez
Resumo de risco
Com base em seu mecanismo de ação, DANYELZA® (naxitamabe) pode causar lesão fetal quando administrado a mulheres grávidas (vide item 2. Resultados de Eficácia]. Não há dados disponíveis sobre o uso de DANYELZA® (naxitamabe) em mulheres grávidas e não foram realizados estudos de reprodução animal com DANYELZA® (naxitamabe). Os anticorpos monoclonais IgG1 são transportados através da placenta de uma forma linear à medida que a gravidez progride, com a maior quantidade transferida durante o terceiro trimestre. Aconselhar as mulheres grávidas sobre o potencial risco para o feto.
Na população geral dos EUA, o risco de base estimado de defeitos congênitos importantes e aborto em gestações clinicamente reconhecidas é 2% a 4% e 15% a 20%, respectivamente.
Categoria de risco na gravidez: C.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Lactação
Resumo de risco
Não há dados sobre a presença de naxitamabe no leite humano ou seus efeitos na criança em aleitamento materno ou sobre a produção de leite, no entanto, IgG humana está presente no leite humano. Devido ao potencial de reações adversas graves em uma criança em aleitamento materno por DANYELZA® (naxitamabe), aconselhar as mulheres a não amamentar durante o tratamento e por 2 meses após a dose final de DANYELZA® (naxitamabe).
Mulheres e Homens com Potencial Reprodutivo
DANYELZA® (naxitamabe) pode causar dano fetal quando administrado a mulheres grávidas (vide item "Uso em Populações Especiais").
Exame de gravidez
Verifique o estado de gravidez em mulheres com potencial reprodutivo antes de iniciar DANYELZA® (naxitamabe).
Contracepção
Mulheres
Aconselhar as mulheres com potencial reprodutivo a utilizar contracepção eficaz durante tratamento e por 2 meses após a dose final de DANYELZA® (naxitamabe).
Uso Pediátrico
A segurança e a eficácia de DANYELZA® (naxitamabe), em combinação com sargramostim, para o tratamento de neuroblastoma de alto risco recidivado ou refratário nos ossos ou na medula óssea, em pacientes que apresentaram resposta parcial, resposta mínima ou doença estável após a terapia anterior, foram estabelecidas em pacientes pediátricos com 1 ano de idade ou mais.
A segurança e a efetividade não foram estabelecidas em pacientes pediátricos com menos de 1 ano de idade.
Efeitos sobre a capacidade de dirigir e de operar máquinas
Não foram realizados estudos com DANYELZA® (naxitamabe) e os efeitos sobre a capacidade de dirigir e operar máquinas.
Uso geriátrico
Neuroblastoma é, em grande parte, uma doença de pacientes pediátricos e adultos jovens. Os estudos clínicos de DANYELZA® (naxitamabe) em combinação com sargramostim não incluíram pacientes com 65 anos de idade ou mais.
TOXICOLOGIA NÃO CLÍNICA
Carcinogênese, Mutagênese, Comprometimento da Fertilidade
Não foram realizados estudos em animais para avaliar o potencial carcinogênico ou mutagênico de naxitamabe. Não foram realizados estudos dedicados avaliando os efeitos de naxitamabe sobre a fertilidade em animais.
Toxicologia Animal e/ou Farmacologia
Estudos não clínicos sugerem que a dor neuropática induzida por naxitamabe é mediada pela ligação do anticorpo ao antígeno GD2 localizado na superfície das fibras nervosas periféricas e da mielina e subsequente indução de atividade citotóxica imunomediada.
Em um modelo de ratos nus, ocorreu hiperplasia e erosão da mucosa glandular do estômago leves-moderadas, ocasionalmente acompanhadas de inflamação difusa. Foi observada recuperação completa de todos os achados histopatológicos nos estômagos de ratos machos; no entanto, foi observada apenas uma recuperação parcial nos estômagos de ratas fêmeas durante o período de quatro semanas sem droga.
6. INTERAÇÕES MEDICAMENTOSAS
Até o momento, não há dados disponíveis sobre as interações medicamentosas com DANYELZA® (naxitamabe).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Armazenar o frasco-ampola de DANYELZA® (naxitamabe) refrigerado entre 2°C a 8°C no cartucho para proteger da luz até ao momento do uso.
Prazo de validade: 24 meses.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Características físicas e organolépticas:
DANYELZA® (naxitamabe) injeção é uma solução para infusão intravenosa estéril, livre de conservantes, transparente a levemente opalescente e incolor a levemente amarela. Cada frasco-ampola de dose única contém 40 mg de naxitamabe em 10 mL de solução.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance de crianças.
8. POSOLOGIA E MODO DE USAR
Dose recomendada
A dose recomendada de DANYELZA® (naxitamabe) é 3 mg/kg/dia (até 150 mg/dia) nos Dias 1, 3 e 5 de cada ciclo de tratamento, administrado como uma infusão intravenosa após diluição, em combinação com sargramostim via subcutânea, conforme mostrado na Tabela 3. Consulte as informações de prescrição do sargramostim para informações sobre a dosagem recomendada.
Os ciclos de tratamento são repetidos a cada 4 semanas até a resposta completa ou resposta parcial, seguidos por 5 ciclos adicionais a cada 4 semanas. Os ciclos subsequentes podem ser repetidos a cada 8 semanas. Descontinuar DANYELZA® (naxitamabe) e sargramostim por progressão da doença ou toxicidade inaceitável.
Administrar medicações pré-infusão e tratamento de suporte, conforme apropriado, durante a infusão.
O esquema de administração recomendado para cada ciclo de tratamento é descrito abaixo e na Tabela 3:
• Dias -4 a 0: administrar sargramostim 250 mg/m2/dia por injeção subcutânea, iniciando 5 dias antes da infusão de DANYELZA® (naxitamabe).
• Dias 1 a 5: administrar sargramostim 500 mg/m2/dia por injeção subcutânea. Administrar pelo menos 1 hora antes da administração de DANYELZA® (naxitamabe) nos Dias 1, 3 e 5.
• Dias, 1, 3 e 5: administrar DANYELZA® (naxitamabe) 3 mg/kg/dia (até 150 mg/dia) por infusão intravenosa.
Dose perdida
Se uma dose de DANYELZA® (naxitamabe) for esquecida, administrar a dose esquecida na semana seguinte até o Dia 10. Administrar sargramostim 500 mg/m2/dia no primeiro dia de infusão de DANYELZA® (naxitamabe), no dia anterior e no dia da segunda e da terceira infusão, respectivamente (isto é, um total de 5 dias com 500 mg/m2/dia).
Pré-medicações e medicações de suporte
Manejo da dor antes e durante a infusão (vide item 5. Advertências e Precauções):
• Cinco dias antes da primeira infusão de DANYELZA® (naxitamabe) em cada ciclo, iniciar um ciclo de 12 dias (Dia -4 ao Dia 7) de medicação profilática para dor neuropática, como gabapentina.
• Administrar opioides orais 45-60 minutos antes do início de cada infusão de DANYELZA® (naxitamabe) e opioides intravenosos adicionais conforme necessário para dor disruptiva durante a infusão.
• Considere o uso de cetamina para dor que não é adequadamente controlada por opioides.
Pré-medicação: reduzir o risco de reações relacionadas à infusão e náuseas/vômitos (vide itens 5. Advertências e Precauções e 9. Reações Adversas).
• Administrar corticosteroides intravenosos (por exemplo, metilprednisolona 2 mg/kg com dose máxima de 80 mg ou dose de corticosteroide equivalente) 30 minutos a 2 horas antes da primeira infusão de DANYELZA® (naxitamabe). Administrar pré- medicação com corticosteroide para as infusões subsequentes se tiver ocorrido uma reação grave à infusão com a infusão anterior ou durante o ciclo anterior.
• Administrar um anti-histamínico, um antagonista de H2, acetaminofeno e um antiemético 30 minutos antes de cada infusão.
Modificações de dose de acordo com as reações adversas
As modificações de dose recomendadas para DANYELZA® (naxitamabe) para reações adversas são apresentadas na Tabela 4.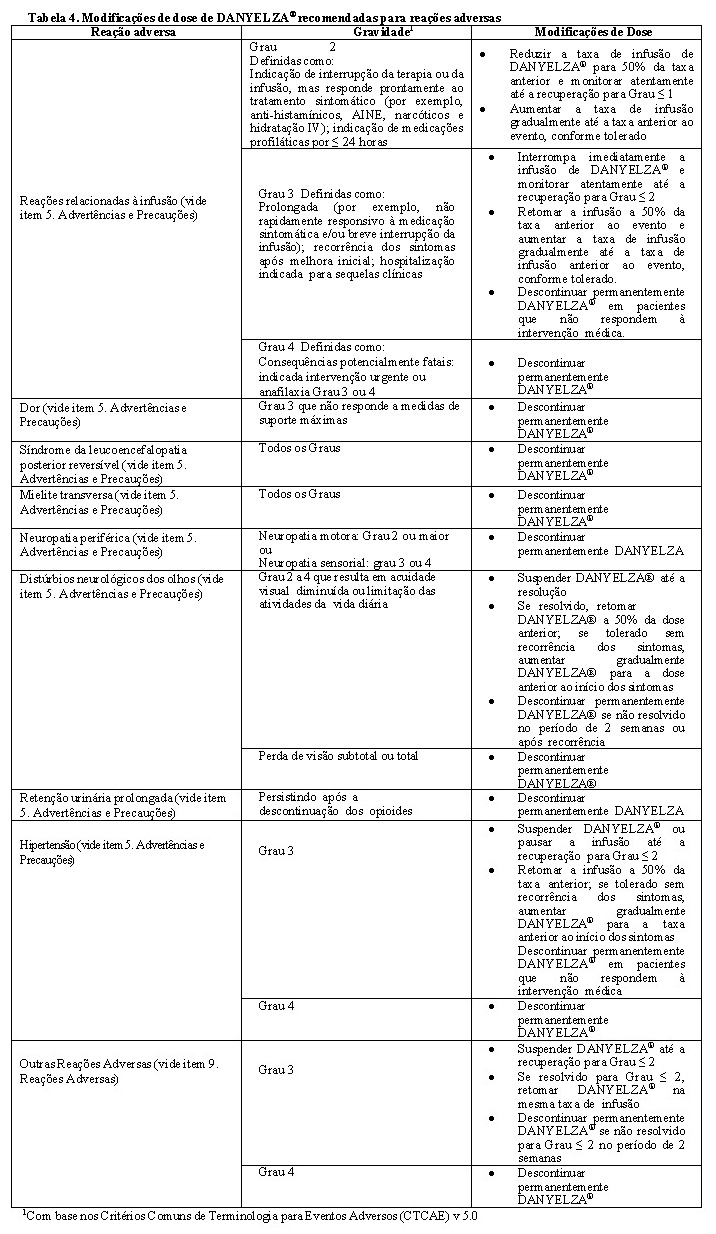
Instruções para preparação
• Use técnica asséptica apropriada.
• Inspecionar visualmente o frasco-ampola quanto a matéria particulada e descoloração/mancha antes da administração. Descartar o frasco-ampola se a solução estiver manchada, turva ou contiver matéria particulada.
• Adicione quantidades apropriadas de Albumina 5% (Humana) e Cloreto de Sódio 0,9% solução para injeção a uma bolsa de infusão intravenosa vazia e estéril grande o suficiente para conter o volume necessário para a dose relevante, conforme indicado na Tabela 5. Permitir 5-10 minutos de mistura passiva.
• Retirar a dose necessária de DANYELZA® (naxitamabe) e injetar na bolsa de infusão contendo Albumina 5% (Humana) e Cloreto de Sódio 0,9% solução para injeção. Descartar qualquer porção não utilizada de DANYELZA® (naxitamabe) deixada no frasco-ampola.
As instruções de preparação de DANYELZA® (naxitamabe) estão descritas na Tabela 5.
Se não utilizada imediatamente, armazenar a solução para infusão diluída de DANYELZA® (naxitamabe) em temperatura ambiente (15°C a 25°C) por até 8 horas ou refrigerar (2°C a 8°C) por até 24 horas.
Administração
• Administrar DANYELZA® (naxitamabe) como uma infusão intravenosa diluída conforme recomendado. Não administrar DANYELZA® (naxitamabe) como uma injeção intravenosa ou bolus (vide item 8. Posologia e Modo de Usar).
• Para a primeira infusão (Ciclo 1, Dia 1), administrar DANYELZA® (naxitamabe) via intravenosa durante 60 minutos. Para as infusões subsequentes, administrar DANYELZA® (naxitamabe) via intravenosa durante 30 a 60 minutos, conforme tolerado. (vide item 8. Posologia e Modo de Usar).
• Observar os pacientes por um mínimo de 2 horas após cada infusão.
9. REAÇÕES ADVERSAS
As seguintes reações adversas clinicamente significativas também estão descritas em outras partes da bula:
• Reações graves relacionadas à infusão (vide item 5. Advertências e Precauções)
• Neurotoxicidade (vide item 5. Advertências e Precauções)
• Hipertensão (vide item 5. Advertências e Precauções)
Experiência em Estudos Clínicos
Uma vez que os estudos clínicos são realizados em condições amplamente variadas, as taxas de reações adversas observadas em estudos clínicos de um medicamento não podem ser comparadas diretamente às taxas em estudos clínicos de outro medicamento e podem não refletir as taxas observadas na prática.
A segurança de DANYELZA® (naxitamabe) em combinação com sargramostim foi avaliada em pacientes com neuroblastoma de alto risco refratário ou recidivado nos ossos ou na medula óssea que apresentaram resposta parcial, resposta mínima ou doença estável após a terapia inicial ou subsequente e em pacientes que estavam na segunda remissão completa, a partir de dois estudos abertos de braço único, Estudo 201 (n=25) e Estudo 12-230 (n=72). Os pacientes receberam DANYELZA® (naxitamabe) 9 mg/kg/ciclo administrado como três infusões intravenosas de 3 mg/kg (Dias 1, 3 e 5) na primeira semana de cada ciclo. Os pacientes também receberam sargramostim 250 mg/m2/dia via subcutânea nos Dias -4 a 0 e sargramostim 500 mg/m2/dia via subcutânea nos Dias 1 a 5 (vide item 2. Resultados de eficácia).
As reações adversas mais comuns nos Estudos 201 e 12-230 (≥ 25% em qualquer estudo) foram reação relacionada à infusão, dor, taquicardia, vômitos, tosse, náuseas, diarreia, diminuição do apetite, hipertensão, fadiga, eritema multiforme, neuropatia periférica, urticária, febre, cefaleia, reação no local da injeção, edema, ansiedade, edema localizado e irritabilidade. As anormalidades laboratoriais Grau 3 ou 4 mais comuns (≥ 5% em qualquer estudo) foram redução dos linfócitos, redução dos neutrófilos, redução da hemoglobina, redução do número de plaquetas, diminuição de potássio, aumento de alanina aminotransferase, diminuição de glicose, diminuição de cálcio, diminuição de albumina, diminuição de sódio e diminuição de fosfato.
Estudo 201
No Estudo 201, entre os 25 pacientes que receberam DANYELZA® (naxitamabe) em combinação com sargramostim, 12% foram expostos por 6 meses ou mais e nenhum foi exposto por mais de um ano.
Reações adversas graves ocorreram em 32% dos pacientes que receberam DANYELZA® (naxitamabe) em combinação com sargramostim.
As reações adversas graves em mais de um paciente incluíram reação anafilática (12%) e dor (8%). A descontinuação permanente de DANYELZA® (naxitamabe) devido a uma reação adversa ocorreu em 12% dos pacientes. As reações adversas que resultaram na descontinuação permanente de DANYELZA® (naxitamabe) incluíram reação anafilática (8%) e depressão respiratória (4%).
Interrupções da administração de DANYELZA® (naxitamabe) devido a uma reação adversa ocorreram em 84% dos pacientes. As reações adversas que exigiram interrupção da administração em > 10% dos pacientes incluíram hipotensão e broncoespasmo.
A Tabela 6 resume as reações adversas no Estudo 201.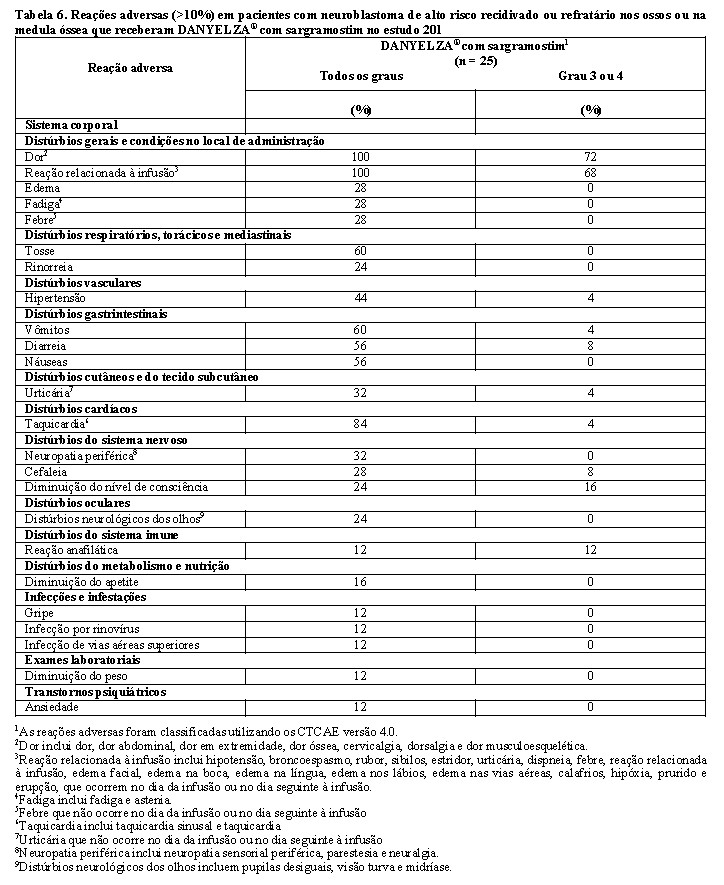
As reações adversas clinicamente relevantes que ocorreram em ≤10% dos pacientes que receberam DANYELZA® (naxitamabe) com sargramostim incluíram edema periférico (8%).
A Tabela 7 resume as anormalidades laboratoriais no Estudo 201.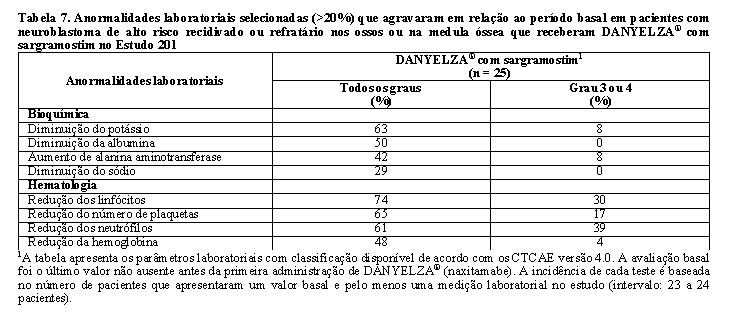
Estudo 12-230
No Estudo 12-230, entre os 72 pacientes que receberam DANYELZA® (naxitamabe) em combinação com sargramostim, 32% foram expostos por 6 meses ou mais e 8% foram expostos por mais de um ano.
Reações adversas graves ocorreram em 40% dos pacientes que receberam DANYELZA® (naxitamabe) em combinação com sargramostim.
As reações adversas graves em > 5% dos pacientes incluíram hipertensão (14%), hipotensão (11%) e febre (8%).
A descontinuação permanente de DANYELZA® (naxitamabe) devido a uma reação adversa ocorreu em 8% dos pacientes. Quatro (6%) pacientes descontinuaram permanentemente DANYELZA® (naxitamabe) devido à hipertensão e um (1,4%) paciente descontinuou devido à SLPR.
A Tabela 8 resume as reações adversas no Estudo 12-230.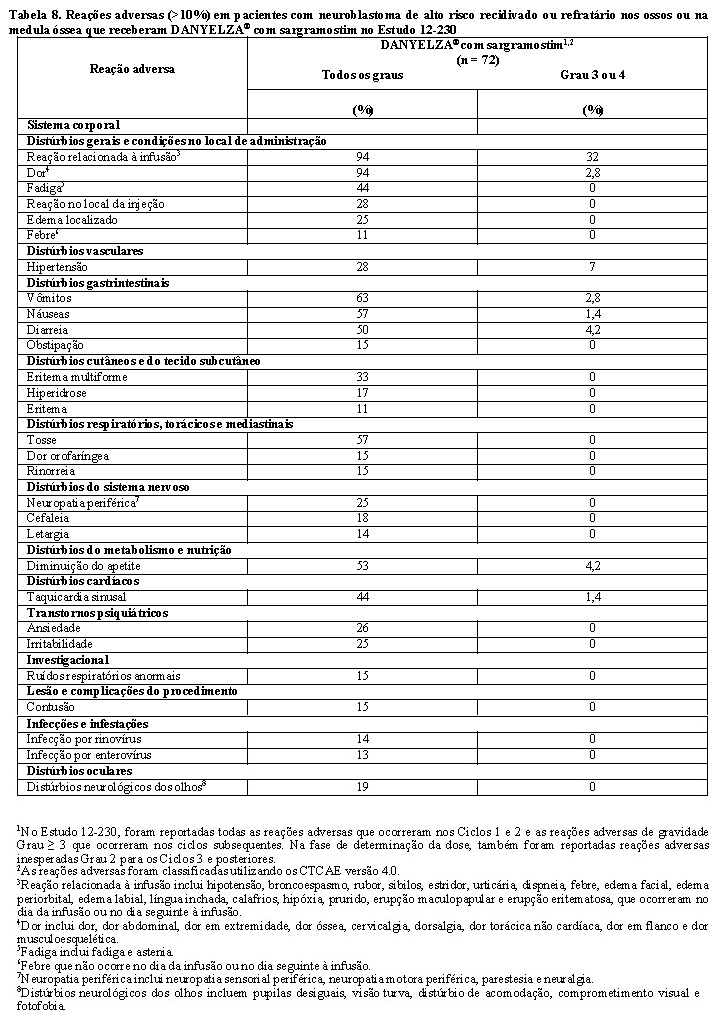
As reações adversas clinicamente relevantes em ≤10% dos pacientes que receberam DANYELZA® (naxitamabe) com sargramostim incluíram apneia (4,2%), hipopneia (2,8%), edema generalizado (2,8%), edema periférico (8,3%) e infecção relacionada ao dispositivo (4,2%).
A Tabela 9 resume as anormalidades laboratoriais no Estudo 12-230.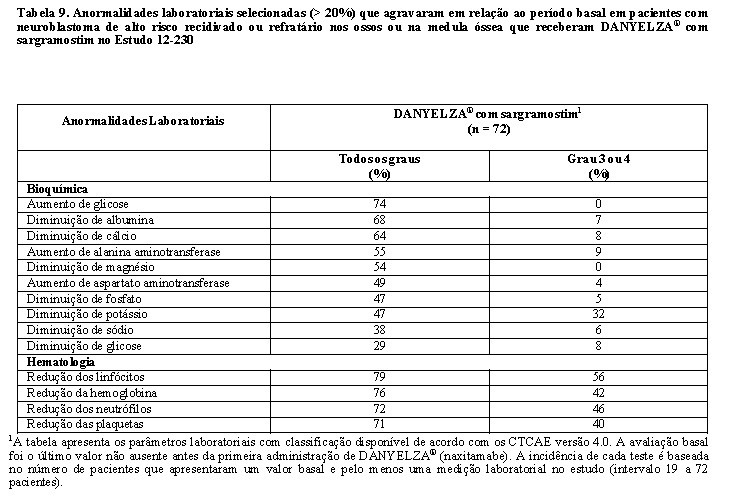
Imunogenicidade
Assim como com todas as proteínas terapêuticas, há um potencial para imunogenicidade. A detecção da formação de anticorpo antidroga (ADA) é altamente dependente da sensibilidade e da especificidade do ensaio. Além disso, a incidência observada de positividade de ADA (incluindo anticorpo neutralizante) em um ensaio pode ser influenciada por vários fatores, incluindo a metodologia do ensaio, o manuseio da amostra, o momento da coleta da amostra, as medicações concomitantes e a doença subjacente. Por essas razões, a comparação da incidência de ADA nos estudos descritos abaixo com a incidência de ADA em outros estudos ou com outros produtos de naxitamabe pode ser enganosa.
No Estudo 201, 2 dos 24 (8%) pacientes testaram positivo para ADA após o tratamento com DANYELZA® (naxitamabe).
No Estudo 12-230, 27 dos 117 pacientes (23%) testaram positivo para ADA após tratamento com DANYELZA por um ensaio que não era totalmente validado; portanto, a incidência de ADA pode não ser confiável.
Experiência Pós-comercialização/Relatos Espontâneos
As seguintes reações adversas foram identificadas a partir dos relatórios de acesso expandido com o uso de DANYELZA® (naxitamabe). Uma vez que estas reações são reportadas voluntariamente a partir de uma população de tamanho incerto, nem sempre é possível calcular de modo confiável sua frequência ou estabelecer uma relação causal com a exposição à medicação.
Neurológicos: Mielite transversa.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Até o momento, não há dados disponíveis sobre a superdosagem com DANYELZA® (naxitamabe).
Em caso de intoxicação, ligue 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
Registro n°: 1.2214.0124
VENDA SOB PRESCRIÇÃO MÉDICA.
USO RESTRITO A ESTABELECIMENTO DE SAÚDE.
Esta bula foi aprovada em 18/01/2024