CRISCY
CRISTÁLIA
somatropina
Hormonoterapia.
Apresentações.
Pó Liofilizado para Solução Injetável 4 UI (1,33 mg):
Embalagem com 1 frasco-ampola de pó liofilizado para solução injetável e 1 frasco-ampola de solução diluente;
Pó Liofilizado para Solução Injetável 12 UI (4,00 mg):
Embalagem com 1 frasco-ampola de pó liofilizado para solução injetável e 1 frasco-ampola de solução diluente;
Pó Liofilizado para Solução Injetável 16 UI (5,33 mg):
Embalagem com 1 frasco-ampola de pó liofilizado para solução injetável e 1 frasco-ampola de solução diluente;
Pó Liofilizado para Solução Injetável 30 UI (10,13 mg):
Embalagem com 1 frasco-ampola de pó liofilizado para solução injetável e 1 frasco-ampola de solução diluente;
USO SUBCUTÂNEO
USO ADULTO E PEDIÁTRICO
Composição.

Informações técnicas.
1. INDICAÇÕES
Este medicamento é indicado para:
Crianças
No tratamento do distúrbio de crescimento em crianças devido à secreção insuficiente do hormônio de crescimento ou associado à síndrome de Turner.
No distúrbio de crescimento (altura atual < -2,5 DP e altura ajustada pelos dados dos pais < -1 DP) em crianças de baixa estatura nascidas PIG (pequenos para a idade gestacional), com peso e/ou estatura ao nascimento abaixo de -2 DP, que não apresentaram catch-up (recuperação do crescimento em altura < 0 DP durante o último ano) até os 4 anos de idade ou mais.
Pacientes que apresentam síndrome de Prader-Willi, com o objetivo de melhorar o crescimento e a composição corpórea. O diagnóstico da síndrome de Prader-Willi deve ser confirmado através de teste genético apropriado.
No tratamento de baixa estatura idiopática, que é definida como altura abaixo de 2 SDS da altura média para determinada idade e gênero, associada a taxas de crescimento que provavelmente não permitam alcançar a altura adulta normal em pacientes pediátricos, cujas epífises não estejam fechadas e cujo diagnóstico exclui outras causas de baixa estatura que possam ser observadas ou tratadas por outros meios.
Adultos
Na terapia de reposição em adultos com deficiência de hormônio de crescimento acentuada. Insuficiência grave de hormônio de crescimento na idade adulta pode ser devido à doença hipofisária hipotalâmica conhecida e com deficiência de no mínimo um hormônio hipofisário que não seja a prolactina. Estes pacientes devem ser submetidos a um teste de estímulo com a finalidade de diagnóstico de deficiência de hormônio de crescimento. Em pacientes com deficiência de hormônio de crescimento isolada desde a infância (sem evidência de doença hipofisária hipotalâmica ou irradiação craniana), são recomendados dois testes de estímulo, exceto para aqueles que apresentam baixa concentração de IGF-I ( < 2 DP) que pode ser considerado o primeiro teste. O ponto de corte para o teste de estímulo deve ser rigoroso.
2. RESULTADOS DE EFICÁCIA
CRISCY é um medicamento biológico desenvolvido pela via da comparabilidade (biossimilar). O programa de desenvolvimento do produto foi projetado para demonstrar a comparabilidade entre CRISCY e GENOTROPIN®. A comparabilidade do CRISCY e do medicamento comparador GENOTROPIN®, em termos de eficácia clínica, foi demonstrada no Estudo CERES, um Estudo Clínico de Fase III em pacientes com deficiência no crescimento.
ESTUDOS COMPARATIVOS
Estudo clínico de fase I - A farmacocinética e a farmacodinâmica do r-hGH fabricado pelo Cristália foi avaliada de forma comparativa ao r-hGH fabricado pelos Laboratórios Pfizer Ltda, Genotropin®, em um estudo fase 1 cruzado, randomizado, aberto, de dois tratamentos, dois períodos, duas sequências e dose única subcutânea, em voluntários adultos, sadios, de ambos os gêneros. O estudo clínico fase 1 teve como parâmetro primário a comparação da farmacocinética dos produtos após administração de dose única subcutânea de 12,8 UI de r-hGH e como secundários, a comparação dos perfis farmacodinâmicos dos biomarcadores IGF-1 e IGFBP-3, avaliação de segurança pelos relatos de ocorrência e intensidade de eventos adversos e pela avaliação da tolerância local pela inspeção visual do local e relato de dor local (Ver item 3. Características Farmacológicas - Propriedades farmacocinéticas para mais informações).
Ref: Toffoletto O, Afiune J, Thiemann JE, Khandave SS, Patel S, Rodrigues DG. Comparative pharmacokinetic and pharmacodynamic evaluation between a new biosimilar and reference recombinant human growth hormone. Growth Horm IGF Res. outubro de 2016;30-31:31-6.
Estudo clínico de fase III (Estudo Ceres).
O estudo clínico de fase III (Estudo Ceres), de não inferioridade, nacional, multicêntrico, randomizado, investigador-cego, com grupo controle ativo, teve como objetivo primário avaliar a eficácia do r-hGH Cristália comparado ao Genotropin®, após 12 meses de tratamento em crianças pré-púberes, com distúrbio no crescimento devido à deficiência do hormônio do crescimento e sem histórico de tratamento anterior. O desfecho de eficácia primário foi à diferença da velocidade de crescimento (VC) entre os grupos de tratamento e o desfecho de eficácia secundário foi à variação do escore z de altura (escore z de altura após 12 meses de tratamento - escore z de altura no início do tratamento). Os participantes no estudo foram randomizados em uma proporção 1:1 para rhGH Cristália (33 mg/kg/dia) ou Genotropin? (33 mg/kg/dia). A randomização foi estratificada por idade (corte: < 10 anos; > 10 anos de idade) e teste de estímulo de GH (corte: ≤ 5 ng/mL; > 5 e ≤7 ng/mL). Os grupos de tratamento foram comparáveis em termos das características demográficas e antropométricas basais e dos resultados do teste de estímulo de GH basal, diferindo significativamente em termos dos valores médios da altura do pai (superior no grupo Cristália). Apesar desta diferença, os grupos não diferiram significativamente em termos do escore z para o alvo familiar, cujo cálculo tem como base a altura do pai, bem como a altura da mãe. A velocidade média de crescimento após 12 meses de tratamento foi de 9,7 cm/ano no grupo Cristália e de 9,5 cm/ano no grupo Genotropin®. De acordo com o modelo ANCOVA que considerou o tratamento, o teste GH (≤5 ng/mL vs. > 5 e ≤7 ng/mL) e a idade (≤10 vs > 10 anos) como covariáveis, a diferença média estimada entre os grupos foi 0,16 cm/ano, favorável ao grupo Cristália, com um intervalo de confiança de 95 % a variar entre -0,72 cm/ano e 1,03 cm/ano. Uma vez que o limite inferior do intervalo de confiança a 95 % não excede a margem de d = -2 cm/ano estabelecida, a não inferioridade foi verificada para o grupo Cristália. A mesma conclusão é obtida considerando as populações ITT e PP e com base nas análises de sensibilidade. Similarmente, o escore Z, 12 meses após o início do tratamento foi considerado semelhante entre os grupos de tratamento, independentemente do método de análise e população (ITT e PP), provando-se a não inferioridade do r-hGH Cristália.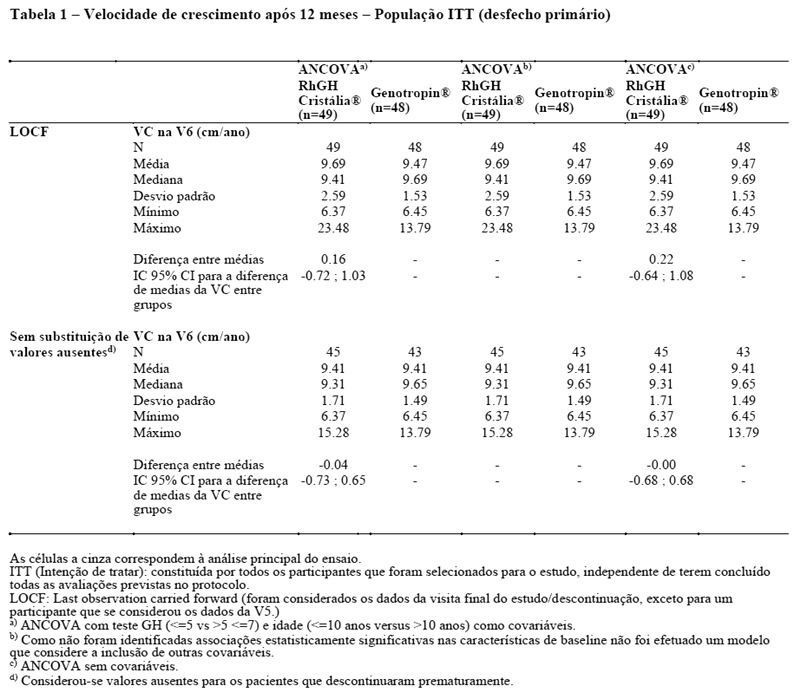
Ref: Cristália Produtos Químicos Farmacêuticos Ltda. Estudo clínico para comparar o hormônio do crescimento humano recombinante Cristália (r-hGH Cristália) versus Genotropin® em crianças pré-púberes com déficit de crescimento devido à deficiência do hormônio do crescimento (Estudo Ceres). Código do estudo: CRT-076. Fase de desenvolvimento clínico: III.
ESTUDOS CLÍNICOS - realizados pelo produto biológico comparador Genotropin®
Deficiência do hormônio de crescimento em adultos (GHDa)
Genotropin® pó liófilo injetável foi comparado com placebo em seis ensaios clínicos randomizados envolvendo um total de 172 pacientes adultos com GHD. Estes ensaios incluíram um período de tratamento duplo cego de 6 meses, durante o qual 85 pacientes receberam Genotropin® e 87 pacientes receberam placebo, seguido por um período de tratamento aberto no qual os pacientes participantes receberam Genotropin® por um período de até 24 meses. Genotropin® foi administrado por injeção subcutânea diária numa dose de 0,04 mg/kg/semana no primeiro mês de tratamento e 0,08 mg/kg/semana nos meses seguintes.
Alterações benéficas na composição corporal foram observadas no final do período de tratamento de 6 meses para os pacientes que receberam Genotropin® comparado aos pacientes que receberam placebo. A massa corporal magra, água corporal total e relação massa magra/gorda aumentaram enquanto que a massa corporal gorda total e a circunferência da cintura diminuíram. Estes efeitos na composição corporal foram mantidos quando o tratamento foi continuado além de 6 meses. A densidade mineral óssea diminuiu após 6 meses de tratamento, mas retornou aos valores basais após 12 meses de tratamento.
Deficiência do hormônio de crescimento em crianças (GHDc)
Dois estudos avaliaram a eficácia e segurança de Genotropin® em crianças com deficiência de hormônio de crescimento (GHD). Bierich, em 1987, avaliou 77 crianças, 49 sem tratamento prévio e 28 previamente tratadas com GH hipofisário. A dose de Genotropin® utilizada foi 12 UI/m2 (superfície corpórea)/semana (4 mg/m2/semana), por via subcutânea (SC), 6 vezes por semana. Nos pacientes virgens deste tratamento, a velocidade de crescimento aumentou de 3,7 cm/ano para 14,9 cm/ano nos primeiros 3 meses, estabilizando-se em 12 cm/ano. Nos pacientes previamente tratados, a taxa de crescimento de 5,6 cm/ano, aumentou para 10,7 cm/ano nos primeiros 3 meses e 8,5 cm/ano em 9 a 12 meses. Efeitos colaterais indesejáveis não foram observados em nenhum paciente.
Albertsson-Wikland et. al., 1988, avaliaram 51 crianças, 23 sem tratamento prévio (grupo A) e 28 previamente tratadas com GH hipofisário (grupo B). A dose de Genotropin® utilizada foi de 0,1 UI/Kg/dia (0,033 UI/Kg/dia ou 0,23 mg/Kg/semana). Os resultados observados foram: no grupo A, a velocidade de crescimento aumentou de 4,0 ± 1,1 cm/ano para 10,7 ± 2,3 cm/ano e no grupo B, aumentou de 3,0 ± 1,6 cm/ano para 10,9 ± 1,8 cm/ano. Estes resultados corresponderam a um ganho significativo de 1 DP na altura para a idade cronológica. Relação inversa foi demonstrada entre o ganho na velocidade de crescimento e idade cronológica, demonstrando, desse modo, que o início precoce da terapia, aumenta o ganho em altura.
PIG - Pequenos para a idade gestacional
Pacientes pediátricos nascidos pequenos para a idade gestacional (PIG) que não apresentaram catch-up até 2 anos de idade
A segurança e a eficácia de Genotropin® no tratamento de crianças nascidas pequenas para a idade gestacional (PIG) foram avaliadas em 4 ensaios clínicos randomizados, abertos, controlados. Os pacientes (faixa de idade de 2 a 8 anos) foram observados por 12 meses antes de serem randomizados para Genotropin® (duas doses diferentes em cada estudo, mais frequentemente 0,24 e 0,48 mg/kg/semana) como injeção subcutânea diária ou nenhum tratamento pelos primeiros 24 meses dos estudos. Após 24 meses nos estudos, todos os pacientes receberam Genotropin®.
Pacientes que receberam qualquer dose de Genotropin® apresentaram aumentos significativos no crescimento durante os primeiros 24 meses do estudo comparado com pacientes que não receberam tratamento (veja Tabela 1). Crianças que receberam 0,48 mg/kg/semana demonstraram melhora significativa no escore de desvio padrão da altura (SDS) comparado com crianças tratadas com 0,24 mg/kg/semana. Ambas as doses resultaram em aumento mais lento, porém constante, no crescimento entre os meses 24 e 72 (dados não mostrados).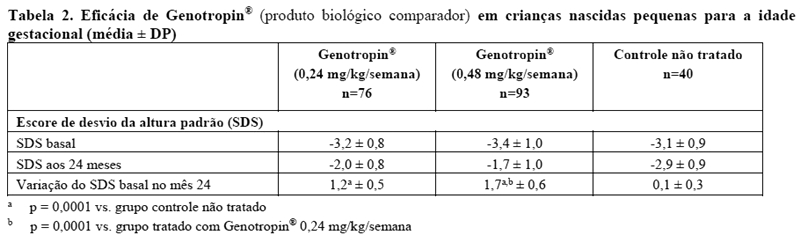
Em estudos clínicos com crianças nascidas pequenas para a idade gestacional (PIG), doses de 0,033 e 0,067 mg/kg de peso corporal por dia têm sido usadas para tratamento até que alcancem a altura final. Em 56 pacientes que foram tratados continuamente e chegaram (próximo) à altura final, a alteração de altura média do início do tratamento foi de +1,90 SDS (0,033 mg/kg de peso corporal por dia) e +2,19 SDS (0,067 mg/kg de peso corporal por dia). Dados da literatura sobre crianças PIG não tratadas e sem catch-up inicial espontâneo sugerem um crescimento tardio de 0,5 SDS. Os dados sobre segurança em longo prazo ainda são limitados.
Síndrome de Turner
Dois ensaios clínicos abertos e randomizados avaliaram a eficácia e segurança de Genotropin® em pacientes com síndrome de Turner com baixa estatura. Os pacientes com síndrome de Turner foram tratados apenas com Genotropin® ou com Genotropin® associado à terapia hormonal (etinilestradiol ou oxandrolona). Um total de 38 pacientes foi tratado somente com Genotropin® nos dois estudos. No Estudo 055, 22 pacientes foram tratados por 12 meses e, no Estudo 092, 16 pacientes foram tratados por 12 meses. Os pacientes receberam Genotropin® numa dose entre 0,13 a 0,33 mg/kg/semana.
Desvio padrão (SDS) de velocidade de crescimento e altura são expressos usando os padrões de Tanner (Estudo 055) ou Sempé (Estudo 092) para crianças normais da mesma idade, bem como o padrão Ranke (ambos os estudos) para pacientes com síndrome de Turner da mesma idade não tratados. Como observado na Tabela 1, os valores de SDS da velocidade de crescimento e da altura foram menores no período basal e após o tratamento com Genotropin®, quando os padrões normativos foram utilizados ao invés do padrão da síndrome de Turner.
Ambos os estudos demonstraram aumentos estatisticamente significativos a partir do basal, em todas as variáveis de crescimento linear (ou seja, velocidade de altura média, SDS de velocidade de crescimento e SDS de altura) após tratamento com Genotropin® (veja Tabela 2). A resposta de crescimento linear foi maior no Estudo 055, no qual os pacientes foram tratados com uma dose maior de Genotropin®.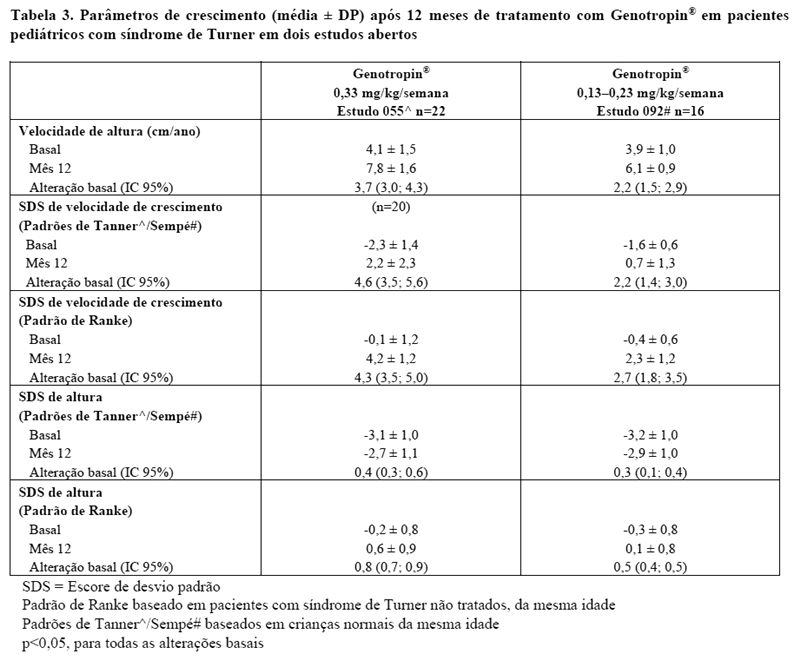
Síndrome de Prader-Willi (PWS)
A segurança e a eficácia de Genotropin® no tratamento de pacientes pediátricos com síndrome de Prader-Willi (PWS) foram avaliadas em dois ensaios clínicos randomizados, abertos, controlados. Os pacientes receberam Genotropin® ou nenhum tratamento durante o primeiro ano dos estudos e todos os pacientes receberam Genotropin® durante o segundo ano. Genotropin® foi administrado como injeção subcutânea diária e a dose foi calculada para cada paciente a cada 3 meses. No Estudo 1, o grupo de tratamento recebeu Genotropin® numa dose de 0,24 mg/kg/semana durante todo o estudo. Durante o segundo ano, o grupo controle recebeu Genotropin® numa dose de 0,48 mg/kg/semana. No Estudo 2 o grupo de tratamento recebeu Genotropin® numa dose de 0,36 mg/kg/semana durante todo o estudo. Durante o segundo ano o grupo controle recebeu Genotropin® numa dose de 0,36 mg/kg/semana.
Pacientes que receberam Genotropin® apresentaram aumentos significativos no crescimento linear durante o primeiro ano do estudo comparado com pacientes que não receberam tratamento (veja Tabela 3). O crescimento linear continuou no segundo ano, quando ambos os grupos receberam tratamento com Genotropin®.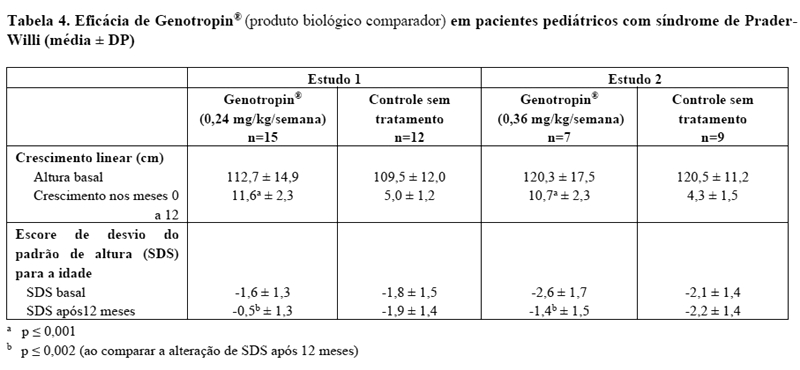
Também foram observadas alterações na composição corporal nos pacientes que receberam Genotropin® (produto biológico comparador, veja Tabela 4). Estas alterações incluíram diminuição na quantidade de massa gorda e aumento de massa corporal magra e da relação tecido magro/gordo, enquanto que as alterações no peso corporal foram semelhantes às observadas em pacientes que não receberam nenhum tratamento. O tratamento com Genotropin® não acelerou a idade óssea comparado a pacientes que não receberam nenhum tratamento.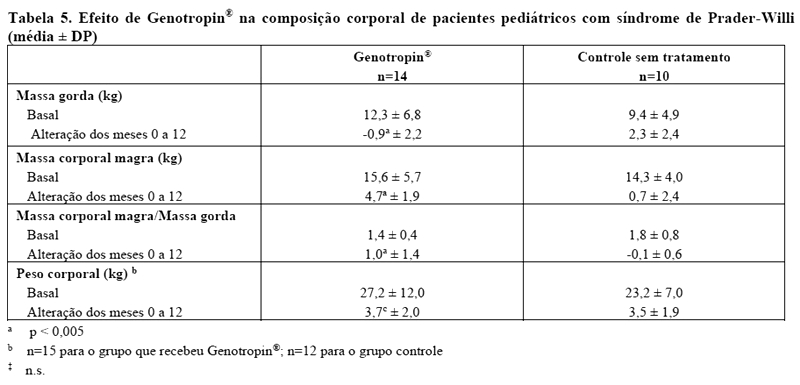
Baixa estatura idiopática
A eficácia e segurança de longo prazo de Genotropin® em pacientes com baixa estatura idiopática foram avaliadas em um estudo clínico randomizado, aberto e que incluiu 177 crianças. Os pacientes incluídos no período basal apresentavam baixa estatura, GH > 10 ng/mL após teste de estímulo e eram pré-puberes (critérios para baixa estatura idiopática foram aplicados retroativamente e mais 126 pacientes foram incluídos). Todos os pacientes foram observados durante 12 meses (evolução da estatura) e posteriormente randomizados para Genotropin® ou conduta expectante. Os pacientes foram acompanhados até atingirem a altura final. Duas doses de Genotropin® foram avaliadas neste estudo: 0,23 mg/kg/semana (0,033 mg/kg/dia) e 0,47 mg/kg/semana (0,067 mg/kg/dia). As características dos pacientes com baixa estatura idiopática pré-puberes na randomização (n = 105) foram: média (± SD): idade cronológica de 11,4 (1,3) anos, altura SDS -2,4 (0,4), velocidade de crescimento SDS -1,1 (0,8), velocidade de crescimento de 4,4 cm (0,9)/ano, IGF-1 SDS -0,8 (1,4). Os pacientes foram tratados durante um período médio de 5,7 anos. Os resultados para a altura final SDS são apresentados por grupo de tratamento, na Tabela 5. A terapia com Genotropin® melhorou a altura final em crianças com baixa estatura idiopática em relação ao grupo controle sem tratamento. O ganho observado na altura média final foi de 9,8 cm para meninas e 5,0 cm para meninos, para ambas as doses combinadas, em relação ao grupo controle sem tratamento. Um ganho de altura de 1 SDS foi observado em 10% dos indivíduos não tratados, 50% dos sujeitos recebendo 0,23 mg/kg/semana e 69% dos indivíduos que receberam 0,47 mg/kg/semana.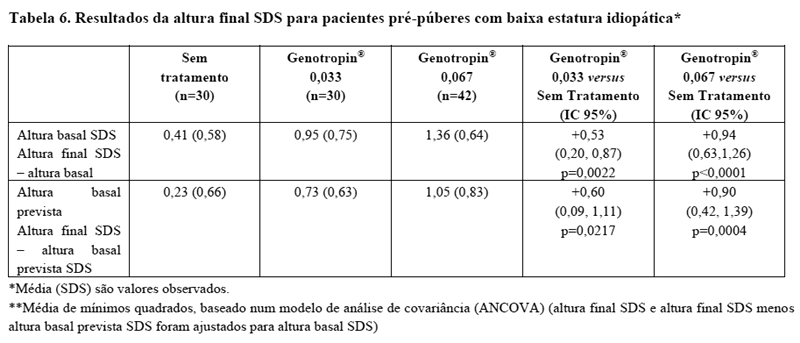
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
O princípio ativo de CRISCY é a somatropina. A somatropina é um hormônio metabólico potente, importante no metabolismo de lípides, carboidratos e proteínas. Em crianças que possuem deficiência de hormônio de crescimento endógeno, a somatropina estimula o crescimento linear e aumenta a velocidade de crescimento. Em adultos, assim como em crianças, a somatropina mantém a composição corpórea normal através do aumento da retenção de nitrogênio e estímulo do crescimento musculoesquelético e da mobilização da gordura corpórea. O tecido visceral adiposo é particularmente responsivo à somatropina. Além do aumento da lipólise, a somatropina diminui a captação de triglicérides para os estoques de gordura corporal. As concentrações séricas de IGF-I (fator de crescimento semelhante à insulina tipo 1) e IGFBP3 (proteína 3 de ligação do fator de crescimento semelhante à insulina) são aumentadas pela somatropina. Além disso, foram demonstradas as seguintes ações da somatropina:
Metabolismo lipídico: a somatropina induz os receptores de colesterol hepáticos LDL e altera o perfil de lípides séricos e lipoproteínas. Em geral, a administração de somatropina para pacientes com deficiência de hormônio de crescimento resulta em reduções nos níveis séricos de LDL e apolipoproteína B. A redução no colesterol total sérico também pode ser observada.
Metabolismo de carboidratos: a somatropina aumenta a produção de insulina, porém a glicemia em jejum geralmente não sofre alterações. Crianças com hipopituitarismo apresentam hipoglicemia de jejum. Esta condição é revertida com o tratamento com somatropina.
Metabolismo mineral e água: a deficiência de hormônio de crescimento está associada à diminuição do volume plasmático e extracelular os quais aumentam rapidamente com o tratamento com somatropina. A somatropina induz retenção de sódio, potássio e fósforo.
Metabolismo ósseo: a somatropina estimula o remodelamento ósseo. A administração de longa duração de somatropina a pacientes com deficiência de hormônio de crescimento com osteopenia resulta em aumento da densidade e do conteúdo mineral ósseo nos sítios de crescimento.
Capacidade física: o tratamento de longa duração com somatropina melhora a força muscular e a capacidade para exercícios físicos. A somatropina também aumenta o débito cardíaco, porém este mecanismo ainda não está claro. Uma diminuição da resistência vascular periférica pode contribuir para este efeito.
Propriedades farmacocinéticas
a) Estudo de biodisponibilidade absoluta do produto biológico comparador Genotropin®
Absorção
A biodisponibilidade da somatropina após administração subcutânea na coxa de 1,3 mg/mL de somatropina (0,03 mg/kg) é de aproximadamente 80% da dose em pacientes adultos com deficiência de hormônio de crescimento, em comparação à dose disponível por via intravenosa. Os resultados foram semelhantes tanto em pacientes masculinos quanto femininos. Biodisponibilidade semelhante foi observada em homens adultos saudáveis.
Em homens adultos saudáveis, uma injeção subcutânea na coxa de 0,03 mg/kg fez com que a extensão de absorção (AUC) de uma concentração de 5,3 mg/mL de somatropina fosse 35% maior do que a concentração de 1,3 mg/mL de somatropina. A média (± desvio padrão) e o pico de níveis séricos (Cmáx) foram 23,0 (± 9,4) ng/mL e 17,4 (± 9,2) ng/mL, respectivamente.
Em estudo similar envolvendo pacientes pediátricos com deficiência de hormônio de crescimento, uma dose de somatropina 5,3 mg/mL produziu uma AUC média 17% maior do que uma dose de somatropina 1,3 mg/mL. Os níveis médios de Cmáx foram 21,0 ng/mL e 16,3 ng/mL, respectivamente.
Pacientes adultos com deficiência de hormônio de crescimento receberam duas únicas doses subcutâneas de 0,03 mg/kg de somatropina numa concentração de 1,3 mg/mL, com intervalo de uma a quatro semanas entre as injeções. Os níveis médios de Cmáx foram 12,4 ng/mL (primeira injeção) e 12,2 ng/mL (segunda injeção), alcançados em aproximadamente seis horas após a administração.
Não há dados de bioequivalência entre as formulações de 1,3 mg/ mL ou 5,3 mg/ mL nem para 12 mg/ mL.
Distribuição
O volume médio de distribuição de somatropina após administração em adultos com deficiência de hormônio de crescimento foi calculado em 1,3 (± 0,8) L/kg.
Metabolismo
O metabolismo de somatropina envolve catabolismo proteico tanto no fígado como nos rins. Em células renais, uma porção dos produtos de degradação retorna à circulação sistêmica. A meia-vida terminal média de somatropina após administração intravenosa em adultos com deficiência de hormônio de crescimento é de aproximadamente 0,4 horas. Entretanto, após a administração subcutânea, a meia-vida obtida é de 2-3 horas. As diferenças observadas ocorrem devido à lenta absorção no local da injeção após a administração subcutânea.
Eliminação
A liberação média de somatropina quando administrada subcutaneamente foi de 0,3 (± 0,11) L/h/kg em 16 pacientes adultos com deficiência de hormônio de crescimento.
b) Estudo de bioequivalência entre o CRISCY e Genotropin®
Estudo clínico de fase I - A farmacocinética e a farmacodinâmica do r-hGH fabricado pelo Cristália foi avaliada de forma comparativa ao r-hGH fabricado pelos Laboratórios Pfizer Ltda, Genotropin®, em um estudo fase 1 cruzado, randomizado, aberto, de dois tratamentos, dois períodos, duas sequências e dose única subcutânea, em voluntários adultos, sadios, de ambos os gêneros. O estudo clínico fase 1 teve como parâmetro primário a comparação da farmacocinética dos produtos após administração de dose única subcutânea de 12,8 UI de r-hGH e como secundários, a comparação dos perfis farmacodinâmicos dos biomarcadores IGF-1 e IGFBP-3, avaliação de segurança pelos relatos de ocorrência e intensidade de eventos adversos e pela avaliação da tolerância local pela inspeção visual do local e relato de dor local.
Os resultados do perfil farmacocinético das formulações do hormônio de crescimento Cristália e produto comparador seguem no gráfico abaixo. Os pontos do gráfico representam a média ± SD de 34 voluntários.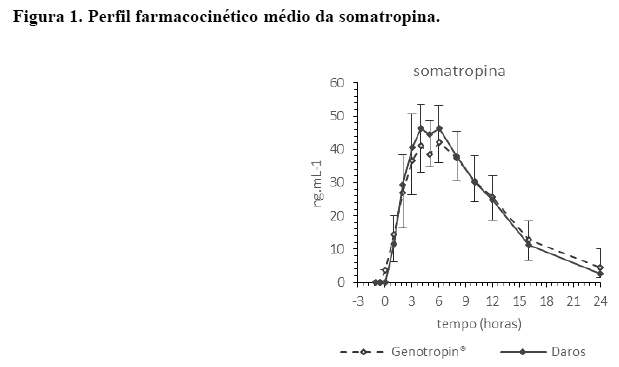
O desfecho primário, incluindo os parâmetros farmacocinéticos após administração de dose única s.c. de 12,8 UI de CRISCY e Genotropin®e comparações entre os produtos estão na tabela 6 e 7.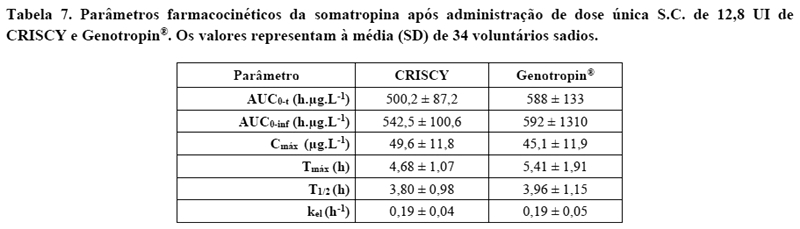
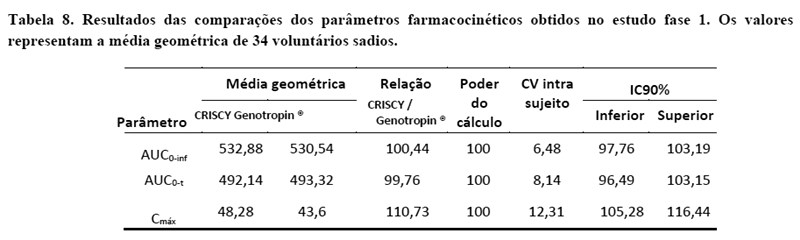
As figuras 2 e 3 e a tabela 8 ilustram o perfil farmacodinâmico das formulações CRISCY e Genotropin® e resume os resultados da comparação entre os parâmetros farmacodinâmicos do IGF-1 e IGFBP-3. Os limites de aceitação para o IC90% são 80,00% (inferior) e 125,00% (superior).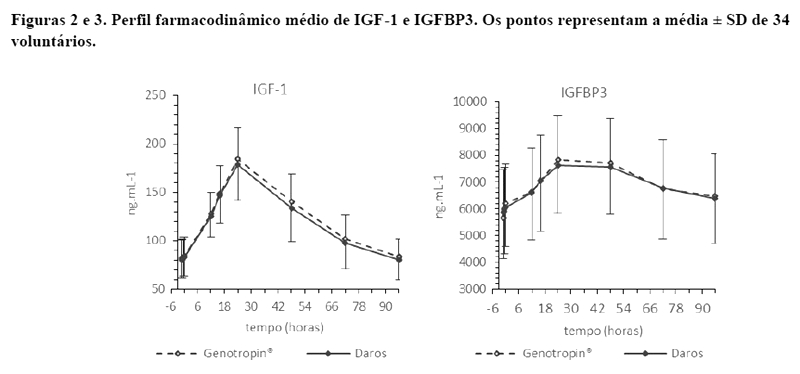
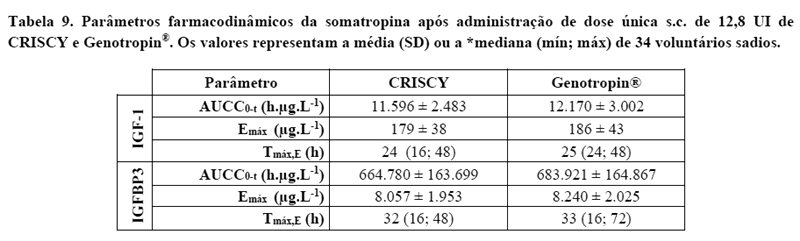
Os resultados mostraram similaridade entre os produtos no aspecto farmacocinético, farmacodinâmico e com relação à segurança.
Ref: Toffoletto O, Afiune J, Thiemann JE, Khandave SS, Patel S, Rodrigues DG. Comparative pharmacokinetic and pharmacodynamic evaluation between a new biosimilar and reference recombinant human growth hormone. Growth Horm IGF Res. outubro de 2016;30-31:31-6.
Populações especiais (Informações do produto biológico comparador Genotropin®)
Crianças: a farmacocinética de somatropina é similar em pacientes pediátricos e adultos com deficiência de hormônio de crescimento.
Gênero: não foram realizados estudos de gênero em pacientes pediátricos com deficiência de hormônio de crescimento; entretanto, em pacientes adultos com deficiência de hormônio de crescimento a biodisponibilidade absoluta da somatropina parece ser similar em homens e mulheres.
Raça: a farmacocinética da somatropina em pacientes de diferentes raças não foi estudada.
Insuficiência renal ou hepática: informações sobre a farmacocinética da somatropina em pacientes com insuficiência renal ou hepática é deficiente ou incompleta.
Dados pré-clínicos de segurança (Informações do produto biológico comparador Genotropin®)
Não foi observado efeito clinicamente relevante em estudos de toxicidade geral, tolerância local e genotoxicidade. Estudos de genotoxicidade in vitro e in vivo sobre mutações pontuais e sobre induções de aberrações cromossômicas foram negativos.
Foi observado aumento na fragilidade dos cromossomos em um estudo in vitro com linfócitos de pacientes, após tratamento de longa duração com somatropina e após a adição do fármaco radiomimético bleomicina. O significado clínico deste achado não está claro.
Em outro estudo, não foi verificado aumento de anormalidades cromossômicas nos linfócitos de pacientes que receberam tratamento de longa duração com somatropina.
4. CONTRAINDICAÇÕES
• Neoplasias
A somatropina é contraindicada a pacientes que possuam qualquer evidência de atividade neoplásica e a pacientes com crescimento não controlado de tumores intracranianos benignos. O tratamento antitumoral deve estar finalizado antes do início da terapia com somatropina.
• Síndrome de Prader-Willi em Crianças
A somatropina é contraindicada em pacientes com síndrome de Prader-Willi gravemente obesos, com história de obstrução das vias aéreas superiores ou apneia do sono, ou com comprometimento respiratório grave. Houve relatos de morte súbita quando a somatropina foi usada em tais pacientes.
• Malignidade Ativa
Em geral, a somatropina é contraindicada na presença de malignidade ativa. Qualquer doença maligna preexistente deve estar inativa e o tratamento finalizado antes de se instituir a terapia com somatropina. A somatropina deve ser descontinuada se houver evidência de atividade recorrente. Como a deficiência de hormônio do crescimento pode ser um sinal precoce da presença de um tumor hipofisário (ou, raramente, de outros tumores cerebrais), a presença de tais tumores deve ser descartada antes do início do tratamento. A somatropina não deve ser utilizada em doentes com qualquer evidência de progressão ou recorrência de um tumor intracraniano subjacente.
• Epífises Consolidadas
A somatropina não deve ser utilizada para promover crescimento em crianças com epífises consolidadas.
• Doença Crítica Aguda
O tratamento com somatropina é contraindicado em pacientes com doença crítica aguda por complicações após cirurgia cardíaca aberta, cirurgia abdominal, trauma acidental múltiplo ou insuficiência respiratória aguda.
• Hipersensibilidade
A somatropina é contraindicada em pacientes com hipersensibilidade à somatropina ou a qualquer componente da fórmula.
• Retinopatia Diabética
A somatropina é contraindicada em pacientes com retinopatia diabética proliferativa ativa ou não proliferativa grave.
Categoria de Risco: C.
Este medicamento não deve ser utilizado em mulheres grávidas sem orientação médica ou do cirurgião-dentista.
5. ADVERTÊNCIAS E PRECAUÇÕES
5.1 Intolerância à Glicose e Diabetes Mellitus
A somatropina reduz a sensibilidade à insulina e, portanto, os pacientes devem ser observados quanto à intolerância à glicose. Em raros casos, a terapia com somatropina pode produzir uma intolerância à glicose suficiente para preencher os critérios diagnósticos de diabetes mellitus tipo 2. O risco de desenvolvimento da diabetes durante o tratamento com somatropina é maior em pacientes com outros fatores de risco para a diabetes mellitus tipo 2, como obesidade, histórico familiar de diabetes, tratamento com esteroides ou distúrbio prévio de intolerância à glicose. Nos pacientes com diabetes mellitus pré-existente, pode ser necessário ajuste da dose da terapia hipoglicemiante ao se iniciar a terapia com somatropina.
5.2 Hipotireoidismo
Geralmente, os níveis de hormônios tireoidianos periféricos se mantêm dentro dos valores normais de referência para indivíduos saudáveis durante o tratamento com somatropina. Entretanto, foi observada conversão aumentada de T4 para T3, o que pode resultar na redução da concentração sérica de T4 e no aumento da concentração sérica de T3. Os efeitos da somatropina nesses níveis hormonais podem ser de relevância clínica em pacientes com hipotireoidismo central subclínico, nos quais o hipotireoidismo pode, teoricamente, se desenvolver. Inversamente, pode ocorrer hipertireoidismo leve em pacientes que recebem terapia de reposição com tiroxina. Portanto, recomenda-se avaliar a função tireoidiana após o início do tratamento com somatropina e após os ajustes de dose.
5.3 Hipoadrenalismo
A introdução ao tratamento com somatropina pode resultar na inibição da 11b-hidroxiesteróide desidrogenase tipo 1 (11b-HSD-1) e na redução das concentrações séricas de cortisol. Em pacientes tratados com somatropina, o hipoadrenalismo central (secundário), anteriormente não diagnosticado, pode ser desmascarado e pode ser necessária a substituição de glicocorticoides. Além disso, os pacientes tratados com terapia de substituição com glicocorticoides para o hipoadrenalismo previamente diagnosticado podem necessitar de um aumento das suas doses de manutenção ou dose de estress, após o início do tratamento com somatropina (vide item 6. Interações medicamentosas).
5.4 Estrogênio Oral
Se uma mulher que toma a somatropina iniciar a terapia oral com estrogênios, a dose de somatropina pode necessitar de ser aumentada para manter os níveis séricos do fator de crescimento semelhante à insulina-I (IGF-I) dentro do intervalo adequado à idade normal. Por outro lado, se uma mulher sob somatropina interromper a terapia oral com estrogênios, a dose de somatropina poderá ter de ser reduzida para evitar o excesso de hormônio do crescimento e / ou efeitos secundários (vide item 6. Interações medicamentosas).
5.5 Recidiva de Malignidade
Nos casos de deficiência do hormônio de crescimento secundária ao tratamento de doenças neoplásicas, recomenda-se especial atenção aos sinais de possível recidiva de malignidade.
5.6 Deslizamento da Epífise Femoral Proximal em Pacientes Pediátricos
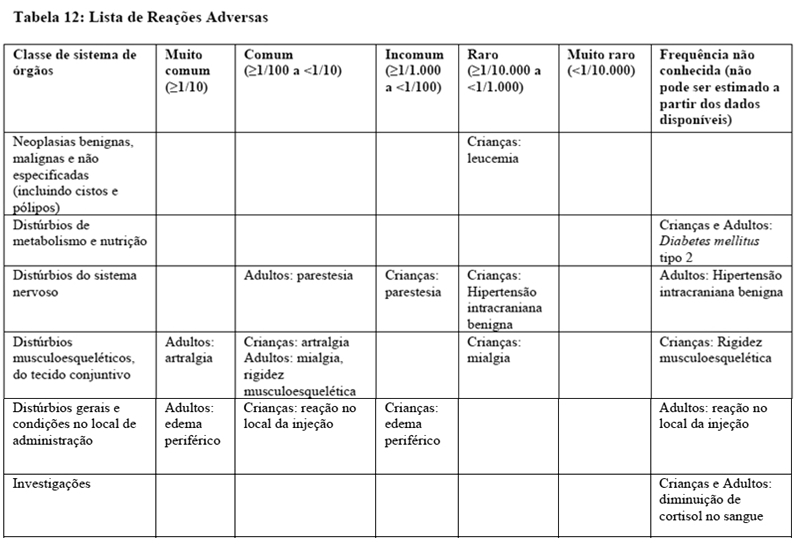
Pode ocorrer, com maior frequência, deslizamento da epífise femoral proximal em pacientes com distúrbios endócrinos, incluindo deficiência de hormônio de crescimento. Toda criança que desenvolver claudicação durante o tratamento com somatropina deve ser avaliada (vide item 9. Reações Adversas).
5.7 Hipertensão Intracraniana
No caso de cefaleia grave ou recorrente, alterações visuais, náusea e/ou vômitos, recomenda-se fundoscopia para detecção de papiledema. Caso seja confirmado o papiledema, deve ser considerado o diagnóstico de hipertensão intracraniana benigna e o tratamento com hormônio de crescimento deve ser descontinuado, se apropriado. Até o momento, não há evidências suficientes para orientar a continuação ou não da terapia com hormônio de crescimento em pacientes com hipertensão intracraniana resolvida. Porém, a experiência clínica demonstrou que a reintrodução da terapia é possível frequentemente sem recorrência de hipertensão intracraniana. Se o tratamento com este hormônio for reiniciado, exige-se monitoração cuidadosa para sintomas de hipertensão intracraniana.
5.8 Síndrome de Prader-Willi em Crianças
Em pacientes com síndrome de Prader-Willi, o tratamento deve ser sempre acompanhado por uma dieta de restrição calórica.
Foi relatada morte associada ao uso de hormônio de crescimento em pacientes com síndrome de Prader-Willi que apresentaram um ou mais dos seguintes fatores de risco: obesidade grave, histórico de insuficiência respiratória, apneia do sono ou infecção respiratória não identificada. Pacientes masculinos com um ou mais destes fatores podem ter o risco aumentado. Antes do início do tratamento com somatropina em pacientes com síndrome de Prader-Willi, sinais de obstrução das vias aéreas superiores, apneia do sono ou infecções respiratórias devem ser avaliadas. Caso sejam observadas alterações durante avaliação da obstrução das vias aéreas superiores, a criança deve ser encaminhada a um otorrinolaringologista para o tratamento e resolução do distúrbio respiratório antes do início do tratamento com somatropina.
Antes do início do tratamento com somatropina, a apneia do sono deve ser avaliada, através de métodos reconhecidos como polissonografia ou oximetria noturna; o paciente deve ser monitorado em caso de suspeita de apneia do sono.
Caso os pacientes apresentem obstrução das vias aéreas superiores (incluindo início ou aumento de ronco) durante o tratamento com somatropina, este deve ser interrompido. E deve ser realizada uma nova avaliação com o otorrinolaringologista. Todos os pacientes com síndrome de Prader-Willi devem ser monitorados caso haja suspeita de apneia do sono.
Os pacientes devem ser monitorados quanto a sinais de infecção respiratória, que devem ser diagnosticados assim que possível e tratados agressivamente.
Escoliose é comumente observada em pacientes com síndrome de Prader-Willi. Pode ocorrer progressão de escoliose em pacientes que apresentam crescimento rápido. Uma vez que a somatropina aumenta a taxa de crescimento, os médicos devem estar atentos a essa anormalidade, que pode se manifestar durante a terapia com hormônio de crescimento. Desta forma, sinais de escoliose devem ser monitorados durante o tratamento. Apesar disso, o tratamento com hormônio de crescimento não demonstrou aumentar a incidência ou gravidade da escoliose.
A experiência com tratamentos prolongados em pacientes adultos ou com síndrome de Prader-Willi é limitada.
5.9 Crianças de Baixa Estatura Nascidas PIG (Pequenas para a Idade Gestacional)
Devem ser consideradas outras razões médicas ou tratamentos que possam explicar o distúrbio de crescimento em crianças com baixa estatura nascidas PIG antes do início do tratamento com somatropina.
Em crianças nascidas PIG é recomendável avaliar a insulina e glicemia em jejum antes do início do tratamento e anualmente após o início do mesmo. Em pacientes com risco aumentado de diabetes mellitus (por exemplo, histórico familiar de diabetes, obesidade, resistência grave à insulina, acantosis nigricans) deve ser realizado o teste oral de tolerância à glicose. Caso o diabetes seja comprovado, a somatropina não deve ser administrada.
Em crianças PIG, é recomendada a avaliação dos níveis de IGF-I antes do início do tratamento e após isso, 2 vezes por ano. Caso os níveis de IGF-I em avaliações repetidas excedam + 2 DP comparadas às referências de acordo com a idade e o status puberal, a razão IGF-I/ IGFBP-3 pode ser utilizada para considerar ajuste de dose.
A experiência no tratamento em pacientes nascidos PIG perto do início da puberdade é limitada. Portanto, o início do tratamento nesta idade não é recomendado. A experiência em pacientes com síndrome de Silver-Russel também é limitada.
O ganho em altura em pacientes de baixa estatura nascidos PIG tratados com hormônio de crescimento pode ser perdido caso o tratamento seja interrompido antes que a altura final seja atingida.
5.10 Insuficiência Renal
Em pacientes com insuficiência renal crônica, a função renal deve estar 50% abaixo do normal antes da instituição da terapia com somatropina. Para se verificar o distúrbio de crescimento, o crescimento deve ser acompanhado por um ano antes da instituição do tratamento. Uma terapia conservadora para insuficiência renal deve ser estabelecida e mantida durante o tratamento com somatropina. O tratamento conservador para insuficiência renal deve ser estabelecido e mantido durante o tratamento com hormônio de crescimento. Deve-se descontinuar o tratamento com somatropina em caso de transplante renal.
5.11 Doença Crítica Aguda
Aumento da mortalidade em pacientes com doença crítica aguda devido a complicações após cirurgia cardíaca aberta, cirurgia abdominal ou trauma acidental múltiplo, ou aqueles com insuficiência respiratória aguda foi relatado após o tratamento com quantidades farmacológicas de somatropina (vide item 4. Contraindicações). Dois ensaios clínicos controlados por placebo em pacientes adultos com deficiência de hormônio não relacionado ao crescimento (n = 522) com estas condições em unidades de cuidados intensivos revelaram um aumento significativo da mortalidade (42% vs. 19%) entre pacientes tratados com somatropina (doses de 5,3 a 8 mg/dia) em comparação com aqueles que receberam placebo. A segurança da continuação do tratamento com somatropina em pacientes que recebem dos es de reposição para as indicações aprovadas e que desenvolvem concomitantemente estas doenças não foi estabelecida. Portanto, o potencial benefício da continuação do tratamento com somatropina em pacientes com doenças críticas agudas deve ser ponderado em relação ao risco potencial.
5.12 Baixa Estatura Idiopática
Pacientes com baixa estatura idiopática, com a progressão da idade cronológica, a velocidade de crescimento diminui naturalmente, com ou sem o uso de somatropina. Em pacientes pediátricos, a falha em aumentar a velocidade de crescimento, particularmente durante o 1° ano da terapia, indica a necessidade de uma avaliação da adesão ao tratamento e avaliação de outras causas de falha de crescimento, tais como hipotireoidismo, subnutrição, idade óssea avançada e anticorpos contra o rhGH.
5.13 Reações Graves de Hipersensibilidade
Foram notificadas reações graves de hipersensibilidade sistê