COLEDUE-R
COSMED
rosuvastatina + ezetimiba
Hipocolesterolemiante.
Apresentações.
Cápsula dura. Embalagens contendo 10 ou 30 cápsulas duras de 10mg + 5mg. Embalagens contendo 10 ou 30 cápsulas duras de 10mg + 10mg. Embalagens contendo 10 ou 30 cápsulas duras de 10mg + 20mg.
VIA DE ADMINISTRAÇÃO: ORAL
USO ADULTO
Composição.
Cada cápsula dura de 10mg + 5mg contém: ezetimiba 10mg, rosuvastatina cálcica (equivalente a 5mg de rosuvastatina) 5,209mg, excipientes q.s.p. 1 cápsula dura (lactose monoidratada, hipromelose, croscarmelose sódica, laurilsulfato de sódio, celulose microcristalina, estearilfumarato de sódio, lactose, crospovidona, talco, dióxido de silício, dióxido de titânio, triacetina e óxido de ferro vermelho).
Cada cápsula dura de 10mg + 10mg contém: ezetimiba 10mg, rosuvastatina cálcica (equivalente a 10mg de rosuvastatina) 10,417mg, excipientes q.s.p. 1 cápsula dura (lactose monoidratada, hipromelose, croscarmelose sódica, laurilsulfato de sódio, celulose microcristalina, estearilfumarato de sódio, lactose, crospovidona, talco, dióxido de silício, dióxido de titânio, triacetina e óxido de ferro vermelho).
Cada cápsula dura de 10mg + 20mg contém: ezetimiba 10mg, rosuvastatina cálcica (equivalente a 20mg de rosuvastatina) 20,834mg, excipientes q.s.p. 1 cápsula dura (lactose monoidratada, hipromelose, croscarmelose sódica, laurilsulfato de sódio, celulose microcristalina, estearilfumarato de sódio, lactose, crospovidona, talco, dióxido de silício, dióxido de titânio, triacetina e óxido de ferro vermelho).
Informações técnicas.
1. INDICAÇÕES
Coledue R é indicado como terapia adjuvante à dieta, em pacientes considerados como de alto ou muito alto risco cardiovascular, quando a resposta à dieta e aos exercícios é inadequada em pacientes adultos com hipercolesterolemia primária (familiar heterozigótica ou não-familiar) ou com dislipidemia mista. Em pacientes adultos com hipercolesterolemia Coledue R é indicado para redução do LDL-colesterol, colesterol total e triglicérides elevados, diminuição de ApoB, não HDL-C, das razões LDL-C/HDL-C, não HDL-C/HDL-C, ApoB/Apo A-I, C-total/HDL-C e aumento de HDL-C.
2. RESULTADOS DE EFICÁCIA
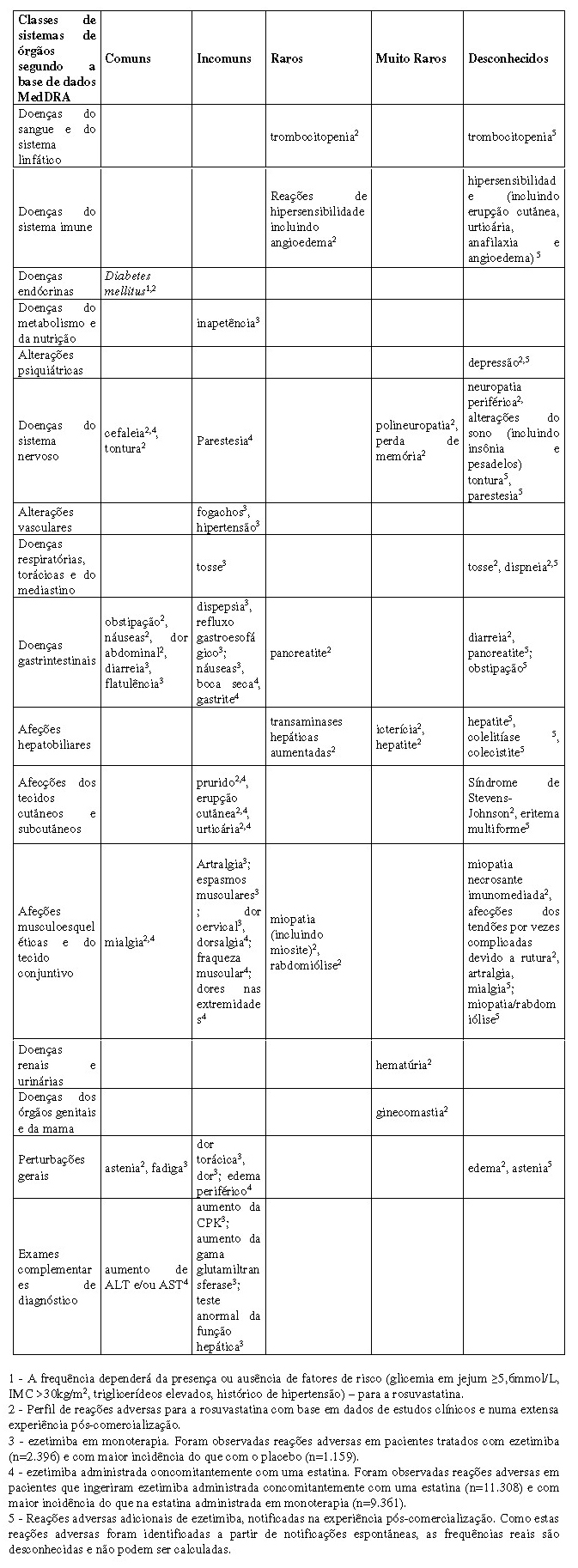
Um estudo clínico aleatorizado, duplo-cego, com grupos paralelos, com a duração de 6 semanas avaliou a segurança e eficácia de ezetimiba (10mg) adicionada à terapêutica fixa de rosuvastatina vs. aumento da dose de 5 para 10mg ou de 10 para 20mg (n=440). Nesse estudo foram incluídos 440 pacientes com risco moderadamente alto/alto de doença coronariana. Os dados agrupados demonstraram que a ezetimiba adicionada à rosuvastatina 5mg ou 10mg reduziu o colesterol LDL em 21%. Por outro lado, a duplicação da dose de rosuvastatina para 10mg ou 20mg reduziu o colesterol LDL em 5,7% (diferença entre grupos de 15,2%, p < 0,001). Individualmente, a terapêutica de ezetimiba adicionada de rosuvastatina 5mg reduziu o colesterol LDL mais do que a rosuvastatina 10mg (diferença de 12,3%, p < 0,001), e a terapêutica de ezetimiba adicionada de rosuvastatina 10mg reduziu o colesterol LDL mais do que a rosuvastatina 20mg (diferença de 17,5%, p < 0,001) (Bays, et al. 2011)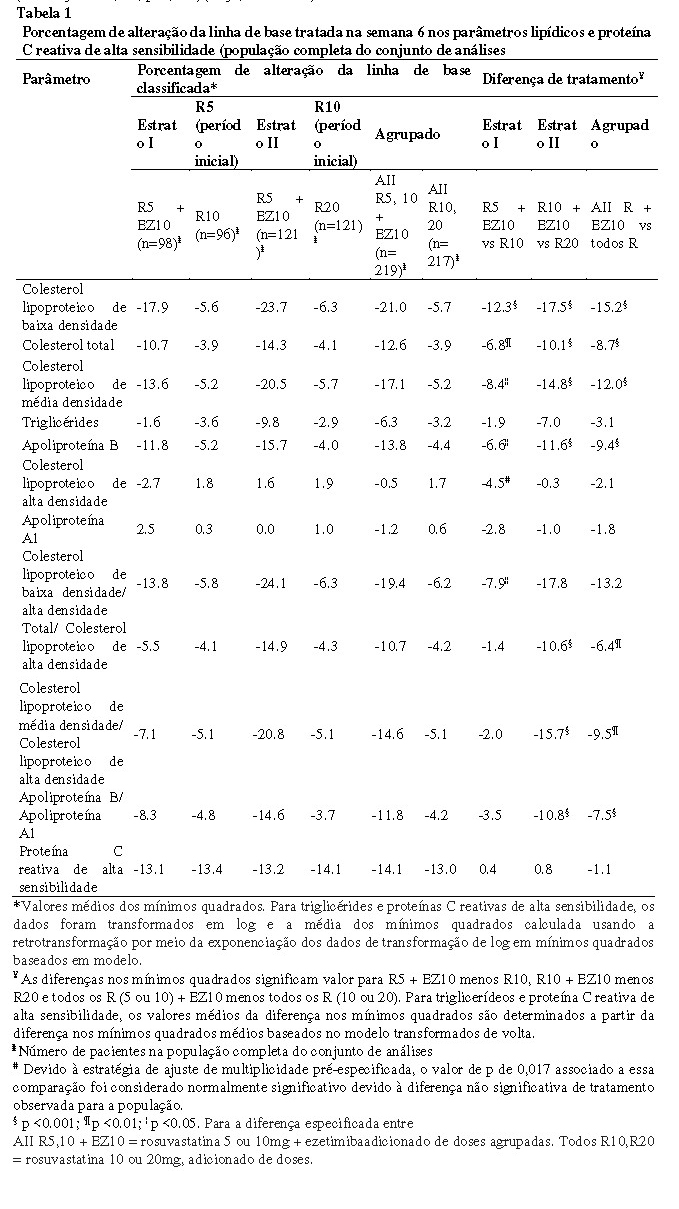
Um estudo aleatorizado, de regime aberto e com a duração de 12 semanas investigou o nível de redução de colesterol LDL em cada braço de tratamento (rosuvastatina 10mg adicionado de ezetimiba 10mg, rosuvastatina 20mg/ezetimiba 10mg, sinvastatina 40/ezetimiba 10mg, sinvastatina 80/ezetimiba 10mg). Nesse estudo foram incluídos 833 pacientes com alto risco cardiovascular. A redução desde os valores basais com as combinações de rosuvastatina de dose baixa foi de 59,7%, significativamente superior às combinações de sinvastatina de dose baixa, 55,2% (p < 0,01). O tratamento com uma combinação de rosuvastatina de dose elevada reduziu o colesterol LDL em 63,5% em comparação com uma redução de 57,4% com a combinação de sinvastatina de dose elevada (p < 0,001) (Ballantyne, et al. 2014)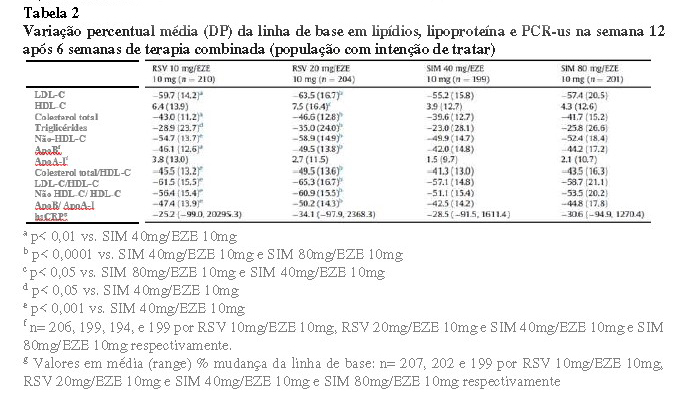
Referências bibliográficas:
Ballantyne CM, et al. Efficacy, safety and effect on biomarkers related to cholesterol and lipoprotein metabolism of rosuvastatin 10 or 20mg plus ezetimibe 10mg vs. simvastatin 40 or 80mg plus ezetimibe 10mg in high-risk patients: Results of the GRAVITY randomized study. Atherosclerosis. 2014;232(1):86
93. Bays HE, et al. Safety and efficacy of ezetimibe added on to rosuvastatin 5 or 10mg versus up-titration of rosuvastatin in patients with hypercholesterolemia (the ACTE Study). Am J Cardiol. 2011;108(4):523-30.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Terapêutica de combinação de rosuvastatina e ezetimiba. A utilização concomitante de 10mg de rosuvastatina e 10mg de ezetimiba resultou num aumento de 1,2 vezes na AUC da rosuvastatina em pacientes com hipercolesterolemia. No entanto, não é possível excluir uma interação farmacodinâmica, em termos de efeitos adversos, entre a rosuvastatina e a ezetimiba.
rosuvastatina
A rosuvastatina cálcica é administrada por via oral na forma ativa, com picos de níveis plasmáticos ocorrendo 5 horas após a administração. A absorção aumenta linearmente com a faixa de dose. A meia vida é de 19 horas e não aumenta com a elevação da dose. A biodisponibilidade absoluta é de 20%. Há um acúmulo mínimo com dose única diária repetida. A rosuvastatina sofre metabolismo de primeira passagem no fígado, que é o local primário da síntese de colesterol e da depuração de LDL-C. Aproximadamente 90% da rosuvastatina liga-se às proteínas plasmáticas, principalmente à albumina. Mais de 90% da atividade inibitória para a HMG-CoA redutase circulante é atribuída ao princípio ativo. A rosuvastatina sofre metabolismo limitado (aproximadamente 10%), principalmente para a forma Ndesmetila, e 90% são eliminados como droga inalterada nas fezes, sendo o restante excretado na urina.
Linearidade: a exposição sistêmica da rosuvastatina aumenta em proporção à dose. Não existem alterações nos parâmetros farmacocinéticos após múltiplas doses diárias.
ezetimiba
Absorção: após a administração oral, a ezetimiba é rapidamente absorvida e extensivamente conjugada para um glucuronido fenólico farmacologicamente ativo (glucuronido de ezetimiba). A média das concentrações plasmáticas máximas (Cmax) ocorre no período de 1 a 2 horas para o glucuronido de ezetimiba e no período de 4 a 12 horas para a ezetimiba. A biodisponibilidade absoluta de ezetimiba não pode ser determinada, uma vez que o composto é praticamente insolúvel em meio aquoso adequado para injeção. A administração concomitante de alimentos (refeições com alto teor de gordura ou sem gordura) não teve qualquer efeito na biodisponibilidade oral de ezetimiba. A ezetimiba pode ser administrada com ou sem alimentos.
Distribuição: a ezetimiba e o glucuronido de ezetimiba ligam-se às proteínas plasmáticas humanas em 99,7% e em 88 a 92%, respetivamente.
Metabolismo: a ezetimiba é metabolizada principalmente no intestino delgado e no fígado através da conjugação em glucuronido (uma reação de fase II) com subsequente excreção biliar. Em todas as espécies estudadas foi observado um metabolismo oxidativo mínimo (uma reação de fase I). A ezetimiba e o glucuronido de ezetimiba são os principais compostos derivados do fármaco detectados no plasma, constituindo aproximadamente 10 a 20% e 80 a 90% do fármaco total no plasma, respetivamente. Quer a ezetimiba quer o glucuronido de ezetimiba são eliminados lentamente do plasma, evidenciando-se uma significativa recirculação entero-hepática. A meia-vida de ezetimiba e do glucuronido de ezetimiba é de, aproximadamente, 22 horas.
Eliminação: após a administração oral de 14C-ezetimiba (20mg) a seres humanos, a ezetimiba total representou aproximadamente 93% da radioatividade total no plasma. Ao longo de um período de colheita de 10 dias, foram detectados aproximadamente 78% e 11% da radioatividade administrada, respetivamente, nas fezes e na urina. Após 48 horas, os níveis de radioatividade não eram detectáveis no plasma.
Farmacocinética em populações especiais
rosuvastatina
-Idade e sexo: não houve efeito clinicamente relevante associado à idade ou sexo na farmacocinética da rosuvastatina em adultos. A exposição em crianças e adolescentes com hipercolesterolemia heterozigótica familiar aparenta ser similar ou menor que em adultos com dislipidemia.
-Raça: estudos farmacocinéticos mostram uma elevação de aproximadamente duas vezes na mediana da Área Sob a Curva (ASC) em descendentes asiáticos comparados com caucasianos. Uma análise da farmacocinética da população não revelou diferenças clinicamente relevantes na farmacocinética entre caucasianos, hispânicos e negros ou grupos de afro-caribenhos.
-Insuficiência renal: em um estudo realizado em indivíduos com graus variáveis de insuficiência renal, a doença renal de leve a moderada apresentou pouca influência nas concentrações plasmáticas da rosuvastatina. Entretanto, indivíduos com insuficiência grave (depuração de creatinina < 30mL/min) apresentaram um aumento de 3 vezes na concentração plasmática em comparação com voluntários sadios.
-Insuficiência hepática: em um estudo realizado em indivíduos com graus variáveis de insuficiência hepática, não houve evidência de aumento da exposição à rosuvastatina, exceto em 2 indivíduos com doença hepática mais grave (graus 8 e 9 de Child-Pugh). Nestes indivíduos, a exposição sistêmica foi aumentada em no mínimo 2 vezes em comparação aos indivíduos com grau menor de Child-Pugh.
-Polimorfismos genéticos: a disponibilidade dos inibidores da HMG-CoA redutase, incluindo a rosuvastatina, envolve OATP1B1 e as proteínas transportadoras BCRP. Em pacientes com polimorfismos genéticos em SLCO1B1 (OATP1B1) e/ou ABCG2 (BCRP) existe um risco de maior exposição à rosuvastatina. Polimorfismos individuais de SLCO1B1 c.521CC e ABCG2 c.421AA estão associados com uma exposição (ASC) à rosuvastatina aproximadamente 1,6 ou 2,4 vezes maior, respectivamente, em comparação com os genótipos SLCO1B1 c.521TT ou ABCG2 c.421CC.
Dados de segurança pré-clínica
Os dados pré-clínicos não revelam danos especiais em humanos, tendo como base estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade, potencial carcinogênico e toxicidade reprodutiva.
ezetimiba
Idade e sexo: as concentrações plasmáticas de ezetimiba total são aproximadamente 2 vezes superiores nos idosos (≥65 anos) do que nos jovens (18 a 45 anos). A redução do C-LDL e o perfil de segurança são comparáveis entre indivíduos idosos e jovens tratados com ezetimiba. Desta forma, não é necessário qualquer ajuste posológico nos idosos. As concentrações plasmáticas para a ezetimiba total são ligeiramente superiores (aproximadamente 20%) nas mulheres do que nos homens. A redução do C-LDL e o perfil de segurança são comparáveis entre homens e mulheres tratados com ezetimiba. Por conseguinte, não é necessário qualquer ajuste posológico com base no sexo do paciente.
Insuficiência renal: após a administração de uma dose única de 10mg de ezetimiba em pacientes com doença renal grave (n=8; CrCl média ≤30mL/min/1,73m2), a AUC média de ezetimiba total aumentou aproximadamente 1,5 vezes, em comparação com os indivíduos saudáveis (n=9). Este resultado não é considerado relevante em termos clínicos. Não é necessário qualquer ajuste posológico para pacientes com insuficiência renal. Um paciente envolvido neste estudo (transplantado renal e poli medicado, incluindo ciclosporina) apresentou uma exposição 12 vezes superior à ezetimiba total.
Insuficiência hepática: após a administração de uma única dose de 10mg de ezetimiba, a AUC média para a ezetimiba total aumentou aproximadamente 1,7 vezes em pacientes com insuficiência hepática leve (pontuação de 5 ou 6 na escala de Child-Pugh), em comparação com os indivíduos saudáveis. Num estudo com a duração de 14 dias, com doses múltiplas (10mg uma vez por dia) realizado em pacientes com insuficiência hepática moderada (pontuação de 7 a 9 na escala de Child-Pugh), a AUC média para a ezetimiba total aumentou aproximadamente 4 vezes no dia 1 e no dia 14, em comparação com os indivíduos saudáveis. Não é necessário qualquer ajuste posológico para pacientes com insuficiência hepática leve. Devido ao desconhecimento dos efeitos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave (pontuação > 9 na escala de Child-Pugh). Este medicamento não é recomendado nestes pacientes.
População pediátrica: Este medicamento não tem indicação para população pediátrica
Propriedades farmacodinâmicas
rosuvastatina
Mecanismo de ação: a rosuvastatina é um inibidor seletivo e competitivo da HMG-CoA redutase, a enzima limitante da taxa de conversão da 3-hidroxi-3-metilglutaril coenzima A em mevalonato, um precursor do colesterol. O principal local de ação da rosuvastatina é o fígado, o órgão alvo na redução do colesterol. A rosuvastatina aumenta o número de receptores hepáticos de LDL na superfície celular, potenciando a captação e o catabolismo de LDL e inibindo a síntese hepática de VLDL, reduzindo, desta forma, o número total de partículas de VLDL e LDL.
Efeitos farmacodinâmicos: a rosuvastatina reduz os níveis elevados de colesterol-LDL, colesterol total e triglicerídeos e aumenta o nível de colesterol-HDL. Reduz ainda a ApoB, o C-não-HDL, C-VLDL, TGVLDL e aumenta a ApoA-I. A rosuvastatina reduz também a relação de C-LDL/C-HDL, C-Total/C-HDL e C-não-HDL/C-HDL e de ApoB/ApoA-I. O efeito terapêutico é obtido dentro de uma semana após o início do tratamento e obtém-se 90% da resposta máxima em 2 semanas. A resposta máxima é geralmente obtida após 4 semanas, mantendo-se subsequentemente.
ezetimiba
A ezetimiba é uma nova classe de compostos hipolipemiantes que inibem seletivamente a absorção intestinal de colesterol e esteróis vegetais relacionados. A ezetimiba é ativa por via oral e possui um mecanismo de ação que difere das outras classes de compostos hipocolesterolemiantes (ex.: estatinas, sequestrantes dos ácidos biliares [resinas], derivados do ácido fíbrico e estanois vegetais). O alvo molecular da ezetimiba é o transportador esterol Niemann-PiCPK C1-Like 1 (NPC1L1), que é o responsável pela absorção intestinal de colesterol e de fitoesterois. A ezetimiba fixa-se na borda em escova do intestino delgado e inibe a absorção de colesterol, conduzindo a uma diminuição do aporte de colesterol intestinal para o fígado; as estatinas diminuem a síntese hepática do colesterol e, em conjunto, estes diferentes mecanismos originam uma redução complementar do colesterol. Num estudo clínico com duração de 2 semanas realizado em 18 pacientes hipercolesterolêmicos, a ezetimiba inibiu a absorção intestinal de colesterol em 54%, em comparação com o placebo. Foram realizados vários estudos pré-clínicos para determinar a seletividade de ezetimiba na inibição da absorção de colesterol. A ezetimiba inibiu a absorção de colesterol-[14C] sem qualquer efeito na absorção de triglicerídeos, ácidos graxos, ácidos biliares, progesterona, etinilestradiol ou de vitaminas lipossolúveis A e D. Estudos epidemiológicos estabeleceram que a morbidade e mortalidade cardiovascular variam diretamente de acordo com o nível de C-total e CLDL e inversamente com o nível de C-HDL.
4. CONTRAINDICAÇÕES
Coledue R é contraindicado:
• em pacientes com hipersensibilidade a qualquer um dos componentes da fórmula.
• em pacientes com doença hepática ativa incluindo elevações persistentes e inexplicáveis das transaminases séricas e qualquer elevação das transaminases séricas excedendo 3 vezes o Limite Superior da Normalidade (LSN).
• durante a gravidez e amamentação e em mulheres com potencial para engravidar que não adotam medidas contraceptivas apropriadas.
• em pacientes com comprometimento renal grave (depuração da creatinina < 30mL/min).
• em pacientes com miopatia.
Categoria de risco na gravidez X: Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
5. ADVERTÊNCIAS E PRECAUÇÕES
Rosuvastatina
Fígado
Como outros inibidores da HMG-CoA redutase, rosuvastatina cálcica deve ser usada com cautela em pacientes que consomem quantidades excessivas de álcool e/ou que tenham uma história de doença hepática. É recomendado que os testes de enzimas hepáticas sejam realizados antes e por 12 semanas após
o início da terapia com Coledue R e no caso de qualquer elevação da dose, e depois periodicamente (ex.: semestralmente).
Sistema musculoesquelético
Como com outros inibidores da HMG-CoA redutase, foram relatados efeitos musculoesqueléticos, como mialgia, miopatia e, raramente, rabdomiólise em pacientes tratados com rosuvastatina. Assim como outros inibidores da HMG-CoA redutase, a frequência de rabdomiólise no uso pós-comercialização é maior com as doses mais altas administradas. Pacientes que desenvolverem quaisquer sinais ou sintomas sugestivos de miopatia devem ter os seus níveis de creatinoquinase (CPK) medidos. O tratamento com Coledue R deve ser interrompido se os níveis de CPK estiverem notadamente elevados ( > 10 vezes o LSN) ou se houver diagnóstico ou suspeita de miopatia. Houve relatos muito raros de uma miopatia necrotizante imunomediada caracterizada clinicamente por fraqueza muscular proximal persistente e elevação da creatinoquinase sérica durante o tratamento ou após a descontinuação de estatinas, incluindo a rosuvastatina. Testes neuromusculares e sorológicos adicionais podem ser necessários. Tratamento com agentes imunossupressores podem ser requeridos. Nos estudos com a rosuvastatina cálcica não houve evidência de aumento de efeitos musculoesqueléticos na administração concomitante com qualquer terapia. Entretanto, foi observado um aumento da incidência de miosite e miopatia em pacientes que estavam recebendo outros inibidores da HMG-CoA redutase junto com ciclosporina, derivados do ácido fíbrico, incluindo genfibrozila, ácido nicotínico, antifúngicos do grupo azóis e antibióticos macrolídeos. Coledue R deve ser prescrito com precaução em pacientes com fatores de predisposição para miopatia, tais como, insuficiência renal, idade avançada e hipotireoidismo, ou situações em que pode ocorrer um aumento nos níveis plasmáticos (Vide item 3. Características farmacológicas - Propriedades farmacodinâmicas e 6 Interações medicamentosas). O uso de Coledue R deve ser temporariamente interrompido em qualquer paciente com uma condição aguda grave sugestiva de miopatia ou que predispõe ao desenvolvimento de insuficiência renal secundária à rabdomiólise (ex.: sepse; hipotensão; cirurgia de grande porte; trauma; alterações metabólicas, endócrinas e eletrolíticas graves; ou convulsões não controladas).
Ezetimiba
Enzimas Hepáticas: em estudos controlados para avaliar a coadministração de ezetimiba e uma estatina, foram observadas elevações consecutivas das transaminases (≥3 vezes o LSN). É recomendado que os testes de enzimas hepáticas sejam realizados antes e por 12 semanas após o início da terapia e no caso de qualquer elevação da dose, e depois periodicamente (ex.: semestralmente).
Insuficiência Hepática: uma vez que os efeitos da maior exposição à ezetimiba em pacientes com insuficiência hepática moderada ou grave são desconhecidos, Coledue R não é recomendado para esses pacientes. Após uma dose única de 10 mg de ezetimiba, a média da área sob a curva (AUC) para ezetimiba total aumentou aproximadamente 1,7 vez em pacientes com insuficiência hepática leve (escore Child-Pugh de 5 ou 6), em comparação com indivíduos saudáveis. Em um estudo de doses múltiplas de 14 dias de duração (10 mg/dia) em pacientes com insuficiência hepática moderada (escore de Child-Pugh de 7 a 9), a AUC média para ezetimiba total aumentou aproximadamente 4 vezes no 1° e no 14° dia em comparação com indivíduos saudáveis. Não é necessário nenhum ajuste de dose para pacientes com insuficiência hepática leve. Em razão dos efeitos desconhecidos da exposição aumentada à ezetimiba em pacientes com insuficiência hepática moderada ou grave (escore de Child-Pugh > 9), Coledue R não é recomendada para esses pacientes.
Musculoesquelético: em estudos clínicos, não foi verificado excesso de miopatia ou rabdomiólise associados a ezetimiba em comparação com o braço controle (placebo ou estatina isoladamente). Entretanto, miopatia e rabdomiólise são reações adversas conhecidas das estatinas e de outros fármacos redutores de lípides. Em estudos clínicos, a incidência de CPK > 10 vezes o LSN foi de 0,2% para ezetimiba versus 0,1% para o placebo, e de 0,1% para ezetimiba coadministrado com uma estatina versus 0,4% para as estatinas isoladamente. Na experiência pós-comercialização com ezetimiba, foram relatados casos de miopatia e rabdomiólise, independentemente da causalidade. A maioria dos pacientes que desenvolveram rabdomiólise recebiam uma estatina antes de iniciar o tratamento com ezetimiba. No entanto, a rabdomiólise foi relatada muito raramente com a monoterapia com ezetimiba ou com a adição de ezetimiba a agentes conhecidamente associados a risco aumentado de rabdomiólise. Todos os pacientes que iniciam tratamento com Coledue R devem ser alertados sobre o risco de miopatia e instruídos a relatar imediatamente qualquer dor, sensibilidade ou fraqueza muscular inexplicada. Coledue R deve ser imediatamente descontinuado se houver suspeita de ou for comprovada a miopatia. A presença desses sintomas e um nível de creatina fosfoquinase (CPK) > 10 vezes o LSN indica miopatia.
Fibratos: a coadministração de Coledue R com fibratos não foi estudada, portanto, a coadministração de Coledue R e fibratos não é recomendada (veja o item 6. Interações medicamentosas).
Ciclosporina: deve-se ter cautela ao prescrever Coledue R para pacientes que utilizam ciclosporina. As concentrações de ciclosporina devem ser monitoradas nesses pacientes (veja o item 6. Interações medicamentosas).
Anticoagulantes: se Coledue R for acrescentado ao tratamento com varfarina, outro anticoagulante cumarínico ou fluindiona, a Razão Normalizada Internacional (International Normalized Ratio -INR) deve ser adequadamente monitorada (veja o item 6. Interações medicamentosas).
Pacientes idosos: a concentração plasmática da ezetimiba total é, aproximadamente, 2 vezes mais elevada nos indivíduos idosos (? 65 anos de idade) em relação aos jovens (18 a 45 anos de idade). A redução de
LDL-C e o perfil de segurança são comparáveis em indivíduos idosos e jovens que recebem ezetimiba. Não é necessário, entretanto, ajuste posológico para pacientes idosos. Como idade avançada (≥65 anos) é um fator predisponente para miopatia por estatina, Coledue R deve ser prescrito com cautela a idosos. Sexo: a concentração plasmática da ezetimiba é pouco maior ( < 20%) em mulheres do que em homens. A redução do LDL-C e do perfil de segurança é comparável entre homens e mulheres tratados com ezetimiba. Portanto, não é necessário nenhum ajuste de dose com base no sexo.
Uso pediátrico: este medicamento não está indicado abaixo de 18 anos.
Raça: inibidores da HMG-CoA redutase tem sua exposição aumentada em pacientes asiáticos quando comparados com caucasianos (ver itens "3. Características Farmacológicas" e "8. Posologia e Modo de usar"). Metanálise de estudos farmacocinéticos da ezetimiba não mostra diferenças farmacocinéticas entre negros e caucasianos.
Diabetes mellitus
Pacientes tratados com Coledue R podem experimentar aumento dos níveis séricos de HbA1c e da glicemia, que em alguns casos, podem alcançar o limiar para o diagnóstico do diabetes, principalmente em pacientes com alto risco para o desenvolvimento do Diabetes mellitus. (Vide item 9. Reações adversas).
Doença pulmonar intersticial: foram notificados casos excepcionais de doença pulmonar intersticial com algumas estatinas, especialmente com tratamentos de longa duração. Os sintomas observados incluem dispneia, tosse não produtiva e deterioração do estado de saúde em geral (fadiga, perda de peso e febre). Se houver suspeita de desenvolvimento de doença pulmonar intersticial, o tratamento com Coledue R deve ser descontinuado.
Efeito sobre a capacidade de dirigir veículos e operar máquinas: O componente rosuvastatina de Coledue R não tem efeito sedativo. O componente ezetimiba de Coledue R não tem estudos sobre os efeitos na capacidade de dirigir veículos e operar máquinas. Porém, certas reações adversas que foram relatadas com ezetimiba podem afetar a capacidade de alguns pacientes para executar essas tarefas. As respostas individuais dos pacientes com Coledue R podem variar (ver item "9. Reações adversas")".
Insuficiência renal: a doença renal leve a moderada tem pouca influência nas concentrações plasmáticas dos inibidores da HMG-CoA redutase. Como na insuficiência renal grave observou-se aumento de 3 vezes na concentração plasmática do inibidor da HMG-CoA redutase em comparação com o observado em voluntários sadios, Coledue R é contraindicado em pacientes com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2).
Pâncreas: há citações na literatura de aumento do risco de pancreatite pelo uso de estatinas, incluindo rosuvastatina, assim como da ezetimiba. No entanto, é difícil confirmar, de forma consistente, a relação de causalidade
Miopatia secundária a outros agentes hipolipemiantes: pacientes que desenvolveram miopatia induzida por outras estatinas ou ezetimiba não devem receber Coledue R ácido fusídico: na terapia com ácido fusídico (ver item "6. Interações medicamentosas") existe risco de lesão muscular e de rabdomiólise. Eventos musculares, incluindo rabdomiólise, foram relatados na experiência pós-comercialização com a administração concomitante de rosuvastatina e do ácido fusídico. O tratamento com Coledue R e ácido fusídico deve ser evitado.
Gravidez e lactação: categoria de risco na gravidez X: Este medicamento não deve ser utilizado por mulheres grávidas ou que possam ficar grávidas durante o tratamento.
Estudos conduzidos em ratas demonstraram que a ezetimiba é excretada no leite. Não se sabe se a ezetimiba é excretada no leite de seres humanos, portanto Coledue R não deverá ser administrado a nutrizes a não ser que o potencial benefício justifique o provável risco para o lactente. A segurança de rosuvastatina durante a gravidez e a lactação não foi estabelecida. Mulheres com potencial de engravidar devem usar métodos contraceptivos apropriados (vide 4. Contraindicações).
6. INTERAÇÕES MEDICAMENTOSAS
rosuvastatina:
Dados in vitro e in vivo indicam que a rosuvastatina não tem interação clinicamente significativa com o citocromo P450 (como um substrato, inibidor ou indutor). A rosuvastatina é um substrato para determinadas proteínas transportadoras, incluindo o transportador hepático de captação OATP1B1 e o transportador de efluxo BCRP. A administração concomitante de Coledue R com medicamentos que são inibidores destas proteínas transportadoras pode resultar em maior concentração plasmática de rosuvastatina e maior risco de miopatia (vide Tabela 3, "5. Advertências e precauções e 8. Posologia e modo de usar").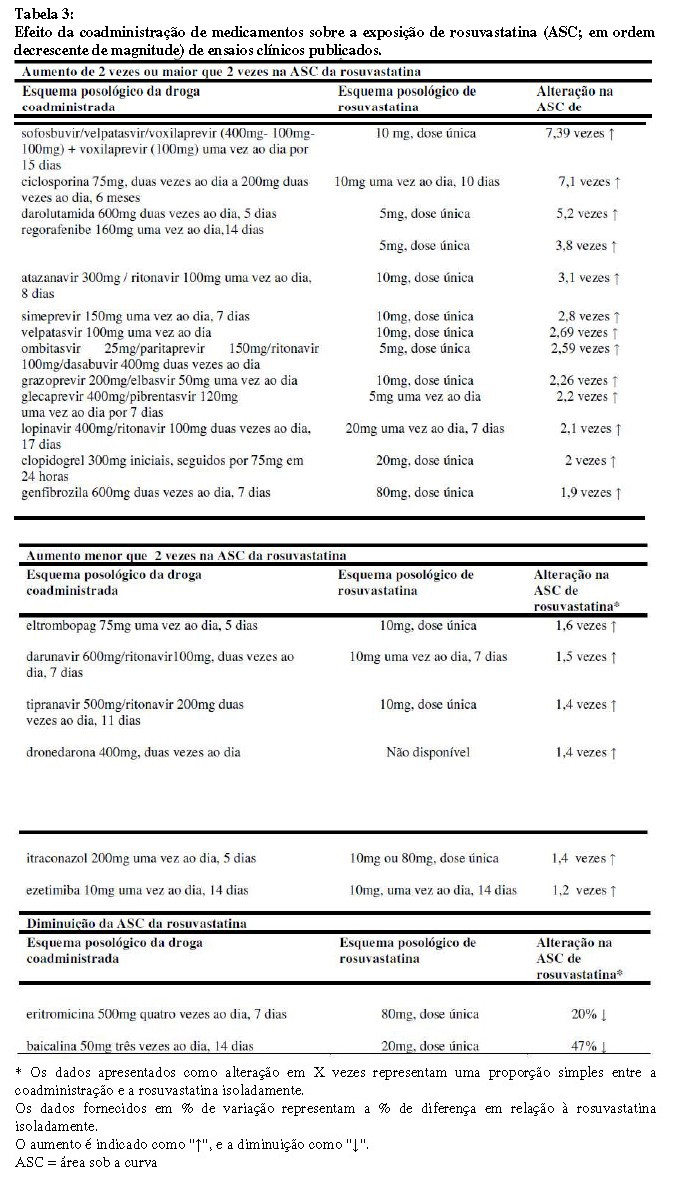
Os seguintes medicamentos/combinações não tiveram um efeito clinicamente significativo na relação ASC da rosuvastatina na coadministração: aleglitazar 0,3 mg, por 7 dias; fenofibrato 67 mg três vezes ao dia, por 7 dias; fluconazol 200 mg uma vez ao dia, por 11 dias; fosamprenavir 700 mg/ritonavir 100 mg duas vezes ao dia, por 8 dias; cetoconazol 200 mg duas vezes ao dia, por 7 dias; rifampicina 450 mg uma vez ao dia, por 7 dias; silimarina 140 mg três vezes ao dia, por 5 dias.
Interações que requerem ajuste da dose de Coledue R (vide também Tabela 3).
Quando é necessária a coadministração de Coledue R com outros medicamentos que conhecidamente aumentam a exposição à rosuvastatina, a dose deste medicamento deve ser ajustada. É recomendado que o médico consulte as informações relevantes dos medicamentos quando considerar administrar esses medicamentos concomitantemente com Coledue R. Deve-se iniciar com uma dose de 5mg uma vez dia de rosuvastatina cálcica se o aumento esperado na exposição (ASC) for de aproximadamente 2 vezes ou maior. A dose máxima diária de Coledue R deve ser ajustada e então a exposição esperada de rosuvastatina provavelmente não excederá aquela de uma dose diária de 40 mg de rosuvastatina cálcica administrada sem medicamentos que possam interagir, por exemplo, uma dose de 5mg de rosuvastatina cálcica com ciclosporina (aumento de 7,1 vezes na exposição), uma dose de 10mg de rosuvastatina cálcica com ritonavir/atazanavir combinados (aumento de 3,1 vezes) e uma dose de 20mg de rosuvastatina cálcica com genfibrozila (aumento de 1,9 vezes). Se for observado que o medicamento aumenta a ASC da rosuvastatina em menos de duas vezes, a dose inicial não precisa ser diminuída, mas deve-se ter cuidado ao aumentar a dose de Coledue R acima de 20 mg. Inibidores de protease: A coadministração de Coledue R com certos inibidores de protease ou combinação de inibidores de protease pode aumentar a exposição à rosuvastatina (ASC) em até 7 vezes (consulte a Tabela 4). O ajuste da dose é necessário dependendo do nível de efeito na exposição à rosuvastatina (vide
8. Posologia e modo de usar e 5. Advertências e Precauções).
Interação com outros medicamentos
Antiácidos: a administração simultânea de Coledue R com uma suspensão de antiácido contendo hidróxido de alumínio e hidróxido de magnésio resultou em diminuição da concentração plasmática da rosuvastatina de aproximadamente 50%. Este efeito foi reduzido quando o antiácido foi administrado 2 horas após Coledue R. A relevância clínica desta interação não foi estudada.
ácido fusídico: estudos de interação com rosuvastatina e ácido fusídico não foram conduzidos. Assim como com outras estatinas, eventos musculares relacionados incluindo rabdomiólise foram relatados na experiência pós-comercialização com a administração concomitante de rosuvastatina e ácido fusídico. Os pacientes devem ser rigorosamente monitorados e a suspensão temporária do tratamento com Coledue R pode ser apropriada.
Efeito da rosuvastatina sobre medicamentos coadministrados
varfarina: a farmacocinética da varfarina não é significativamente afetada após a coadministração com Coledue R. Entretanto, como com outros inibidores da HMG-CoA redutase, a coadministração de rosuvastatina cálcica e varfarina pode resultar em um aumento da razão internacional normalizada (INR) em comparação com a varfarina isoladamente. Em pacientes em tratamento com antagonistas da vitamina K, recomenda-se a monitorização da INR, tanto no início quanto no término do tratamento com Coledue R ou após ajuste de dose.
fenofibrato / derivados do ácido fíbrico: embora nenhuma interação farmacocinética entre rosuvastatina e fenofibrato tenha sido observada, uma interação farmacodinâmica pode ocorrer. A genfibrozila, o fenofibrato e outros ácidos fíbricos, incluindo o ácido nicotínico, podem aumentar o risco de miopatia quando administrados concomitantemente com inibidores da HMG-CoA redutase (vide "Advertências e Precauções").
ciclosporina: a coadministração de rosuvastatina com ciclosporina não resultou em alterações significativas na concentração plasmática da ciclosporina. O uso concomitante da ciclosporina pode aumentar o risco de miopatia/rabdomiólise. Se for necessária a coadministração com Coledue R, a dose de rosuvastatina deve ser limitada a 5mg e de ezetimiba a 10mg, uma vez ao dia.
Outros medicamentos: não houve interações clinicamente significativas com contraceptivo oral, digoxina, ezetimiba ou fenofibrato. Em estudos clínicos, a rosuvastatina cálcica foi coadministrada com agentes anti-hipertensivos, agentes antidiabéticos e terapia de reposição hormonal. Esses estudos não demonstraram evidência de interações adversas clinicamente significativas. Apesar de estudos clínicos terem demonstrado que a rosuvastatina em monoterapia não reduz a concentração de cortisol plasmático basal ou prejudique a reserva adrenal, deve-se ter cautela se este medicamento for administrado concomitantemente com fármacos que podem diminuir os níveis ou a atividade de hormônios esteroidais endógenos, tais como cetoconazol, espironolactona e cimetidina.
ezetimiba:
Em estudos pré-clínicos, demonstrou-se que a ezetimiba não induz enzimas de metabolização do citocromo P-450. Não foram observadas interações farmacocinéticas clinicamente relevantes entre a ezetimiba e os medicamentos reconhecidamente metabolizados pelos citocromos P-450 1A2, 2D6, 2C8, 2C9 e 3A4 ou Nacetiltransferase. A ezetimiba não exerceu efeito sobre a farmacocinética dos seguintes compostos: dapsona, dextrometorfano, digoxina, anticoncepcionais orais (etinilestradiol e levonorgestrel), glipizida, tolbutamida ou midazolam durante a coadministração. A cimetidina, coadministrada com a ezetimiba, não exerceu efeito sobre a biodisponibilidade da ezetimiba.
Antiácidos: a administração concomitante de antiácidos reduziu a taxa de absorção da ezetimiba, embora não tenha exercido efeito sobre a biodisponibilidade. Essa redução da taxa de absorção não é considerada clinicamente relevante.
colestiramina: a administração concomitante de colestiramina reduziu a AUC média da ezetimiba total (ezetimiba + glicuronídeo de ezetimiba) em aproximadamente 55%. A redução adicional do LDL-C pelo acréscimo da ezetimiba à colestiramina pode ser minimizada por essa interação.
ciclosporina: em um estudo que envolveu oito pacientes submetidos a transplante renal, com clearance de creatinina > 50 mL/min e que estavam recebendo dose estável de ciclosporina, uma única dose de 10mg de ezetimiba resultou em aumento de 3,4 vezes (variação de 2,3 a 7,9 vezes) da AUC média da ezetimiba total em comparação com uma população de controle sadia de outro estudo (n= 17). Em um estudo diferente, um paciente submetido a transplante renal com insuficiência renal grave (clearance de creatinina de 13,2mL/min/1,73m2) que estava recebendo diversos medicamentos, inclusive ciclosporina, apresentou exposição 12 vezes maior à ezetimiba total em comparação com os controles de comparação. Em um estudo cruzado de dois períodos, a administração diária de 20mg de ezetimiba durante 8 dias com uma única dose de 100mg de ciclosporina no 7° dia a 20 indivíduos saudáveis resultou em aumento de 15%, em média, na AUC da ciclosporina (variação de 10% de redução a 51% de aumento) em comparação a uma dose única de 100mg de ciclosporina isoladamente (veja item 5. Advertências e Precauções).
Fibratos: a segurança e a eficácia da ezetimiba coadministrada com fenofibrato foram avaliadas em um estudo clínico (veja os itens 9. Reações Adversas e, Coadministração com Fenofibrato); a coadministração da ezetimiba com outros fibratos não foi estudada. Os fibratos podem aumentar a excreção biliar de colesterol, levando à colelitíase. Em um estudo pré-clínico conduzido em cães, a ezetimiba aumentou as concentrações de colesterol na vesícula biliar. A coadministração de Coledue R com fibratos não foi estudada, portanto, a coadministração de Coledue R e fibratos não é recomendada. Fenofibrato: em um estudo farmacocinético, a administração concomitante do fenofibrato aumentou a concentração total da ezetimiba em aproximadamente 1,5 vez. A coadministração de Coledue R com fenofibrato não foi estudada, portanto, a coadministração de Coledue R e fenofibrato não é recomendada.
genfibrozila: em um estudo farmacocinético, a administração concomitante de genfibrozila aumentou a concentração total de ezetimiba em aproximadamente 1,7 vez. Esse aumento não é considerado clinicamente significativo. Não há dados clínicos disponíveis. Estatinas: não foram observadas interações farmacocinéticas clinicamente importantes quando a ezetimiba foi coadministrada com atorvastatina, sinvastatina, pravastatina, lovastatina, fluvastatina ou rosuvastatina.
Anticoagulantes: a administração concomitante de ezetimiba (10mg em dose única diária) não apresentou efeito significativo sobre a biodisponibilidade da varfarina e sobre o tempo de protrombina em um estudo que incluiu doze adultos saudáveis do sexo masculino. Houve relatos pós-comercialização de Razão Normalizada Internacional (INR) aumentada em pacientes para os quais ezetimiba foi adicionado à varfarina ou à fluindiona. A maioria desses pacientes também estava recebendo outros medicamentos (veja
o item 5. Advertências e Precauções).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Conservar em temperatura ambiente (entre 15 e 30°C).
Prazo de Validade: 24 meses.
Número de lote e datas de fabricação e validade: vide embalagem. Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Coledue R 10mg + 5mg apresenta-se como cápsula gelatinosa dura, de corpo e tampa alaranjados, contendo 1 comprimido revestido rosa e 2 comprimidos brancos. Coledue R 10mg + 10mg apresenta-se como cápsula gelatinosa dura, de corpo e tampa amarelos, contendo 2 comprimidos revestidos rosas e 2 comprimidos brancos. Coledue R 10mg + 20mg apresenta-se como cápsula gelatinosa dura, de corpo branco e tampa verde, contendo 4 comprimidos revestidos rosas e 2 comprimidos brancos.
Antes de usar, observe o aspecto do medicamento. Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
O paciente deve estar recebendo uma dieta hipolipemiante adequada e deve continuá-la durante o tratamento com Coledue R. Coledue R deve ser administrado por via oral, uma vez ao dia, com ou sem a ingestão de alimentos. A dose inicial recomendada é de Coledue R 10mg/5 mg ou Coledue R 10mg/10 mg, uma vez por dia, com ou sem a ingestão de alimentos. A terapia deve ser individualizada, podendo ser aumentada até a dose máxima de Coledue R 10mg/20 mg uma vez por dia, de acordo com os níveis lipídicos desejados, o objetivo recomendado da terapia e a resposta do paciente. Se necessário, o ajuste de dose pode ser feito em intervalos de 4 semanas. A dose máxima diária de Coledue R 10mg/20 mg não deve ser excedida. Para pacientes com hipercolesterolemia grave (incluindo hipercolesterolemia familiar heterozigótica) ou aqueles pacientes que necessitam atingir metas agressivas de redução de LDL-C, podese considerar uma dose inicial de Coledue R 10mg/20 mg por dia.
Populações especiais Idosos: utiliza-se a faixa de dose habitual.
Pacientes com insuficiência renal: a faixa de dose habitual se aplica a pacientes com insuficiência renal de leve a moderada. Coledue R é contraindicado para pacientes com insuficiência renal grave (TFGe < 30 mL/min/1,73 m2) Pacientes com insuficiência hepática: não é necessário ajuste posológico para pacientes com insuficiência hepática leve. O tratamento com Coledue R não é recomendado para pacientes com insuficiência hepática moderada ou grave. Foi observado aumento da exposição sistêmica à rosuvastatina em pacientes com insuficiência hepática grave, portanto, o uso de doses superiores a 10/10mg em pacientes com doenças hepáticas deve ser cuidadosamente considerado. Raça: não há dados específicos relativ