CONTRAVE
MERCK
naltrexona
Tratamento da obesidade.
Apresentações.
Embalagem com 120 comprimidos revestidos de liberação prolongada.
USO ORAL
USO ADULTO
Composição.
Cada comprimido revestido de liberação prolongada contém 8 mg de cloridrato de naltrexona e 90 mg de cloridrato de bupropiona. Excipientes: cloridrato de cisteína monoidratado, celulose microcristalina, hiprolose, estearato de magnésio, lactose, lactose monoidratada, crospovidona, azul de indigotina 132 laca de alumínio, hipromelose, edetato dissódico di-hidratado, dióxido de silício, álcool polivinílico, dióxido de titânio, macrogol, talco.
Informações técnicas.
1. INDICAÇÕES
CONTRAVE® é indicado como adjuvante de dieta hipocalórica e aumento da atividade física para controle crônico de peso em adultos com índice de massa corporal (IMC) inicial de:
- 30 kg/m2 ou acima (obeso) ou
- 27 kg/m2 a 30 kg/m2 (sobrepeso) na presença de pelo menos uma comorbidade relacionada ao peso (por exemplo, hipertensão, diabetes mellitus tipo 2 ou dislipidemia).
Limitações de uso:
- O efeito de CONTRAVE® na morbidade e mortalidade cardiovascular não foi estabelecido.
- Não foram estabelecidas a segurança e a eficácia de CONTRAVE® em combinação com outros produtos destinados à perda de peso, incluindo medicamentos de prescrição, medicamentos isentos de prescrição e preparações à base de plantas.
2. RESULTADOS DE EFICÁCIA
Os efeitos de CONTRAVE® na perda de peso em conjunto com a redução da ingestão calórica e aumento da atividade física foram estudados em estudos duplo-cegos, controlados por placebo (faixa de IMC 27 a 45 kg/m2) com durações de estudo de 16 a 56 semanas, randomizados para naltrexona (16 a 50 mg/dia) e/ou bupropiona (300 a 400 mg/dia) ou placebo.
Efeito na perda de peso e manutenção do peso
Quatro estudos multicêntricos, duplo-cegos, controlados com placebo, de 56 semanas para obesidade (CONTRAVE® Obesity Research, ou COR-I, COR-II, COR-BMOD e COR-Diabetes) foram conduzidos para avaliar o efeito de CONTRAVE® em conjunto com mudança do estilo de vida em 4.536 pacientes randomizados para CONTRAVE® ou placebo. Os estudos COR-I, COR-II e COR-BMOD incluíram pacientes com obesidade (IMC 30 kg/m2 ou acima) ou sobrepeso (IMC 27 kg/m2 ou acima) e pelo menos uma comorbidade (hipertensão ou dislipidemia). O estudo COR-Diabetes envolveu pacientes com IMC maior que 27 kg/m2 com diabetes tipo 2 com ou sem hipertensão e/ou dislipidemia.
O tratamento foi iniciado com um período de titulação de dose de três semanas, seguido de aproximadamente 1 ano de tratamento contínuo. Os pacientes foram instruídos a tomar CONTRAVE® com alimentos. COR-I e COR-II incluíram um programa consistindo em uma dieta reduzida em calorias, resultando em uma diminuição aproximada de 500 kcal/dia na ingestão calórica, aconselhamento comportamental e aumento da atividade física. O COR-BMOD incluiu um programa intensivo de mudança comportamental, consistindo em 28 sessões de aconselhamento em grupo durante 56 semanas, bem como uma dieta prescrita e esquema de exercícios. O COR-Diabetes avaliou pacientes com diabetes tipo 2 que não atingiram o objetivo glicêmico de HbA1c menor que 7%, tanto com agentes antidiabéticos orais quanto com dieta e exercícios isolados. Da população total desses quatro estudos, 24% tinham hipertensão, 54% tinham dislipidemia no início do estudo e 10% tinham diabetes tipo 2.
Além do COR-Diabetes, que incluiu apenas pacientes com diabetes tipo 2, as características demográficas dos pacientes foram semelhantes nos quatro estudos. Para as quatro populações experimentais combinadas, a idade média foi de 46 anos, 83% eram do sexo feminino, 77% eram caucasianos, 18% eram negros e 5% eram outras raças. No início do estudo, o IMC médio foi de 36 kg/m2 e a circunferência média da cintura foi de 110 cm.
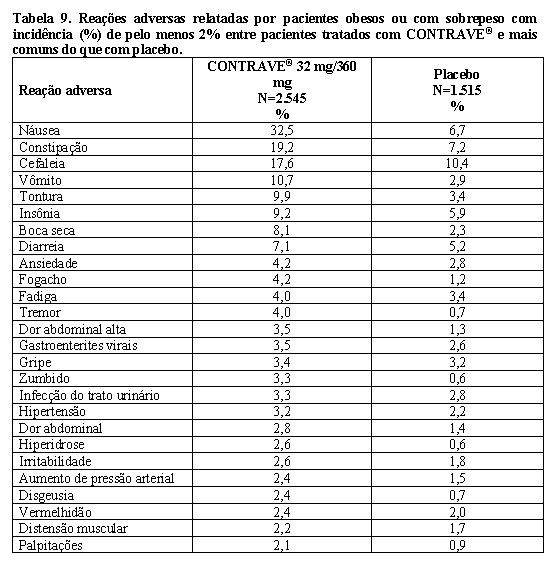
Uma porcentagem substancial de pacientes randomizados retirou-se dos estudos antes da semana 56: 45% do grupo placebo e 46% do grupo CONTRAVE®. A maioria desses pacientes interrompeu dentro das primeiras 12 semanas de tratamento. Aproximadamente 24% dos pacientes tratados com CONTRAVE® e 12% dos pacientes tratados com placebo descontinuaram o tratamento devido a uma reação adversa (ver "Reações adversas").
Os desfechos co-primários foram a alteração percentual do peso corporal inicial e a proporção de pacientes que atingiram pelo menos uma redução de 5% no peso corporal. No estudo COR-I de 56 semanas, a alteração média no peso corporal foi de -5,4% entre os pacientes randomizados para CONTRAVE® 32 mg/360 mg, comparado com -1,3% entre os pacientes designados para placebo (População com Intenção de Tratar [ITT]), como mostrado na Tabela 1 e Figura 1. Neste estudo, a redução de pelo menos 5% do peso corporal em relação ao valor basal ocorreu com maior frequência em pacientes tratados com CONTRAVE® 32 mg/360 mg em comparação com placebo (42% vs. 17%; Tabela 1). Os resultados de COR-BMOD e COR-Diabetes são mostrados na Tabela 1 e Figuras 2 e 3.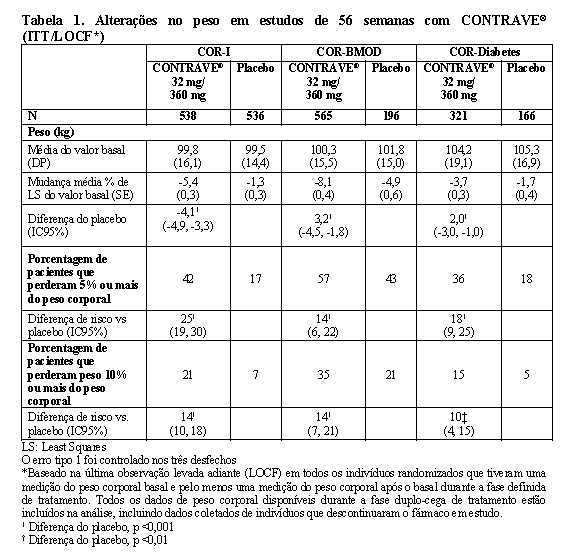
As percentagens de pacientes que atingiram a perda de, pelo menos, 5% ou pelo menos 10% do peso corporal desde o basal foram superiores entre os randomizados para CONTRAVE®, comparativamente ao placebo, nos quatro estudos de obesidade (Tabela 1).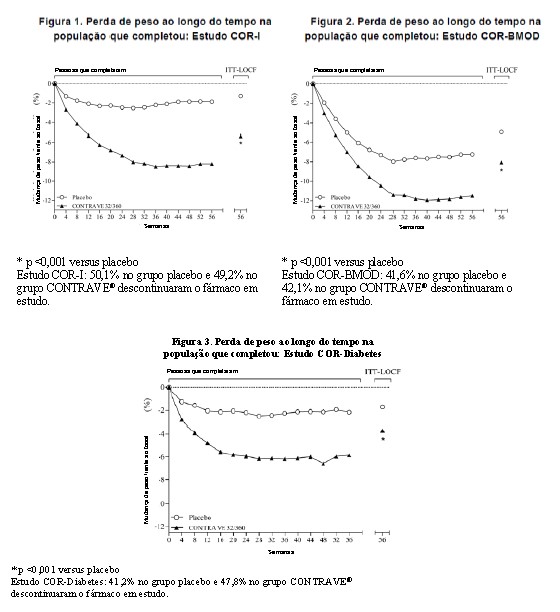
Efeito nos parâmetros cardiovasculares e metabólicos
Alterações nos parâmetros cardiovasculares e metabólicos associados à obesidade são apresentadas para COR-I e COR-BMOD (Tabela 2). Alterações na pressão sanguínea média e frequência cardíaca são descritas em outra seção (ver "Advertências e precauções").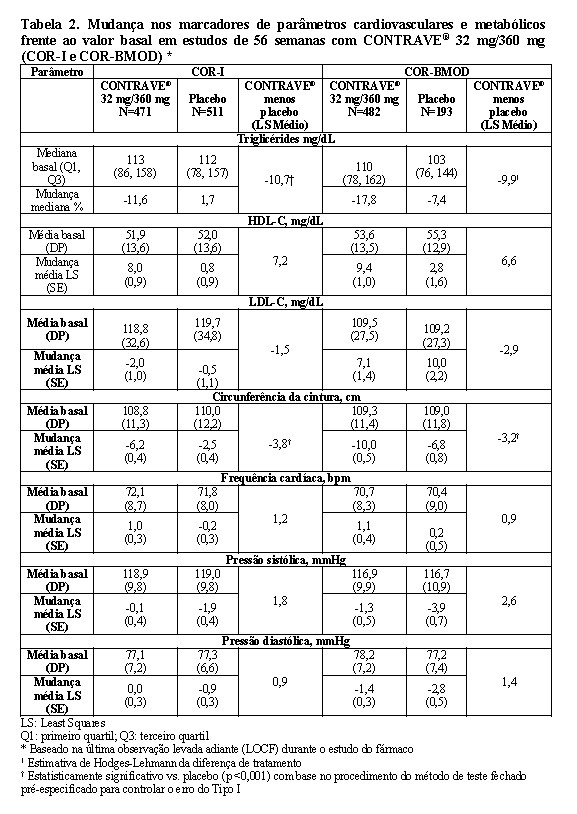
Efeito do CONTRAVE® nos parâmetros cardiometabólicos e na antropometria em pacientes com diabetes mellitus tipo 2
As alterações no controle da glicemia observadas desde o início até a semana 56 entre os pacientes com diabetes tipo 2 e obesidade, randomizados para CONTRAVE® 32 mg/360 mg ou placebo são apresentadas na Tabela 3.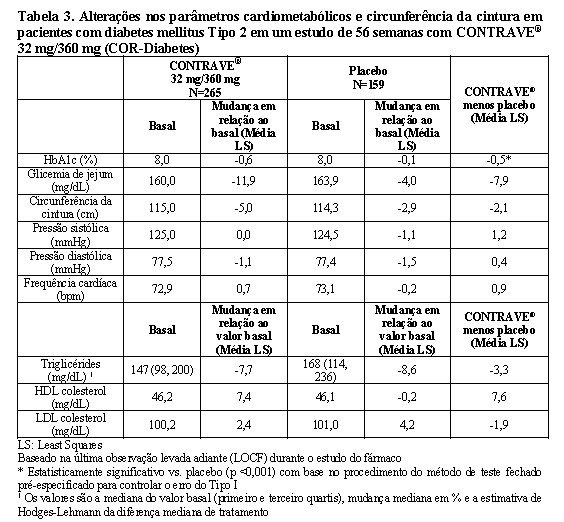
Efeito na composição corporal
Em um subconjunto de 124 pacientes (79 CONTRAVE®, 45 placebo), a composição corporal foi medida usando a absorciometria por raios-X de dupla energia (DEXA). A avaliação DEXA mostrou que a massa gorda corporal média total diminuiu em 4,7 kg (11,7%) no grupo CONTRAVE® vs. 1,4 kg (4,3%) no grupo placebo na semana 52/LOCF (diferença de tratamento, -3,3 kg [-7,4%], p < 0,01).
Referências:
1. Greenway FL, Fujioka K, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376(9741):595-605.
2. Apovian CM, Aronne L, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring). 2013;21(5):935-943.
3. Wadden TA, Foreyt JP, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: The COR-BMOD trial. Obesity (Silver Spring). 2011;19(1):110-120.
4. Hollander P, Gupta AK, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36(12):4022-4029.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
CONTRAVE® tem dois componentes ativos: naltrexona, um antagonista opioide, e bupropiona, um inibidor relativamente fraco da recaptação neuronal de dopamina e norepinefrina. Estudos não-clínicos sugerem que a naltrexona e a bupropiona têm efeitos em duas áreas distintas do cérebro envolvidas na regulação da ingestão de alimentos: o hipotálamo (centro regulador do apetite) e o circuito mesolímbico da dopamina (sistema de recompensa). Os efeitos neuroquímicos exatos de CONTRAVE® que levam à perda de peso não são totalmente compreendidos.
Farmacodinâmica
Combinadas, a bupropiona e a naltrexona aumentaram, in vitro, a taxa de disparos dos neurônios hipotalâmicos da pro-opiomelanocortina (POMC), que estão associados à regulação do apetite.
A combinação de bupropiona e naltrexona também reduziu a ingestão de alimentos quando injetada diretamente na área tegmental ventral do circuito mesolímbico em camundongos, uma área associada à regulação das vias de recompensa.
Farmacocinética
Absorção
Naltrexona
Após administração oral única de CONTRAVE® (dois comprimidos de 8 mg de naltrexona/90 mg de bupropiona) a indivíduos saudáveis, a concentração plasmática máxima média de naltrexona (Cmáx) foi de 1,4 ng/mL, o tempo até a concentração máxima (Tmáx) foi de 2 horas e a extensão da exposição (ASC0-inf) foi de 8,4 ng.h/mL.
Bupropiona
Após administração oral única de CONTRAVE® (dois comprimidos de 8 mg de naltrexona/90 mg de bupropiona) a indivíduos saudáveis, a concentração plasmática máxima média de bupropiona (Cmáx) foi de 168 ng/mL, o tempo até a concentração plasmática máxima (Tmáx) foi de três horas e a extensão da exposição (ASC0-inf) foi de 1.607 ng.h/mL.
Efeito dos alimentos na absorção
Quando CONTRAVE® foi administrado com uma refeição rica em gorduras, a ASC e a Cmáx da naltrexona aumentaram 2,1 vezes e 3,7 vezes, respetivamente, e a ASC e a Cmáx da bupropiona aumentaram 1,4 vezes e 1,8 vezes, respetivamente. No estado de equilíbrio, o efeito do alimento aumentou a ASC e a Cmáx da naltrexona em 1,7 vezes e 1,9 vezes, respetivamente, e aumentou a ASC e a Cmáx da bupropiona em 1,1 vezes e 1,3 vezes, respetivamente. Assim, o CONTRAVE® não deve ser tomado com refeições com alto teor de gordura, devido aos significativos aumentos na exposição sistêmica à bupropiona e à naltrexona.
Distribuição
Naltrexona
A naltrexona é 21% ligada às proteínas plasmáticas. O volume aparente médio de distribuição da naltrexona no estado de equilíbrio (Vss/F) é de 5.697 litros.
Bupropiona
A bupropiona é 84% ligada às proteínas plasmáticas. O volume aparente médio de distribuição da bupropiona no estado de equilíbrio (Vss/F) é de 880 litros.
Metabolismo e excreção
Naltrexona
O principal metabolito da naltrexona é o 6-beta-naltrexol. Acredita-se que a atividade da naltrexona seja o resultado tanto do composto original quanto do metabólito 6-beta-naltrexol. Embora menos potente, o 6-beta-naltrexol é eliminado mais lentamente e, portanto, circula em concentrações muito mais elevadas do que a naltrexona. A naltrexona e o 6-beta-naltrexol não são metabolizados pelas enzimas do citocromo P450 e estudos in vitro indicam que não há potencial para inibição ou indução de isoenzimas importantes.
A naltrexona e seus metabólitos são excretados principalmente pelo rim (53% a 79% da dose). A excreção urinária de naltrexona inalterada é responsável por menos de 2% de uma dose oral. A excreção urinária de 6-beta-naltrexol inalterado e conjugado é responsável por 43% de uma dose oral. A depuração renal da naltrexona varia de 30 a 127 mL/min, sugerindo que a eliminação renal é principalmente por filtração glomerular. A depuração renal do 6-beta-naltrexol varia de 230 a 369 mL/min, sugerindo um mecanismo adicional de secreção tubular renal. A excreção fecal é uma via de eliminação menor.
Após administração oral única de comprimidos de CONTRAVE® a indivíduos saudáveis, a meia-vida de eliminação (T1/2) foi de aproximadamente 5 horas para a naltrexona. Após a administração duas vezes por dia de CONTRAVE®, a naltrexona não se acumulou e a sua cinética pareceu ser linear.
No entanto, em comparação com naltrexona, o 6-beta-naltrexol se acumula em maior extensão (taxa de acumulação ~ 3).
Bupropiona
A bupropiona é extensamente metabolizada em três metabólitos ativos: hidroxibupropiona, treoidrobupropiona e eritroidrobupropiona. Os metabólitos têm uma eliminação mais longa do que a bupropiona e se acumulam em maior extensão. Após a administração de bupropiona, mais de 90% da exposição é resultado de metabólitos. Os achados in vitro sugerem que o CYP2B6 é a principal isoenzima envolvida na formação da hidroxibupropiona, enquanto as isoenzimas do citocromo P450 não estão envolvidas na formação dos outros metabólitos ativos.
A bupropiona e seus metabólitos inibem o CYP2D6. A ligação às proteínas plasmáticas da hidroxibupropiona é semelhante à da bupropiona (84%), enquanto que os outros dois metabolitos têm aproximadamente metade desta ligação.
Após a administração oral de 200 mg de 14C-bupropiona em seres humanos, 87% e 10% da dose radioativa foram recuperados na urina e nas fezes, respectivamente. A fração da dose oral de bupropiona excretada inalterada foi de 0,5%, um achado consistente com o amplo metabolismo da bupropiona.
Após administração oral única de comprimidos de CONTRAVE® a indivíduos saudáveis, a meia-vida de eliminação (T½) foi de aproximadamente 21 horas para a bupropiona. Após a administração duas vezes por dia de CONTRAVE®, os metabolitos de bupropiona e, em menor grau, de bupropiona inalterada, acumulam-se e atingem as concentrações do estado de equilíbrio em aproximadamente uma semana.
Populações específicas
Gênero
A análise agrupada dos dados de CONTRAVE® não sugeriu diferenças clinicamente significativas nos parâmetros farmacocinéticos da bupropiona ou da naltrexona com base no sexo.
Raça
A análise agrupada dos dados de CONTRAVE® não sugeriu diferenças clinicamente significativas nos parâmetros farmacocinéticos da bupropiona ou da naltrexona com base na raça.
Idosos
A farmacocinética do CONTRAVE® não foi avaliada na população geriátrica. Os efeitos da idade sobre a farmacocinética da naltrexona ou bupropiona e seus metabólitos não foram totalmente caracterizados. Uma exploração das concentrações de bupropiona no estado de equilíbrio em vários estudos de eficácia sobre a depressão envolvendo pacientes tratados numa faixa de 300 a 750 mg/dia, em um esquema de três tomadas diárias, não revelou relação entre a idade (18 a 83 anos) e concentração plasmática de bupropiona. Um estudo farmacocinético de dose única demonstrou que a distribuição da bupropiona e dos seus metabolitos em idosos foi semelhante à dos indivíduos mais jovens. Esses dados sugerem que não há efeito significativo da idade sobre a concentração de bupropiona; entretanto, outro estudo farmacocinético, de dose única e múltipla, sugeriu que os idosos estão em maior risco de acúmulo de bupropiona e seus metabólitos (ver "Uso em populações específicas").
Fumantes
Uma análise agrupada dos dados de CONTRAVE® não revelou diferenças significativas nas concentrações plasmáticas de bupropiona ou naltrexona em fumantes em comparação com não fumantes. Os efeitos do tabagismo sobre a farmacocinética da bupropiona foram estudados em 34 voluntários saudáveis do sexo masculino e feminino; 17 eram tabagistas crônicos e 17 não tabagistas. Após a administração oral de uma dose única de 150 mg de bupropiona, não houve diferença estatisticamente significativa na Cmáx, meia-vida, Tmáx, ASC ou depuração da bupropiona ou dos seus metabolitos ativos entre fumantes e não-fumantes.
Insuficiência hepática
Os dados farmacocinéticos de CONTRAVE® em pacientes com insuficiência hepática não estão disponíveis.
As seguintes informações estão disponíveis para os princípios ativos individuais:
Naltrexona
Observou-se um aumento na ASC de naltrexona de aproximadamente 5 e 10 vezes em pacientes com cirrose hepática compensada e descompensada, respectivamente, em comparação com indivíduos com função hepática normal. Esses dados também sugerem que alterações na biodisponibilidade da naltrexona estão relacionadas à gravidade da doença hepática.
Bupropiona
O efeito da insuficiência hepática na farmacocinética da bupropiona foi caracterizado em dois estudos de dose única, um em pacientes com doença hepática alcoólica e um segundo em pacientes com cirrose leve a grave.
O primeiro estudo mostrou que a meia-vida da hidroxibupropiona foi significativamente maior em oito pacientes com doença hepática alcoólica do que em oito voluntários saudáveis (32±14 horas vs. 21±5 horas, respectivamente). Embora não estatisticamente significativas, as ASCs para bupropiona e hidroxibupropiona foram mais variáveis e tenderam a ser maiores (entre 53% e 57%) em pacientes com doença hepática alcoólica. As diferenças na meia-vida da bupropiona e dos outros metabólitos nos dois grupos de pacientes foram mínimas.
O segundo estudo clínico não demonstrou diferenças estatisticamente significativas na farmacocinética da bupropiona e dos seus metabolitos ativos em nove indivíduos com cirrose hepática leve a moderada, em comparação com oito voluntários saudáveis. No entanto, foi observada uma maior variabilidade em alguns dos parâmetros farmacocinéticos da bupropiona (ASC, Cmáx e Tmáx) e nos seus metabolitos ativos (t½) em indivíduos com cirrose hepática leve a moderada. Em indivíduos com cirrose hepática grave, foram observadas alterações significativas na farmacocinética da bupropiona e seus metabólitos (Tabela 4).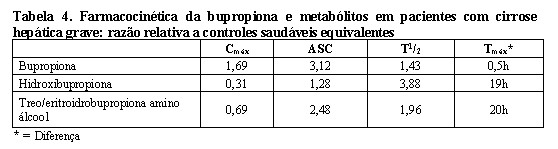
A dose de CONTRAVE® deve ser reduzida em pacientes com insuficiência hepática (ver "Posologia e administração" e "Uso em populações específicas".
Insuficiência renal
Não foi realizado um estudo farmacocinético dedicado para CONTRAVE® em indivíduos com insuficiência renal. As seguintes informações estão disponíveis para os princípios ativos individuais:
Naltrexona
Estão disponíveis informações limitadas para naltrexona em pacientes com insuficiência renal moderada a grave. Em um estudo de sete pacientes com doença renal em estágio terminal que necessitavam de diálise, as concentrações plasmáticas máximas de naltrexona foram elevadas em pelo menos 6 vezes em comparação com indivíduos saudáveis.
Bupropiona
Estão disponíveis informações limitadas para bupropiona em pacientes com insuficiência renal moderada a grave. Uma comparação entre estudos com indivíduos normais e pacientes com insuficiência renal terminal demonstrou que os valores de Cmáx e ASC da bupropiona foram comparáveis nos dois grupos, enquanto que os metabólitos hidroxibupropiona e treohidrobupropiona tiveram um aumento de 2,3 e 2,8 vezes, respectivamente, na ASC para pacientes com insuficiência renal terminal. Um segundo estudo, comparando indivíduos normais e pacientes com insuficiência renal moderada a grave (TFG 30,9 ± 10,8 mL/min), mostrou que a exposição após uma dose única de 150 mg de bupropiona foi aproximadamente 2 vezes maior em pacientes com função renal comprometida, enquanto os níveis dos metabólitos hidroxibupropiona e treo/eritroidrobupropiona (combinados) foram semelhantes nos dois grupos. A eliminação da bupropiona e/ou dos principais metabolitos da bupropiona pode ser reduzida pelo comprometimento da função renal.
A dose de CONTRAVE® deve ser reduzida em pacientes com insuficiência renal moderada ou grave. O uso de CONTRAVE® não é recomendado em pacientes com insuficiência renal terminal (ver "Posologia e administração" e Uso em populações específicas").
Interações medicamentosas
Avaliação in vitro de interações medicamentosas
Em concentrações terapeuticamente relevantes, naltrexona e 6-beta-naltrexol não são os principais inibidores das isoformas de CYP CYP1A2, CYP2B6, CYP2C8, CYP2E1, CYP2C9, CYP2C19, CYP2D6 ou CYP3A4. Tanto a naltrexona quanto o 6-beta-naltrexol não são os principais indutores das isoformas de CYP CYP1A2, CYP2B6 ou CYP3A4.
A bupropiona e seus metabólitos (hidroxibupropiona, eritroidrobupropiona, treoidrobupropiona) são inibidores da CYP2D6.
Estudos in vitro sugerem que a paroxetina, a sertralina, a norfluoxetina, a fluvoxamina e o nelfinavir inibem a hidroxilação da bupropiona.
Bupropiona (CI50 9,3 mcM) e seus metabólitos, hidroxibupropiona (CI50 82 mcM) e a treohidrobupropiona e a eritroidrobupropiona (mistura 1:1; CI50 7,8 mcM) inibiram o transportador orgânico renal OCT2 a um nível clinicamente relevante. É provável que concentrações sistêmicas de fármaco substrato transportado por OCT2 aumentem como resultado da depuração renal reduzida quando coadministrada com CONTRAVE®.
Efeitos da naltrexona/bupropiona na farmacocinética de outros fármacos
A interação medicamentosa entre CONTRAVE® e os substratos da CYP2D6 (metoprolol) ou outros fármacos (atorvastatina, gliburida, lisinopril, nifedipino, valsartana) foi avaliada. Além disso, a interação medicamentosa entre a bupropiona, um componente do CONTRAVE®, e substratos de CYP2D6 (desipramina) ou outros fármacos (citalopram, lamotrigina) também foi avaliada.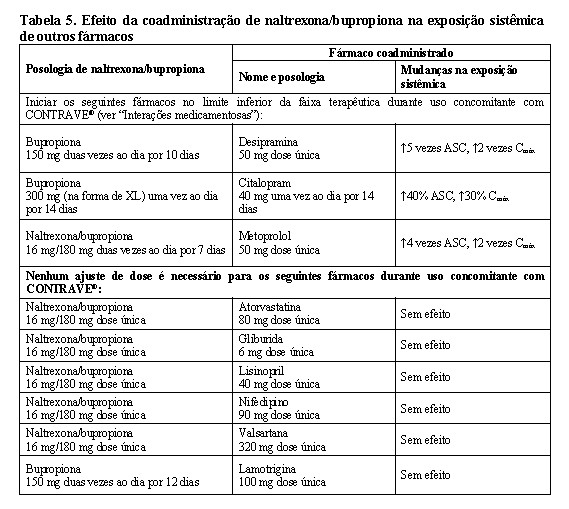
Digoxina: Os dados da literatura mostraram que a exposição à digoxina foi reduzida quando uma dose oral única de 0,5 mg de digoxina foi administrada 24 horas após uma dose oral única de 150 mg de bupropiona de libertação prolongada em voluntários saudáveis.
Efeitos de outros fármacos na farmacocinética da naltrexona/bupropiona
Interações medicamentosas entre inibidores da CYP2B6 (ticlopidina, clopidogrel, prasugrel), indutores da CYP2B6 (ritonavir, lopinavir) e bupropiona (um dos componentes de CONTRAVE®), ou entre outros fármacos (atorvastatina, gliburida, metoprolol, lisinopril, nifedipino, valsartana) e CONTRAVE® foram avaliados. Embora não seja sistematicamente estudada, carbamazepina, fenobarbital ou fenitoina podem induzir o metabolismo da bupropiona.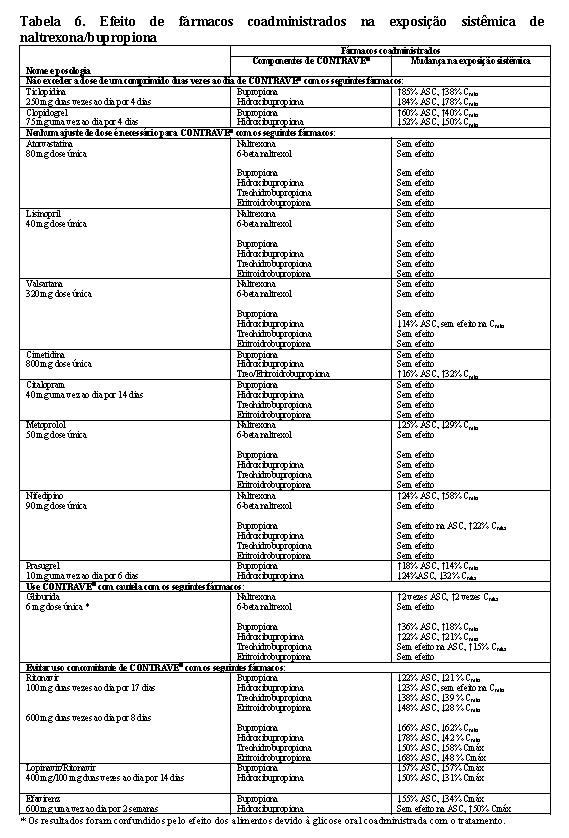
TOXICOLOGIA NÃO-CLÍNICA
Carcinogênese, mutagênese, prejuízo da fertilidade
Estudos para avaliar carcinogênese, mutagênese ou prejuízo da fertilidade com os fármacos combinados de CONTRAVE® não foram conduzidos. Os achados a seguir são de estudos realizados individualmente com naltrexona e bupropiona. Os potenciais efeitos carcinogênicos, mutagênicos e de fertilidade do metabólito 6-beta-naltrexol são desconhecidos. As margens de segurança foram estimadas usando a exposição da área de superfície corporal (mg/m2) com base em um peso corporal de 100 kg.
Em um estudo de carcinogenicidade de dois anos com naltrexona em ratos, houve pequenos aumentos no número de mesoteliomas testiculares em machos e tumores de origem vascular em machos e fêmeas. A incidência de mesotelioma em machos que receberam naltrexona em uma dose de 100 mg/kg/dia (aproximadamente 50 vezes a dose terapêutica recomendada em mg/m2 para a dose de manutenção de naltrexona para CONTRAVE®) foi de 6%, comparado com um histórico de incidência máxima de 4%. A incidência de tumores vasculares em machos e fêmeas que receberam doses dietéticas de 100 mg/kg/dia foi de 4%, mas apenas a incidência no sexo feminino foi aumentada em comparação com um controle histórico de incidência máxima de 2%. Não houve evidência de carcinogenicidade em um estudo dietético de dois anos com naltrexona em camundongos machos e fêmeas.
Estudos de carcinogenicidade da bupropiona ao longo da vida foram realizados em ratos e camundongos com doses de até 300 e 150 mg/kg/dia, respetivamente. Estas doses são aproximadamente 15 e 3 vezes a dose humana máxima recomendada (MRHD) do componente de bupropiona em CONTRAVE®, respectivamente, numa base de mg/m2. No estudo com ratos houve um aumento nas lesões proliferativas nodulares do fígado em doses de 100 a 300 mg/kg/dia (aproximadamente 5 a 15 vezes a MRHD do componente bupropiona em CONTRAVE® com base em mg/m2); doses mais baixas não foram testadas. A questão se essas lesões podem ou não ser precursoras de neoplasias do fígado ainda não está resolvida. Lesões hepáticas semelhantes não foram observadas no estudo com camundongos, e nenhum aumento em tumores malignos do fígado e outros órgãos foi visto em ambos os estudos.
Houve evidência limitada de um efeito genotóxico fraco da naltrexona em um ensaio de mutação genética em uma linhagem celular de mamíferos, no ensaio letal recessivo de Drosophila, e em testes não específicos de reparo de DNA com E. coli. No entanto, nenhuma evidência de potencial genotóxico foi observada em uma série de outros testes in vitro, incluindo estudos de mutação genética em bactérias, leveduras ou em uma segunda linhagem celular de mamíferos, um estudo de aberração cromossômica e um estudo para danos no DNA em células humanas. A naltrexona não exibiu clastogenicidade em um estudo in vivo de micronúcleos de camundongo.
A bupropiona produziu uma resposta positiva (duas a três vezes a taxa de mutação do controle) em duas das cinco cepas no teste de mutagenicidade bacteriana de Ames e um aumento nas aberrações cromossômicas em um dos três estudos citogenéticos de medula óssea in vivo em ratos.
A naltrexona administrada oralmente a ratos causou um aumento significativo na pseudogestação e uma diminuição nas taxas de prenhez em ratas com 100 mg/kg/dia (aproximadamente 50 vezes a MRHD do componente naltrexona de CONTRAVE em uma base de mg/m2). Não houve efeito na fertilidade masculina neste nível de dose. A relevância dessas observações para a fertilidade humana não é conhecida.
Um estudo de fertilidade de bupropiona em ratos em doses até 300 mg/kg/dia (aproximadamente 15 vezes a MRHD do componente bupropiona de CONTRAVE® em uma base de mg/m2) não revelou evidência de fertilidade diminuída.
4. CONTRAINDICAÇÕES
CONTRAVE® é contraindicado em caso de:
- Hipertensão não controlada (ver "Advertências e precauções")
- Transtorno convulsivo ou história de convulsões (ver "Advertências e precauções")
- Insuficiência hepatica grave
- Insuficiência renal terminal
- Tumor do sistema nervoso central conhecido
- Antecedentes de distúrbio bipolar
- Uso de outros produtos contendo bupropiona ou naltrexona
- Bulimia ou anorexia nervosa, que aumentam o risco de convulsões (ver "Advertências e precauções")
- Uso crônico de agonistas opioides ou opiáceos (por exemplo, metadona) ou agonistas parciais (por exemplo, buprenorfina) ou retirada aguda de opiáceos (ver "Advertências e precauções" e "Interações medicamentosas"
Pacientes submetidos à descontinuação abrupta de álcool, benzodiazepínicos, barbitúricos e fármacos antiepilépticos (ver "Advertências e precauções" e "Interações medicamentosas")
- Administração concomitante de inibidores da monoaminoxidase (IMAO). Devem decorrer pelo menos 14 dias entre a interrupção da IMAO e o início do tratamento com CONTRAVE®. Existe um risco aumentado de reações hipertensivas quando CONTRAVE® é usado concomitantemente com IMAOs. Iniciar CONTRAVE® em um paciente tratado com IMAOs reversível, como linezolida ou azul de metileno intravenoso, também é contraindicado (ver "Posologia e administração", "Interações medicamentosas")
- Alergia conhecida à bupropiona, naltrexona ou qualquer outro componente de CONTRAVE®.
- Reações anafilactóides/anafiláticas e síndrome de Stevens-Johnson foram relatadas com bupropiona (ver "Advertências e precauções")
- Gravidez (ver "Uso em populações específicas")
5. ADVERTÊNCIAS E PRECAUÇÕES
Comportamento e ideação suicida
CONTRAVE® contém bupropiona, um inibidor da recaptação de dopamina e norepinefrina que é semelhante a alguns medicamentos usados para o tratamento da depressão; portanto, as seguintes precauções relativas a esses produtos devem ser consideradas ao tratar pacientes com CONTRAVE®.
Pacientes com transtorno depressivo maior, tanto adultos quanto pediátricos, podem apresentar piora de sua depressão e/ou o surgimento de ideação e comportamento suicida (suicidalidade) ou mudanças incomuns no comportamento, independentemente de estarem tomando medicamentos antidepressivos, e esse risco pode persistir até que ocorra remissão significativa. O suicídio é um risco conhecido da depressão e de outros distúrbios psiquiátricos, e esses distúrbios em si são os mais fortes indicadores de suicídio. Há uma preocupação de longa data que os antidepressivos possam ter um papel na indução do agravamento da depressão e surgimento de tendências suicidas em certos pacientes durante as fases iniciais do tratamento.
Em estudos clínicos controlados por placebo com CONTRAVE® para o tratamento da obesidade em pacientes adultos, não foram notificados quaisquer suicídios ou tentativas de suicídio em estudos com duração de até 56 semanas com CONTRAVE® (equivalente a doses de bupropiona de 360 mg/dia). Nesses mesmos estudos, a ideação suicida foi relatada por 3 (0,20%) de 1.515 pacientes tratados com placebo em comparação com 1 (0,03%) de 3.239 tratados com CONTRAVE®.
Análises agrupadas de estudos de curto prazo controlados por placebo de fármacos antidepressivos (inibidores seletivos de recaptação de serotonina [ISRSs] e outros) mostram que esses fármacos aumentam o risco de ideação e comportamento suicida em crianças, adolescentes e adultos jovens (idades entre 18 e 24 anos) com transtorno depressivo maior (TDM) e outros transtornos psiquiátricos. Estudos clínicos de curta duração não mostraram um aumento no risco de suicidalidade com antidepressivos em comparação com placebo em adultos com idade superior a 24 anos; houve uma redução com antidepressivos em comparação com placebo em adultos com 65 anos ou mais.
As análises agrupadas de estudos controlados com placebo de fármacos antidepressivos em crianças e adolescentes com TDM, transtorno obsessivo-compulsivo (TOC) ou outros transtornos psiquiátricos incluíram um total de 24 estudos de curto prazo de nove fármacos antidepressivos em mais de 4.400 pacientes. As análises agrupadas de estudos controlados por placebo em adultos com TDM ou outros transtornos psiquiátricos incluíram um total de 295 estudos de curto prazo (duração mediana de dois meses) de 11 fármacos antidepressivos em mais de 77.000 pacientes. Houve considerável variação no risco de suicidalidade entre os fármacos, mas uma tendência a um aumento nos pacientes mais jovens para quase todos os fármacos estudados. Houve diferenças no risco absoluto de suicidalidade entre as diferentes indicações, com maior incidência em TDM. As diferenças de risco (fármaco versus placebo), no entanto, foram relativamente estáveis dentro das faixas etárias e entre as indicações. Essas diferenças de risco (diferença placebo-fármaco no número de casos de suicidalidade por mil pacientes tratados) são apresentadas na Tabela 7.
Nenhum suicídio ocorreu em nenhum dos estudos pediátricos com antidepressivos. Houve suicídios nos estudos com antidepressivos em adultos, mas o número não foi suficiente para se chegar a uma conclusão sobre o efeito do medicamento no suicídio.
Não se sabe se o risco de suicídio aumenta com o uso a longo prazo, ou seja, ao longo de vários meses. No entanto, há evidências substanciais de estudos controlados por placebo em adultos com depressão de que o uso de antidepressivos pode retardar a recorrência da depressão.
Todos os pacientes em tratamento com antidepressivos para qualquer indicação devem ser adequadamente monitorados e observados de perto quanto à piora clínica, tendências suicidas e mudanças incomuns no comportamento, especialmente durante os primeiros meses do tratamento medicamentoso, ou em momentos de mudança de dose, tanto aumento quanto diminuição. Esta advertência se aplica a CONTRAVE®, pois um de seus componentes, bupropiona, é um membro de uma classe de antidepressivos.
Os seguintes sintomas, ansiedade, agitação, ataques de pânico, insônia, irritabilidade, hostilidade, agressividade, impulsividade, acatisia (inquietação psicomotora), hipomania e mania, foram relatados em pacientes adultos e pediátricos em tratamento com antidepressivos para transtorno depressivo maior, bem como para outras indicações, tanto psiquiátricas quanto não psiquiátricas.
Embora não tenha sido estabelecido um nexo causal entre o surgimento de tais sintomas e o agravamento da depressão e/ou o surgimento de impulsos suicidas, existe a preocupação de que tais sintomas possam representar precursores da tendência suicida emergente.
Deve-se considerar a possibilidade de mudar o regime terapêutico, incluindo possivelmente a interrupção do medicamento, em pacientes cuja depressão seja persistentemente pior, ou que estejam apresentando uma tendência suicida emergente ou sintomas que possam ser precursores de agravamento da depressão ou tendência suicida, especialmente se esses sintomas forem graves, de início abrupto, ou se não faziam parte dos sintomas que o paciente apresentava.
Famílias e cuidadores de pacientes em tratamento com antidepressivos para transtorno depressivo ou outras indicações, tanto psiquiátricas quanto não psiquiátricas, devem ser alertados sobre a necessidade de monitorar os pacientes quanto ao surgimento de ansiedade, agitação, irritabilidade, alterações incomuns no comportamento e outros sintomas descritos acima, bem como o surgimento de tendências suicidas, e relatar tais sintomas imediatamente aos médicos. Esse monitoramento deve incluir a observação diária pelos familiares e cuidadores. As prescrições de CONTRAVE® devem ser feitas com a menor quantidade de comprimidos, consistente com um controle adequado do paciente, a fim de reduzir o risco de superdose.
Eventos adversos neuropsiquiátricos e risco de suicídio no tratamento da cessação do tabagismo
CONTRAVE® não está indicado para o tratamento da cessação do tabagismo, mas foram relatados eventos adversos neuropsiquiátricos graves em pacientes tomando bupropiona para a cessação do tabagismo. Esses relatos pós-comercialização incluíram mudanças de humor (incluindo depressão e mania), psicose, alucinações, paranoia, delírios, ideação homicida, agressão, hostilidade, agitação, ansiedade e pânico, bem como ideação suicida, tentativa de suicídio e suicídio consumado (ver "Advertências e precauções"). Alguns pacientes que pararam de fumar podem ter apresentado sintomas de abstinência de nicotina, incluindo humor deprimido. Depressão, raramente incluindo ideação suicida, foi relatada em fumantes que se submeteram à tentativa de cessação do tabagismo sem medicação. No entanto, alguns desses eventos adversos ocorreram em pacientes em uso de bupropiona que continuaram a fumar.
Eventos adversos neuropsiquiátricos ocorreram em pacientes sem e com doença psiquiátrica pré-existente; alguns pacientes apresentaram agravamento de suas doenças psiquiátricas. Os pacientes devem ser observados quanto à ocorrência de eventos adversos neuropsiquiátricos. Os pacientes e cuidadores devem ser orientados de que o paciente deve parar de tomar CONTRAVE® e contatar um médico imediatamente se forem observados agitação, humor deprimido, mudanças de comportamento ou pensamentos não típicos do paciente ou se o paciente desenvolver ideação suicida ou comportamento suicida. Em muitos casos pós-comercialização, a resolução dos sintomas após a descontinuação da bupropiona foi relatada. Contudo, os sintomas persistiram em alguns casos, portanto, o monitoramento e cuidados de suporte devem ser fornecidos até que os sintomas se resolvam.
Depressão, suicídio, tentativa de suicídio e ideação suicida foram relatados na experiência pós-comercialização com naltrexona usada no tratamento da dependência de opioides. Nenhuma relação causal foi demonstrada.
Convulsões
A bupropiona, um componente do CONTRAVE®, pode causar convulsões. O risco de convulsão é dose-dependente.
A incidência de convulsões em pacientes que receberam CONTRAVE® em estudos clínicos foi de aproximadamente 0,1% vs. 0% com placebo. CONTRAVE® deve ser descontinuado e não reiniciado em pacientes que tenham convulsões durante o tratamento com CONTRAVE®.
O risco de convulsões também está relacionado a fatores do paciente, situações clínicas e medicamentos concomitantes que diminuem o limiar convulsivo. Estes riscos devem ser considerados antes de se iniciar o tratamento com CONTRAVE®. CONTRAVE® é contraindicado em pacientes com distúrbio convulsivo, diagnóstico atual ou prévio de anorexia nervosa ou bulimia, ou que estejam passando por interrupção abrupta de álcool, benzodiazepínicos, barbitúricos e fármacos antiepilépticos. Deve-se ter cuidado ao prescrever CONTRAVE® a pacientes com fatores predisponentes que possam aumentar o risco de convulsão, incluindo:
- histórico de traumatismo cranioencefálico ou convulsão prévia, acidente vascular cerebral grave, má-formação arteriovenosa, tumor ou infecção do sistema nervoso central ou distúrbios metabólicos (por exemplo, hipoglicemia, hiponatremia, insuficiência hepática grave e hipóxia)
- uso excessivo de álcool ou sedativos, dependência de cocaína ou estimulantes, ou abstinência de sedativos
- pacientes com diabetes tratados com insulina e/ou medicamentos orais para diabetes (sulfonilureias e meglitinidas) que podem causar hipoglicemia
- administração concomitante de medicamentos que possam reduzir o limiar convulsivo, incluindo outros produtos contendo bupropiona, antipsicóticos, antidepressivos tricíclicos, teofilina, esteroides sistêmicos
Recomendações para reduzir o risco de convulsão: A experiência clínica com bupropiona sugere que o risco de convulsão pode ser minimizado através da adesão às recomendações de posologia recomendadas (ver "Posologia e modo de usar"), em particular:
- a dose diária total de CONTRAVE® não exceder 360 mg do componente de bupropiona (ou seja, quatro comprimidos por dia)
- a dose diária ser administrada em doses divididas (duas vezes por dia)
- a dose ser aumentada gradualmente
- não mais do que dois comprimidos sejam tomados de uma só vez
- a administração concomitante de CONTRAVE® com refeições ricas em gordura deve ser evitada (ver "Posologia e modo de usar" e "Características farmacológicas").
- se uma dose for perdida, o paciente deve esperar até a próxima dose programada para retomar o esquema posológico regular
Pacientes recebendo analgésicos opioides
Vulnerabilidade à superdose com opioides: CONTRAVE® não deve ser administrado a pacientes recebendo opioides de uso crônico, devido ao componente naltrexona, que é um antagonista do receptor opioide (ver "Contraindicações"). Se for necessária terapia crônica com opioides, o tratamento com CONTRAVE® deve ser interrompido. Em pacientes que necessitam de tratamento intermitente com opioides, a terapia com CONTRAVE deve ser temporariamente suspensa e doses menores de opioides podem ser necessárias.
Os pacientes devem ser alertados de que podem estar mais sensíveis aos opioides, mesmo em doses mais baixas,