CYRAMZA
ELI LILLY
ramucirumabe
Anticorpo monoclonal.
Apresentações.
CYRAMZA é apresentado na forma de solução para diluição injetável para uso intravenoso, em frasco-ampola de vidro transparente tipo I, contendo 100 mg de ramucirumabe em 10 mL (10 mg/mL) e 500 mg de ramucirumabe em 50 mL (10 mg/mL).
USO INTRAVENOSO
USO ADULTO
Composição.
Cada mL de solução contém: 10 mg de ramucirumabe.
Excipientes: histidina, cloridrato de histidina monoidratado, cloreto de sódio, glicina, polissorbato 80 e água para injetáveis.
Informações técnicas.
INDICAÇÕES
Câncer Gástrico
CYRAMZA em combinação com paclitaxel
CYRAMZA, em combinação com paclitaxel, é indicado para o tratamento de pacientes adultos com adenocarcinoma gástrico ou da junção gastroesofágica (JGE) avançado, que tenham apresentado progressão da doença após quimioterapia com platina ou fluoropirimidina.
CYRAMZA como agente isolado
CYRAMZA, como agente isolado, é indicado para o tratamento de pacientes adultos com adenocarcinoma gástrico ou da junção gastroesofágica (JGE) avançado, que tenham apresentado progressão da doença após quimioterapia com platina ou fluoropirimidina, nos quais o tratamento com paclitaxel não é apropriado.
Câncer de Pulmão de Não Pequenas Células (NSCLC)
CYRAMZA em combinação com docetaxel
CYRAMZA, em combinação com docetaxel, é indicado para o tratamento de pacientes adultos com câncer de pulmão de não pequenas células metastático ou localmente avançado que tenham apresentado progressão da doença, e que já tenham apresentado falha com quimioterapia prévia baseada em platina.
CYRAMZA em combinação com erlotinibe
CYRAMZA, em combinação com erlotinibe, é indicado para o tratamento de primeira linha de pacientes adultos com câncer de pulmão de não pequenas células metastático, cujos tumores apresentam mutações ativadoras do receptor do fator de crescimento epidérmico (EGFR) do tipo deleções do éxon 19 ou mutações de substituição do éxon 21 (L858R).
Câncer Colorretal (CRC)
CYRAMZA, em combinação com FOLFIRI (irinotecano, ácido folínico e 5-fluoruracila), é indicado para o tratamento de pacientes adultos com câncer colorretal metastático que tenham apresentado progressão da doença após terapia prévia com bevacizumabe, oxaliplatina e fluoropirimidina.
Carcinoma Hepatocelular (HCC)
CYRAMZA, como agente isolado, é indicado no tratamento de pacientes com carcinoma hepatocelular que tenham alfa-fetoproteína (AFP) ≥ 400 ng/mL, após terapia prévia com sorafenibe.
RESULTADOS DE EFICÁCIA
Câncer Gástrico
Ramucirumabe em combinação com paclitaxel
O Estudo RAINBOW foi um estudo multinacional, randomizado, duplo-cego de CYRAMZA mais paclitaxel
versus
placebo mais paclitaxel, que randomizou (1:1) 665 pacientes com câncer gástrico metastático ou localmente avançado incluindo adenocarcinoma da junção gastroesofágica que receberam quimioterapia contendo fluoropirimidina e platina anteriormente. Os pacientes deveriam apresentar progressão da doença durante ou dentro de 4 meses após a última dose da terapia de primeira linha. Os pacientes também deveriam apresentar performance status (PS) do Eastern Cooperative Oncology Group (ECOG) de 0 ou 1. A randomização foi estratificada pela região geográfica, tempo de progressão desde o início da terapia de primeira linha ( < 6 meses versus ≥ 6 meses) e mensurabilidade da doença.
Os pacientes foram randomizados para receber uma infusão intravenosa de CYRAMZA 8 mg/Kg (N=330) ou placebo (N=335) a cada 2 semanas (nos dias 1 e 15) de um ciclo de 28 dias. Pacientes em ambos os braços receberam paclitaxel 80 mg/m2 por infusão intravenosa nos dias 1, 8 e 15 de um ciclo de 28 dias. Antes da administração de cada dose de paclitaxel, os pacientes deveriam ter a função hepática e hematopoiética adequada. A dose de paclitaxel foi permanentemente reduzida em incrementos de 10 mg/m2 para um máximo de duas reduções de dose para toxicidade hematológica de Grau 4 ou toxicidade não hematológica relacionada ao paclitaxel de Grau 3. A principal medida de resultado de eficácia foi a sobrevida global e a medida de resultado de eficácia de apoio foi a sobrevida livre de progressão e taxa de resposta objetiva.
As características basais e demográficas foram similares entre os braços de tratamento. A faixa etária mediana foi de 61 anos de idade; 71% dos pacientes eram do sexo masculino; 61% eram brancos, 35% asiáticos; o PS ECOG foi 0 para 39% dos pacientes, 1 para 61% dos pacientes; 78% dos pacientes tiveram doença mensurável; 79% dos pacientes tiveram câncer gástrico e 21% tiveram adenocarcinoma de JGE. Dois terços dos pacientes apresentaram progressão da doença durante a terapia de primeira linha (67%) e 25% dos pacientes receberam terapia em combinação com platina/fluoropirimidina com antraciclina.
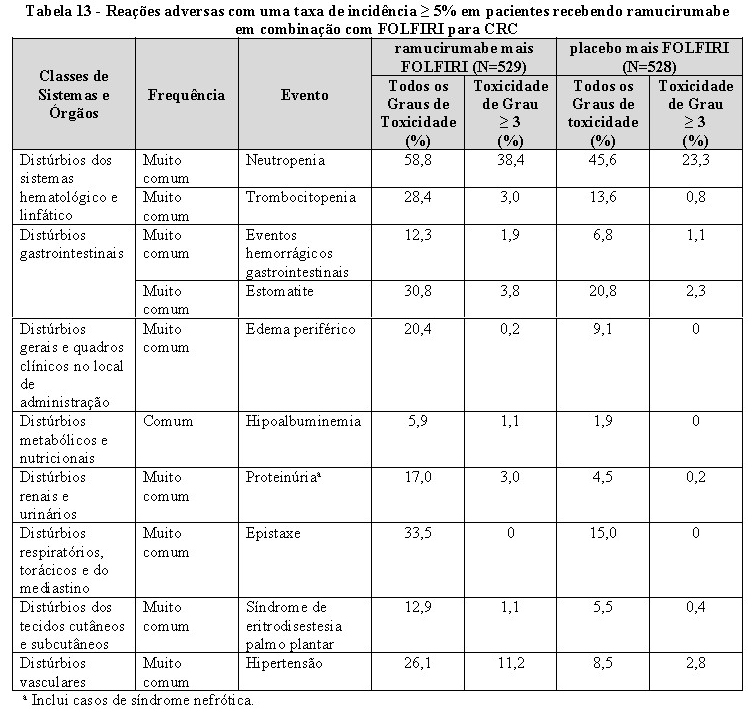
A sobrevida global, sobrevida livre de progressão e taxa de resposta objetiva foram melhoradas de forma estatisticamente significante em pacientes randomizados para receber CYRAMZA mais paclitaxel comparado a pacientes randomizados para receber placebo mais paclitaxel. Os resultados de eficácia são apresentados na Tabela 1.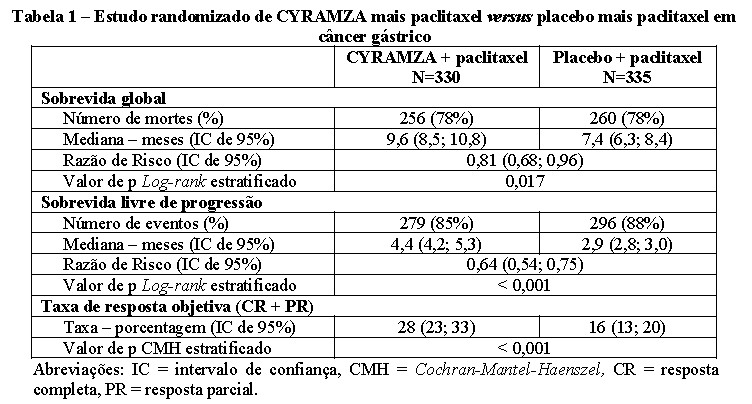
Ramucirumabe como agente isolado
O Estudo REGARD foi um estudo multinacional, randomizado, duplo-cego, multicêntrico de CYRAMZA mais a melhor terapia de suporte (MTS)
versus
placebo mais MTS que randomizou (2:1) 355 pacientes com câncer gástrico metastático ou localmente avançado, incluindo adenocarcinoma da junção gastroesofágica, que receberam quimioterapia contendo fluoropirimidina ou platina anteriormente. A principal medida de resultado de eficácia foi a sobrevida global e a medida de resultado de eficácia de apoio foi a sobrevida livre de progressão. Os pacientes deveriam apresentar progressão da doença dentro de 4 meses após a última dose da terapia de primeira linha para doença metastática ou localmente avançada ou dentro de 6 meses após a última dose da terapia adjuvante. Os pacientes também deveriam apresentar PS ECOG 0 ou 1. Os pacientes receberam infusão intravenosa de solução de CYRAMZA 8 mg/Kg (N=238) ou placebo (N=117) a cada 2 semanas. A randomização foi estratificada pela perda de peso nos 3 meses anteriores (≥10%versus < 10%), região geográfica e local do tumor primário (gástrico versus JGE).
As características basais e demográficas foram similares entre os braços de tratamento. A faixa etária mediana foi de 60 anos de idade; 70% dos pacientes eram do sexo masculino; 77% eram brancos, 16% asiáticos; o PS ECOG foi 0 para 28% dos pacientes e 1 para 72% dos pacientes; 91% dos pacientes tiveram doença mensurável; 75% dos pacientes tiveram câncer gástrico e 25% tiveram adenocarcinoma de JGE. A maioria dos pacientes (85%) apresentou progressão da doença durante ou após a terapia de primeira linha da doença metastática. A quimioterapia prévia para câncer gástrico consistiu de terapia em combinação com platina/fluoropirimidina (81%), regimes contendo fluoropirimidina sem platina (15%) e regimes contendo platina sem fluoropirimidina (4%). No Estudo REGARD, os pacientes receberam uma mediana de 4 doses (faixa de 1-34) de CYRAMZA ou uma mediana de 3 doses (faixa de 1-30) de placebo.
A sobrevida global e a sobrevida livre de progressão foram aumentadas de forma estatisticamente significante em pacientes randomizados para receber CYRAMZA, quando comparado a pacientes randomizados para receber placebo. Os resultados de eficácia são apresentados na Tabela 2.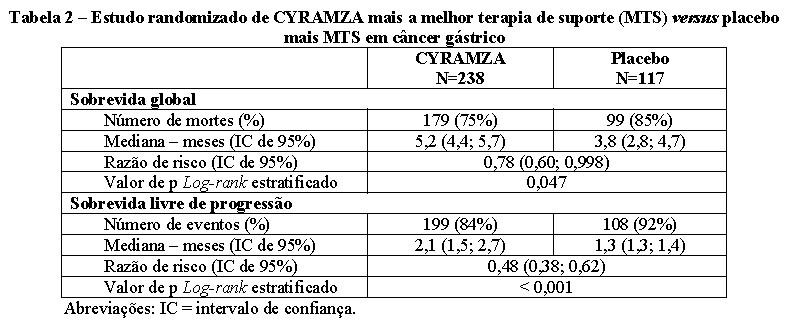
Câncer de Pulmão de Não Pequenas Células (NSCLC)
Ramucirumabe em combinação com docetaxel
O Estudo REVEL foi multinacional, randomizado, duplo-cego, com CYRAMZA mais docetaxel versus placebo mais docetaxel, 1.253 pacientes randomizados (1:1) com NSCLC com progressão da doença durante ou após terapia prévia à base de platina para doença localmente avançada ou metastática. A principal medida de eficácia foi a sobrevida global e as medidas de suporte de resultados de eficácia foram a sobrevida livre de progressão e taxa de resposta objetiva. Os pacientes também deveriam apresentar PS ECOG 0 ou 1. Os pacientes foram randomizados para receber CYRAMZA 10 mg/Kg ou placebo por infusão intravenosa, em associação com o docetaxel 75 mg/m2, ambos a cada 21 dias. Centros na Ásia Oriental administraram uma dose reduzida de docetaxel de 60 mg/m2 a cada 21 dias. Os pacientes que descontinuaram o tratamento combinado, por causa de eventos adversos atribuídos a CYRAMZA/placebo ou docetaxel foram autorizados a continuar a monoterapia com o outro componente do tratamento até a progressão da doença ou toxicidade intolerável. A randomização foi estratificada por região geográfica, sexo, terapia de manutenção anterior e PS ECOG.
As características demográficas e basais foram semelhantes entre os grupos de tratamento. A idade média foi de 62 anos; 67% dos pacientes eram homens; 82% eram brancos e 13% eram asiáticos; 32% apresentavam PS ECOG 0; 73% tinham histologia não-escamosa e 26% tinham histologia escamosa. Em adição à quimioterapia com platina (99%), as terapias anteriores mais comuns foram pemetrexede (38%), gencitabina (25%), taxano (24%) e bevacizumabe (14%). Vinte e dois por cento dos pacientes receberam terapia de manutenção anterior. O status EGFR do tumor era desconhecido para a maioria dos pacientes (65%). Onde o status EGFR do tumor era conhecido (N=445), 7,4% foram positivos para a mutação EGFR (N=33). Não foram coletados dados relacionados ao status de rearranjo tumoral ALK.
A sobrevida global e a sobrevida livre de progressão foram aumentadas de forma estatisticamente significativa em pacientes randomizados para receber CYRAMZA mais docetaxel em comparação com os pacientes randomizados para receber placebo mais docetaxel. A taxa de resposta objetiva (resposta completa + resposta parcial) foi de 23% (IC 95%: 20, 26) para CYRAMZA mais docetaxel e 14% (IC 95%: 11, 17) para placebo mais docetaxel, valor de p < 0,001. Os resultados de eficácia estão apresentados na Tabela 3.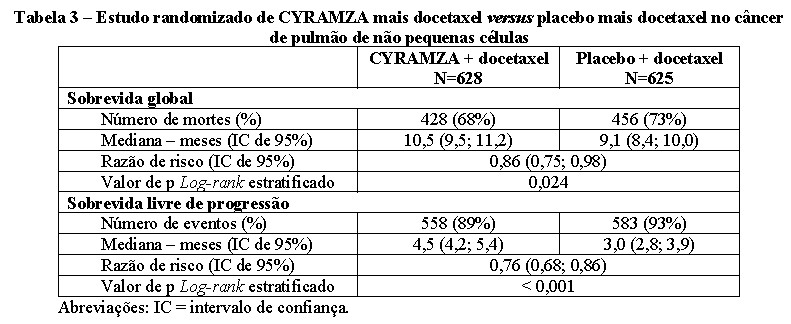
Ramucirumabe em combinação com erlotinibe
O Estudo RELAY foi um estudo global, randomizado, duplo-cego, de Fase 3 de CYRAMZA em combinação com erlotinibe versus placebo em combinação com erlotinibe que randomizou (1:1) 449 pacientes, não tratados previamente, com câncer de pulmão de não pequenas células (NSCLC) metastático com mutações ativadoras do receptor do fator de crescimento epidérmico (EGFR) do tipo deleção no éxon 19 ou mutações de substituição no éxon 21 (L858R), no início do estudo. Os pacientes elegíveis apresentavam PS ECOG 0 ou 1. Pacientes com metástase no sistema nervoso central ou mutações conhecidas no EGFR como T790M, no período basal, foram excluídos do estudo.
Em geral, as características demográficas no período basal foram balanceadas entre os dois braços. A maioria dos pacientes em ambos os braços de tratamento era do sexo feminino (62,9% versus 63,1%); a idade mediana foi de 65 anos (23-89); aproximadamente 75% eram asiáticos (76,8% versus 77,3%); aproximadamente 60% eram nunca fumantes (59,8% versus 61,8%); todos tinham um PS ECOG 0 ou 1; 54,1% tinham deleções no éxon 19, e 45,4% tinham um tipo de mutação do EGFR no éxon 21 (L858R).
Os pacientes tratados com CYRAMZA em combinação com erlotinibe apresentaram uma melhora estatisticamente e clinicamente significativa na sobrevida livre de progressão (PFS) comparado aos pacientes tratados com placebo em combinação com erlotinibe (Tabela 4). Resultados consistentes foram observados entre os subgrupos incluindo deleções no éxon 19 e substituição no éxon 21 (L858R), idade, raça (caucasianos HR: 0,618, asiáticos HR: 0,638) fumantes e nunca fumantes. A taxa de mutações T790M emergentes do tratamento foi semelhante entre o braço de CYRAMZA em combinação com erlotinibe e o braço de placebo mais erlotinibe. Nenhum efeito prejudicial foi observado na sobrevida global de pacientes tratados com CYRAMZA em combinação com erlotinibe comparado a pacientes tratados com placebo mais erlotinibe, quanto aos dados interinos de sobrevida global no momento da análise final da PFS (17,6% de completude). A análise da sobrevida livre de progressão-2 (31,2% de completude) sugeriu que o benefício na PFS foi preservado durante as linhas de tratamento subsequentes. A análise primária de qualidade de vida (QoL) não mostrou diferenças no tempo para a piora dos sintomas conforme Escala de Sintomas de Câncer de Pulmão (LCSS), entre os braços de tratamento, exceto para hemoptise (sangue no escarro), no qual o tempo para a piora foi inferior no braço de CYRAMZA em combinação com erlotinibe. Os resultados de eficácia do estudo RELAY estão apresentados na Tabela 4 e na Figura 1.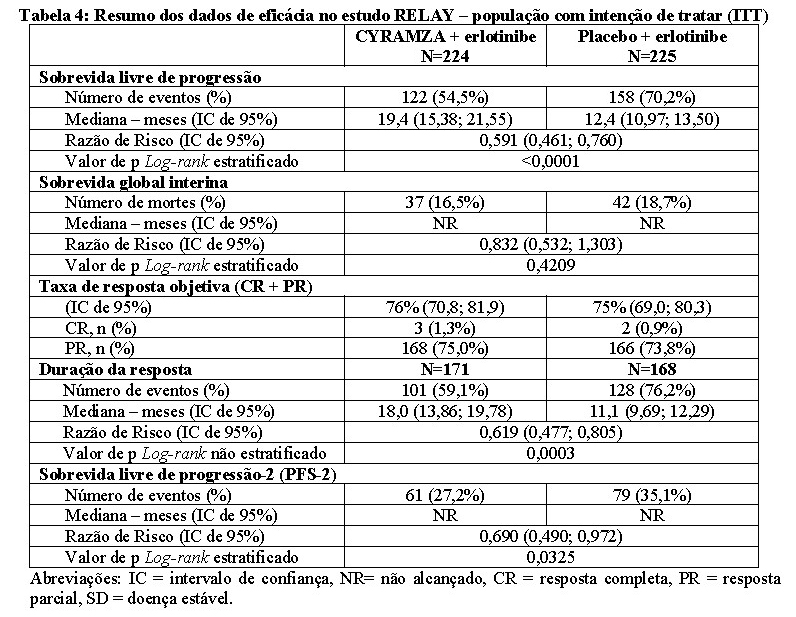
A PFS-2 é definida como o tempo entre a randomização e a segunda progressão da doença (definida como progressão radiológica objetiva ou progressão sintomática após o início de terapia sistêmica anticâncer adicional), ou morte por qualquer causa, o que ocorrer primeiro.
Câncer Colorretal (CRC)
O Estudo RAISE foi multinacional, randomizado, duplo-cego, de CYRAMZA mais FOLFIRI versus placebo mais FOLFIRI, em pacientes com CRC metastático, que tiveram progressão da doença durante ou após terapia prévia com bevacizumabe, oxaliplatina e uma fluoropirimidina. Os pacientes também deveriam apresentar PS ECOG 0 ou 1 e ter a progressão da doença no prazo de até 6 meses após a última dose de terapia de primeira linha. Um total de 1.072 pacientes foram randomizados aleatoriamente (1:1) para receber uma infusão intravenosa de 8 mg/Kg de CYRAMZA (N=536) ou placebo (N=536), em combinação com FOLFIRI: irinotecano 180 mg/m2, administrado por via intravenosa durante 90 minutos e ácido folínico 400 mg/m2, administrado por via intravenosa, simultaneamente, por mais 120 minutos; seguido por bolus de 400 mg/m2 de 5-fluoruracila por via intravenosa ao longo de 2 a 4 minutos; seguido de 2.400 mg/m2 de 5-fluoruracila, administrado por via intravenosa por infusão contínua durante 46 a 48 horas. Os ciclos de tratamento nos dois braços foram repetidos a cada 2 semanas. Os pacientes que descontinuaram um ou mais componentes do tratamento devido a um evento adverso foram autorizados a continuar o tratamento com o(s) outro(s) componente(s) do tratamento até a progressão da doença ou toxicidade inaceitável. A principal medida de eficácia foi a sobrevida global e a medida de suporte de eficácia foi sobrevida livre de progressão. A randomização foi estratificada por região geográfica, status de KRAS do tumor e tempo para progressão da doença após o início do tratamento de primeira linha ( < 6 meses versus ≥ 6 meses).
As características demográficas e basais foram semelhantes entre os grupos de tratamento. A idade média foi de 62 anos; 57% dos pacientes eram homens; 76% eram brancos e 20% asiáticos; 49% apresentavam PS ECOG 0; 49% dos pacientes tinham tumores KRAS mutados; e 24% dos pacientes tiveram < 6 meses de tempo para a progressão da doença após o início do tratamento de primeira linha.
A sobrevida global e a sobrevida livre de progressão foram aumentadas de forma estatisticamente significativa em pacientes randomizados para receber CYRAMZA mais FOLFIRI em comparação com os pacientes randomizados para receber placebo mais FOLFIRI. O efeito do tratamento foi consistente em todos os fatores de estratificação pré-especificados. Os resultados de eficácia estão apresentados na Tabela 5.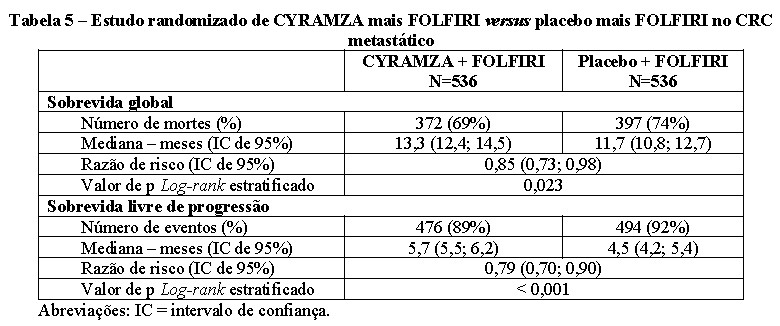
Carcinoma Hepatocelular (HCC)
REACH-2 foi um estudo multinacional, randomizado, duplo-cego, multicêntrico de CYRAMZA em conjunto com o melhor tratamento de suporte (MTS) versus placebo mais MTS, que randomizou pacientes com HCC avançado que tiveram progressão da doença em terapia ou após terapia prévia com sorafenibe, ou que foram intolerantes ao sorafenibe e tinham PS ECOG 0 ou 1. Os pacientes elegíveis eram Child-Pugh A. Além disso, os pacientes eram do estágio B do Barcelona Clinic Liver Cancer (BCLC) e já não eram passíveis de terapia locorregional, ou eram do estágio C do BCLC. Um total de 292 pacientes foram randomizados (2:1) para receber uma infusão intravenosa de CYRAMZA de 8 mg/Kg (N=197) ou placebo (N=95).
Para os pacientes do REACH-2 foi requerido ter uma alfa-fetoproteína (AFP) basal elevada, definida como ≥ 400 ng/mL para ser recrutado. Este limiar de AFP foi determinado com base nos resultados de sobrevida global (OS) de análises pré-especificadas do REACH, um estudo clínico pivotal previamente concluído em pacientes com HCC. Embora o estudo REACH não tenha cumprido o seu objetivo principal de demonstrar um benefício de sobrevida com CYRAMZA em comparação a placebo para a população geral de pacientes (OS HR=0,866; IC 95%: 0,717, 1,046; p=0,139), no subgrupo de pacientes com um patamar de AFP ≥ 400 ng/mL, o tratamento com CYRAMZA levou a uma melhora na OS (HR=0,674; IC 95%: 0,508, 0,895; p=0,0059). O subgrupo de pacientes com AFP basal < 400 ng/mL não apresentou um benefício de sobrevida (OS HR=1,093; IC 95%: 0,836, 1,428; p=0,5059).
No REACH-2, as medidas de resultados de eficácia incluíram a sobrevida global, a sobrevida livre de progressão (PFS) e a taxa de resposta objetiva (ORR). Os sintomas da doença foram avaliados usando o instrumento de Avaliação Funcional de Terapia do Câncer (FACT) - Índice de Sintomas Hepatobiliares (FHSI-8), para avaliar a falta de energia, náusea, dor, perda de peso, dor nas costas, fadiga, icterícia e dor de estômago. A deterioração clinicamente significativa foi pré-especificada como uma diminuição de ≥ 3 pontos na pontuação do índice. A randomização foi estratificada por região geográfica, invasão macrovascular (sim versus não) e PS ECOG (0 versus 1).
Dados demográficos iniciais e características da doença (N=292): a mediana de idade foi de 64 anos; 80% dos pacientes eram homens; 50% asiáticos; 58% tinham PS ECOG 0 e 35% dos pacientes tinham invasão macrovascular, 17% eram intolerantes ao sorafenibe, 37% tinham hepatite B, 26% tinham hepatite C, 24% tinham uso prévio significativo de álcool e 64% tinham terapia locorregional prévia.
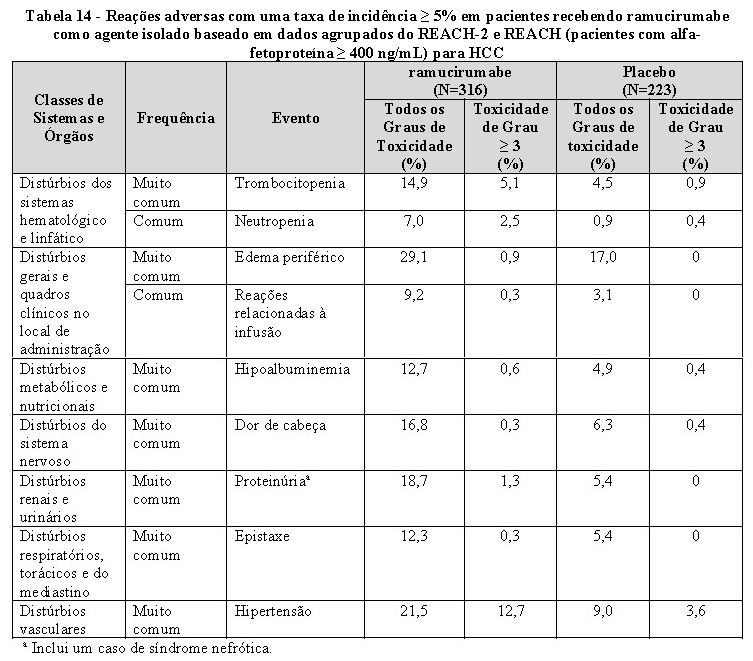
A sobrevida global e a sobrevida livre de progressão demonstraram melhora estatisticamente significativa para pacientes randomizados para receber CYRAMZA, em comparação com pacientes randomizados para receber placebo. A taxa de resposta objetiva foi numericamente maior. Os resultados de eficácia do REACH-2, incluindo OS e PFS, foram consistentes com os do subgrupo REACH AFP ≥ 400 ng/mL.
Para a pontuação total do FHSI-8, foi observada uma tendência para um atraso na deterioração dos sintomas da doença em pacientes randomizados para receber CYRAMZA, em comparação com os pacientes randomizados para receber placebo. O tempo para a deterioração dos sintomas da doença foi maior nos pacientes tratados com CYRAMZA em comparação aos pacientes tratados com placebo para os sintomas de náusea, dor, perda de peso, fadiga e dor nas costas, e similares para os sintomas de dor de estômago, falta de energia e icterícia.
Os resultados de eficácia estão apresentados na Tabela 6.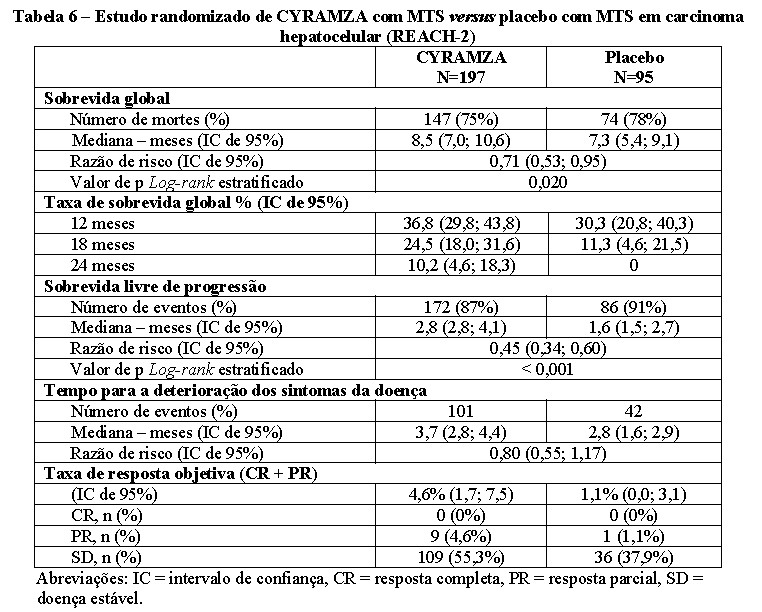
CARACTERÍSTICAS FARMACOLÓGICAS
Descrição
Ramucirumabe é um anticorpo monoclonal recombinante humano do tipo IgG1 que se liga especificamente ao receptor 2 do fator de crescimento endotelial vascular (FCEV). Ramucirumabe tem um peso molecular aproximado de 147 kDa. Ramucirumabe é produzido em células de mamíferos NS0 geneticamente modificadas.
Propriedades farmacodinâmicas
Mecanismo de ação: o receptor 2 do FCEV é o mediador chave da angiogênese induzida pelo FCEV. Ramucirumabe é um anticorpo humano receptor-alvo que se liga especificamente ao receptor 2 do FCEV e bloqueia a ligação do FCEV-A, FCEV-C e FCEV-D. Como resultado, ramucirumabe inibe a ativação do ligante estimulado pelo receptor 2 do FCEV e os seus componentes de sinalização em cascata, incluindo proteínas quinases de ativação mitogênica p44/p42, neutralizando a proliferação e a migração ligante-induzida das células endoteliais humanas.
Propriedades farmacocinéticas
Baseado na análise da farmacocinética da população, as características farmacocinéticas de ramucirumabe são similares entre os pacientes com os vários tipos de câncer.
Absorção: CYRAMZA é somente para administração intravenosa.
Distribuição: o volume médio [% coeficiente de variação (CV%)] de distribuição de ramucirumabe em estado estável foi 5,4 L (15%).
Baseado em dados in vitro e estudos não clínicos in vivo com um anticorpo substituto, espera-se o início de ação do medicamento dentro de horas após a aplicação; entretanto, a eficácia terapêutica é observada ao longo do tempo.
Eliminação: baseado na abordagem da farmacocinética da população, o clearance médio de ramucirumabe foi 0,015 L/h (30%) e a meia vida média foi de 14 dias (20%).
Farmacocinética em populações especiais
A análise da farmacocinética da população sugeriu que a idade, o sexo e a etnia não tinham efeito na farmacocinética de ramucirumabe.
Pacientes idosos: baseado nos resultados da análise da farmacocinética da população, não houve diferença na exposição ao ramucirumabe em pacientes ≥ 65 anos de idade comparado a pacientes < 65 anos de idade.
Pacientes com insuficiência renal: baseado nos resultados da análise da farmacocinética da população, a exposição ao ramucirumabe foi similar em pacientes com insuficiência renal leve [clearance de creatinina calculado (CrCl) ≥ 60 a < 90 mL/min], insuficiência renal moderada (CrCl ≥ 30 a < 60 mL/min) ou insuficiência renal grave (CrCl ≥ 15 a < 30 mL/min) em relação aos pacientes com função renal normal (CrCl ≥ 90 mL/min).
Insuficiência hepática: baseado nos resultados da análise da farmacocinética da população, a exposição ao ramucirumabe foi similar em pacientes com insuficiência hepática leve [bilirrubina total > 1,0-1,5 vezes o limite superior do valor normal (LSN) e qualquer valor de AST (TGO), ou bilirrubina total ≤ 1,0 LSN e AST (TGO) > LSN] ou insuficiência hepática moderada [bilirrubina total > 1,5-3,0 vezes o LSN e qualquer valor de AST (TGO)] comparado aos pacientes com função hepática normal [bilirrubina total e AST (TGO) ≤ LSN]. Não estavam disponíveis dados de farmacocinética para pacientes com insuficiência hepática grave [bilirrubina total > 3 vezes o LSN e qualquer valor de AST (TGO)].
CONTRAINDICAÇÕES
CYRAMZA é contraindicado para pacientes que tenham histórico de reação de hipersensibilidade grave ao ramucirumabe ou a qualquer outro ingrediente usado na formulação.
CYRAMZA é contraindicado se existir evidência radiológica de que o câncer de pulmão tem uma cavidade ou se o câncer de pulmão estiver perto dos grandes vasos.
ADVERTÊNCIAS E PRECAUÇÕES
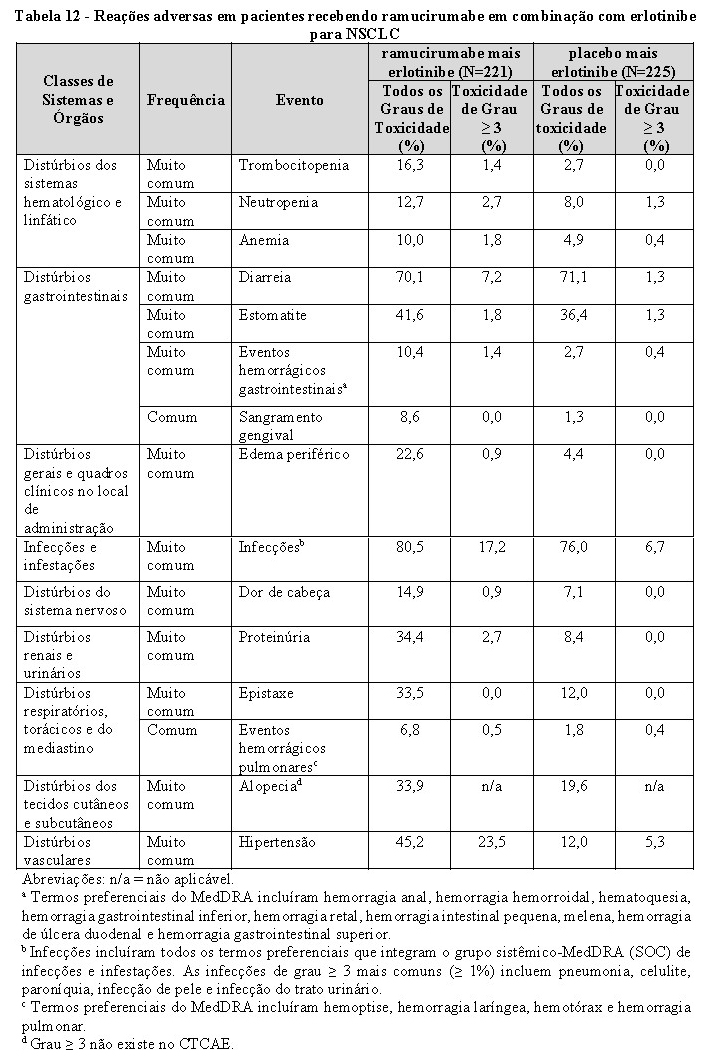
Tumores decorrentes de alterações nos genes EGFR e ALK: pacientes com tumores decorrentes de alterações nos genes EGFR e ALK devem ser tratados com CYRAMZA somente nos casos em que ocorrer progressão da doença após tratamento específico para estas mutações.
Eventos tromboembólicos arteriais (ETAs): ETAs graves, incluindo infarto do miocárdio, parada cardíaca, acidente vascular cerebral e isquemia cerebral foram relatados nos estudos clínicos. Nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA, a incidência de ETA de todos os graus foi de 1-3%. A incidência de ETA de grau 3-5 foi < 1-2%. Descontinuar permanentemente CYRAMZA em pacientes que apresentam ETA grave.
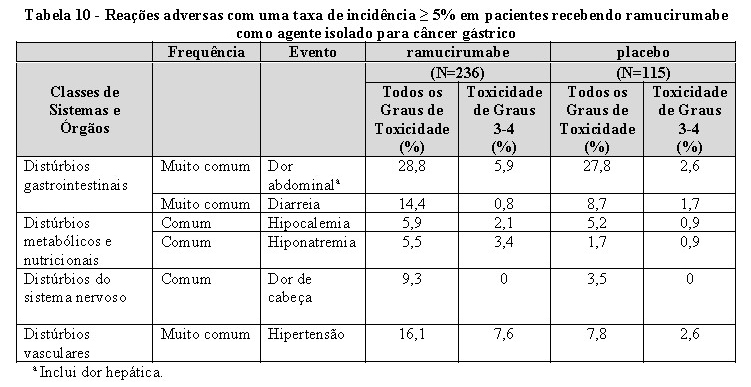
Hipertensão: uma incidência aumentada de hipertensão grave ocorreu em pacientes recebendo CYRAMZA comparado ao placebo. Nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA, a incidência de hipertensão de todos os graus ocorreu entre 11-45%. A incidência de hipertensão de graus 3-5 variou de 6-24%. Na maioria dos casos, a hipertensão foi controlada utilizando o tratamento anti-hipertensivo padrão. A hipertensão preexistente deve ser controlada antes de iniciar o tratamento com ramucirumabe. O monitoramento da pressão arterial é recomendado ao longo da terapia. Suspender temporariamente CYRAMZA no caso de hipertensão grave até que seja controlada com tratamento médico. Descontinuar permanentemente CYRAMZA caso a hipertensão clinicamente significativa não possa ser controlada com terapia anti-hipertensiva (ver POSOLOGIA E MODO DE USAR).
Reações relacionadas à infusão: as reações relacionadas à infusão (RRIs) ocorreram antes de instituir as recomendações de pré-medicação nos estudos clínicos de CYRAMZA. Nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA nos quais a pré-medicação foi recomendada ou requerida, a incidência de reações relacionadas à infusão de todos os graus variou de < 1-9%. A incidência de reações relacionadas à infusão de graus 3-5 foi < 1%. A maioria dos eventos ocorreu durante ou após a primeira ou segunda infusão de ramucirumabe. Monitorar os pacientes durante a infusão quanto a sinais de reações de hipersensibilidade com equipamento de reanimação prontamente disponível. Os sintomas incluíram rigidez/tremores, dorsalgia/espasmos, dores e/ou apertos no peito, calafrios, rubor, dispneia, respiração ruidosa, hipóxia e parestesia. Em casos graves, os sintomas incluíram broncoespasmos, taquicardia supraventricular e hipotensão. Descontinuar imediatamente e permanentemente CYRAMZA no caso de RRIs de Grau 3 ou 4 (ver POSOLOGIA E MODO DE USAR).
Perfurações gastrointestinais: CYRAMZA é uma terapia antiangiogênica e pode aumentar o risco de perfurações gastrointestinais. Casos de perfurações gastrointestinais foram relatados em pacientes tratados com ramucirumabe. Nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA, a incidência de perfurações gastrointestinais de todos os graus e de graus 3-5, variou de < 1-2%. Descontinuar permanentemente CYRAMZA em pacientes que apresentarem perfurações gastrointestinais.
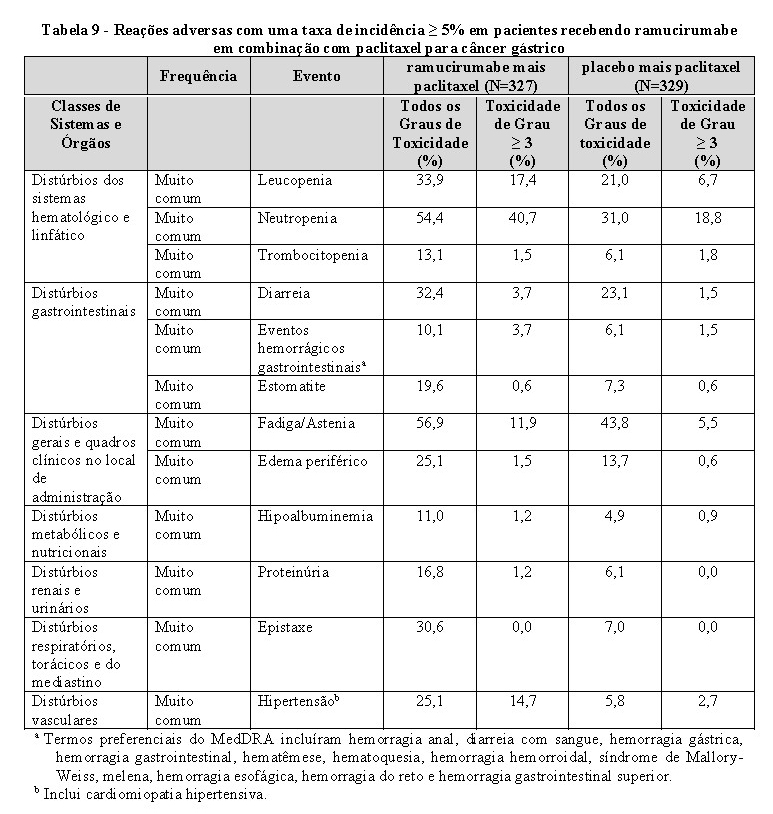
Sangramento grave: CYRAMZA é uma terapia antiangiogênica e pode aumentar o risco de sangramento grave. Nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA, a incidência de hemorragia de todos os graus variou de 13-55%. A incidência de hemorragia de graus 3-5 variou de 2-5%. Hemorragia gastrointestinal grave foi relatada em pacientes que apresentam câncer gástrico tratados com ramucirumabe em combinação com paclitaxel, e em pacientes com câncer colorretal tratados com ramucirumabe em combinação com FOLFIRI. Descontinuar permanentemente CYRAMZA em pacientes que apresentarem sangramento de Grau 3 ou 4.
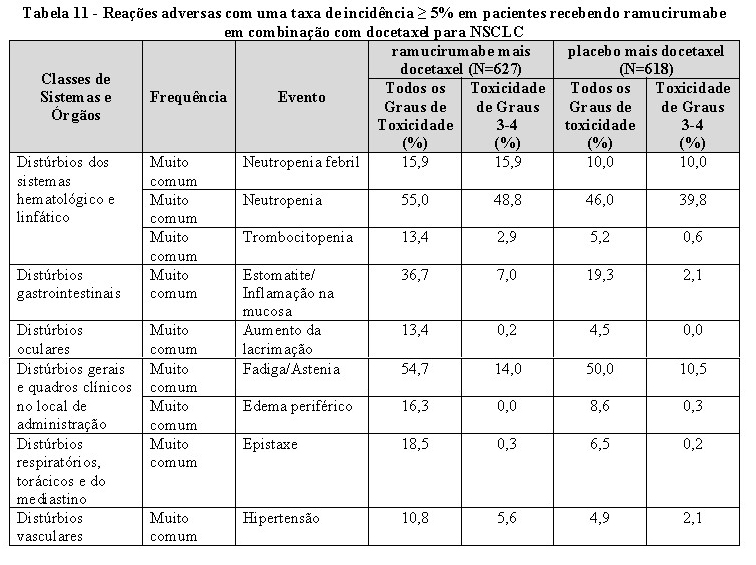
Hemorragia pulmonar em câncer de pulmão de não pequenas células (NSCLC): hemorragia pulmonar grave, incluindo um evento fatal de hemotórax, foi relatada em pacientes com NSCLC tratados com CYRAMZA em combinação com erlotinibe. Pacientes com NSCLC com sangramento pulmonar recente ( > 2,5 mL ou sangue vermelho vivo), bem como pacientes com evidência radiológica de invasão ou contiguidade de grandes vasos sanguíneos por tumor, ou aqueles com cavitação do tumor no início do estudo, independentemente da histologia, foram excluídos do estudo RELAY para NSCLC. Os pacientes que receberam terapia crônica com anti-inflamatórios não esteroidais ou agentes antiplaquetários foram excluídos do RELAY. O uso de ácido acetilsalicílico em doses de até 325 mg/dia foi permitido.
Dificuldade da cicatrização de feridas: o impacto de ramucirumabe não foi avaliado em pacientes com feridas sérias ou que não cicatrizam. Em um estudo conduzido em animais, ramucirumabe não comprometeu a cicatrização de feridas. No entanto, tendo em vista que CYRAMZA é uma terapia antiangiogênica e pode ter o potencial de afetar adversamente a cicatrização de feridas, o tratamento com CYRAMZA deve ser suspenso antes de cirurgia programada. A decisão de retomar o tratamento com CYRAMZA após a intervenção cirúrgica deve ser baseada no julgamento clínico de cicatrização adequada das feridas. Caso um paciente desenvolva complicações na cicatrização da ferida durante a terapia, descontinuar CYRAMZA até a cicatrização total da ferida.
Síndrome da leucoencefalopatia posterior reversível (SLPR): a SLPR foi relatada em < 0,1% dos 2.137 pacientes recrutados nos seis estudos clínicos com CYRAMZA. Os sintomas podem desaparecer ou melhorar em questão de dias, embora alguns pacientes com SLPR possam apresentar sequelas neurológicas contínuas ou morte. Casos de SLPR, incluindo casos fatais, foram raramente reportados em pacientes que receberam ramucirumabe. Os sintomas de SLPR incluíram convulsão, dor de cabeça, náusea/vômito, cegueira e alteração de consciência associada ou não à hipertensão. O diagnóstico de SLPR pode ser confirmado por imagem cerebral (por exemplo, imagem de ressonância magnética). Descontinuar permanentemente CYRAMZA em pacientes que apresentarem SLPR (ver POSOLOGIA E MODO DE USAR).
Insuficiência hepática: utilizar CYRAMZA com cautela em pacientes com cirrose hepática grave (Child-Pugh B ou C), cirrose com encefalopatia hepática, ascites clinicamente significativas em razão de cirrose ou síndrome hepatorrenal. Utilizar somente se os benefícios potenciais do tratamento superarem os riscos potenciais de insuficiência hepática progressiva nesses pacientes. Em pacientes com câncer hepático e cirrose hepática, a encefalopatia hepática foi reportada em uma taxa mais alta em pacientes tratados com ramucirumabe comparado aos pacientes tratados com placebo (ver REAÇÕES ADVERSAS).
Insuficiência cardíaca: em dados agrupados de estudos clínicos com ramucirumabe, a insuficiência cardíaca foi relatada com uma incidência numericamente maior em pacientes recebendo ramucirumabe em combinação com uma variedade de regimes de quimioterapia, ou erlotinibe, em comparação com quimioterapia ou erlotinibe sozinho. Este aumento da incidência não foi observado em pacientes recebendo ramucirumabe em comparação com placebo, em dados agrupados de estudos clínicos como agente isolado. Os pacientes devem ser monitorados quanto a sinais e sintomas clínicos de insuficiência cardíaca durante o tratamento, e a suspensão do tratamento deve ser considerada se houver desenvolvimento de manifestações clínicas.
Fístula: pacientes tratados com CYRAMZA podem apresentar um risco aumentado no desenvolvimento de fístula. O tratamento com CYRAMZA deve ser descontinuado em pacientes que apresentarem fístula.
Proteinúria, incluindo síndrome nefrótica:
nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA, a incidência de proteinúria de todos os graus variou de 3-34%. A incidência de proteinúria de grau ≥ 3 (incluindo 4 pacientes com síndrome nefrótica) variou de < 1-3%. Monitorar a proteinúria através de dipstick urinário e/ou razão creatinina urinária proteica para o desenvolvimento de agravamento de proteinúria durante o tratamento com CYRAMZA. Interromper CYRAMZA para níveis de proteína na urina de 2 ou mais gramas ao longo de 24 horas. Reiniciar CYRAMZA em dose reduzida quando o nível de proteína na urina retornar para menos de 2 gramas ao longo de 24 horas. Interromper permanentemente CYRAMZA para níveis de proteína na urina superiores a 3 gramas ao longo de 24 horas ou presença de síndrome nefrótica (ver POSOLOGIA E MODO DE USAR).
Disfunção da tireoide: monitorar a função da tireoide durante o tratamento com CYRAMZA. Nos seis estudos clínicos, em 2.137 pacientes com vários tipos de câncer tratados com CYRAMZA, a incidência de hipotireoidismo de graus 1-2 variou de < 1-3%; não houve relatos de hipotireoidismo de graus 3-5.
Toxicologia não clínica: não foram realizados estudos em animais para testar ramucirumabe quanto ao potencial de carcinogenicidade, genotoxicidade ou danos à fertilidade. Nos macacos cinomolgos, a patologia anatômica revelou efeitos adversos nas placas epifisárias de crescimento (espessamento e osteocondropatia) em todas as doses testadas (5-50 mg/Kg). A dose semanal mais baixa testada em macacos cinomolgos é 1,2 vezes a dose recomendada de CYRAMZA como agente isolado (8 mg/Kg a cada 2 semanas). Foi demonstrado que a inibição da sinalização de FCEV2 em modelos animais resultou em alterações nos níveis hormonais críticos para a gravidez e, em macacos, um aumento da duração do ciclo folicular. Em um estudo de 39 semanas em animais, as fêmeas de macacos tratadas com ramucirumabe apresentaram aumentos de mineralização folicular do ovário, de forma dose-dependente.
Imunogenicidade: em 25 estudos clínicos, 94/3.059 (3,1%) dos pacientes tratados com CYRAMZA apresentaram teste positivo para anticorpos antirramucirumabe decorrentes do tratamento utilizando o ensaio enzimático imunoabsorvente (ELISA). Os anticorpos neutralizantes foram detectados em 14 dos 94 pacientes que apresentaram teste positivo para anticorpos antirramucirumabe decorrentes do tratamento. No geral, houve uma baixa incidência de anticorpos antirramucirumabe decorrentes do tratamento e anticorpos neutralizantes entre os pacientes tratados com ramucirumabe, e não houve correlação com os resultados de segurança nesses pacientes. Não houve relação entre imunogenicidade e reação relacionada à infusão ou eventos adversos decorrentes do tratamento.
Pacientes geriátricos com câncer de pulmão de não pequenas células (NSCLC): para CYRAMZA usado em combinação com erlotinibe no tratamento de primeira linha de NSCLC com mutações ativadoras do EGFR, os pacientes com 70 anos de idade ou mais, apresentaram uma incidência mais alta de eventos adversos de Grau ≥ 3 e eventos adversos graves de todos os graus em comparação com pacientes com menos de 70 anos de idade.
Uso pediátrico: a segurança e eficácia de CYRAMZA nos pacientes pediátricos não foram estabelecidas.
Efeitos na capacidade de dirigir e utilizar máquinas: não foram conduzidos estudos para determinar os efeitos de ramucirumabe na capacidade de dirigir e usar máquinas.
Uso durante a gravidez, amamentação e em pessoas com potencial reprodutivo: gravidez categoria C - não foram conduzidos estudos em animais especificamente para avaliar o efeito do ramucirumabe na reprodução de fêmeas e desenvolvimento fetal. Não há dados disponíveis sobre o uso de ramucirumabe em mulheres grávidas para informar qualquer risco associado à sua utilização. Os modelos animais ligam angiogênese, FCEV e receptor 2 do FCEV a aspectos críticos da reprodução da fêmea, desenvolvimento embriofetal e desenvolvimento pós-natal. Com base no mecanismo de ação de ramucirumabe (ver Propriedades farmacodinâmicas), existe o potencial de causar danos ao feto. O ramucirumabe irá inibir a angiogênese e pode potencialmente resultar em efeitos adversos durante a gravidez e desenvolvimento pós-natal.
Evitar o uso de ramucirumabe em mulheres grávidas e usar somente caso o benefício potencial à mãe justifique o risco potencial ao feto. Informar às mulheres férteis, grávidas ou que engravidaram durante o tratamento, sobre os riscos potenciais de ramucirumabe ao feto e por seguir com a gravidez. Aconselhar as mulheres a usarem contraceptivos para evitar engravidar enquanto recebem ramucirumabe e por pelo menos 3 meses após a última dose de ramucirumabe (ver Toxicologia não clínica).
Não foram conduzidos estudos para avaliar o impacto de ramucirumabe na produção de leite, sua presença no leite materno ou seus efeitos no recém-nascido lactente.
Não se sabe se ramucirumabe é excretado no leite humano. A IgG humana é excretada no leite humano e, em razão dos riscos potenciais ao recém-nascido lactente, se for amamentar, recomenda-se descontinuar a amamentação ou descontinuar ramucirumabe.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
INTERAÇÕES MEDICAMENTOSAS
Paclitaxel: não foram observadas interações medicamentosas entre ramucirumabe e paclitaxel. A farmacocinética do paclitaxel não foi afetada quando coadministrado com ramucirumabe e a farmacocinética de ramucirumabe não foi afetada quando coadministrado com paclitaxel.
Docetaxel: a farmacocinética do docetaxel não foi afetada quando coadministrado com ramucirumabe.
Irinotecano: a farmacocinética do irinotecano e do seu metabólito ativo, SN-38, não foram afetadas quando coadministrado com ramucirumabe.
Erlotinibe: a farmacocinética do erlotinibe não foi afetada quando coadministrado com ramucirumabe.
Nenhum estudo foi conduzido para investigar possível interação entre CYRAMZA e plantas medicinais, álcool, nicotina e exames laboratoriais e não laboratoriais.
CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Os frascos devem ser armazenados em um refrigerador entre 2°C e 8°C até o momento da utilização. O prazo de validade é de 24 meses quando os frascos são armazenados nesta temperatura e na caixa original protegida da luz.
Mantenha o frasco na caixa do produto para protegê-lo da luz. NÃO CONGELAR OU AGITAR o frasco.
A estabilidade química e física da solução para infusão com cloreto de sódio 0,9% foi demonstrada durante 24 horas quando conservada entre 2°C e 8°C. Do ponto de vista microbiológico, a solução para infusão de CYRAMZA deve ser aplicada imediatamente. Se isso não ocorrer, o tempo e as condições de armazenamento em uso são de responsabilidade do usuário e, normalmente, não devem ultrapassar 24 horas em temperatura entre 2°C e 8°C. NÃO CONGELAR OU AGITAR a solução de infusão de ramucirumabe.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Após preparo, manter de 2°C a 8°C por até 24 horas.
CYRAMZA está disponível como solução para diluição injetável em frasco-ampola de uso único de 10 mL ou 50 mL. CYRAMZA é uma solução estéril, livre de conservante, límpida a ligeiramente opalescente e incolor a ligeiramente amarela sem partículas visíveis. Cada frasco contém 100 mg de ramucirumabe em 10 mL (10 mg/mL) ou 500 mg de ramucirumabe em 50 mL (10 mg/mL). Após a diluição e preparação, ramucirumabe é administrado como infusão intravenosa. O pH é de 5,7 - 6,3.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
POSOLOGIA E MODO DE USAR
Posologia
Não administrar ramucirumabe por push ou bolus em injeção intravenosa.
Utilize somente solução de cloreto de sódio (0,9%) estéril para injeção como diluente. Não utilize dextrose como diluente.
Recomenda-se que o tratamento seja continuado até a progressão da doença subjacente ou até uma toxicidade inaceitável.
Câncer Gástrico
Ramucirumabe como agente isolado
A dose recomendada de ramucirumabe como agente isolado é 8 mg/Kg a cada 2 semanas, administrada por infusão intravenosa durante aproximadamente 60 minutos (velocidade máxima de infusão de 25 mg/min).
Ramucirumabe em combinação com paclitaxel
A dose recomendada de ramucirumabe é 8 m