CALQUENCE
ASTRAZENECA
acalabrutinibe
Inibidor da proteína quinase.
Apresentações.
Cápsulas duras de 100 mg em embalagem com 60 cápsulas.
VIA ORAL
USO ADULTO
Composição.
CALQUENCE 100 mg
Cada cápsula dura contém 100 mg de acalabrutinibe. Excipientes: celulose microcristalina, dióxido de silício, amido, estearato de magnésio e amidoglicolato de sódio. Cápsula dura: gelatina, dióxido de titânio, óxido de ferro amarelo e azul de indigotina. Tinta de impressão: goma laca, óxido de ferro preto, propilenoglicol e hidróxido de amônio.
Informações técnicas.
1. INDICAÇÕES
CALQUENCE (acalabrutinibe) é indicado para o tratamento de pacientes adultos com linfoma de células do manto (LCM) que receberam pelo menos uma terapia anterior. Esta indicação foi aprovada com base na Taxa de Resposta Global (Ver seção 2. Resultados de Eficácia). O benefício clínico deverá ser validado por estudo clínico confirmatório de Fase 3, o qual já está em andamento.
2. RESULTADOS DE EFICÁCIA
Eficácia clínica
A segurança e a eficácia da CALQUENCE no LCM foram avaliadas em um estudo de Fase 2, aberto, multicêntrico e de braço único (ACE-LY-004) com 124 pacientes previamente tratados. Todos os pacientes receberam CALQUENCE 100 mg por via oral duas vezes ao dia até a progressão da doença ou toxicidade inaceitável. O estudo não incluiu pacientes que receberam tratamento anterior com inibidores da BTK (tirosina quinase de Bruton). O desfecho primário foi a taxa de resposta global (TRG) avaliada pelo investigador pela classificação de Lugano para linfoma não Hodgkin (LNH). A duração da resposta (DR) foi uma medida adicional de resultado. Os resultados de eficácia são apresentados na Tabela 1.
A idade média dos pacientes foi 68 anos (variando de 42 a 90 anos); 79,8% eram homens e 74,2% eram caucasianos. Na linha basal, 92,8% dos pacientes tiveram um nível de desempenho na ECOG (Eastern Cooperative Oncology Group - Grupo Oncológico Cooperativo do Leste) de 0 ou 1. O tempo médio desde o diagnóstico foi de 46,3 meses e o número médio de tratamentos anteriores foi de 2 (intervalo de 1 a 5), incluindo 17,7% com transplante prévio de células tronco. Os regimes de tratamento anteriores mais comuns foram baseados em CHOP (ciclofosfamida, adriamicina, vincristina e prednisona) (51,6%) e ARA-C (citarabina) (33,9%). Na linha basal, 37,1% dos pacientes apresentavam pelo menos um tumor com o diâmetro ≥ 5 cm, 72,6% apresentavam envolvimento nodal extra, incluindo 50,8% com envolvimento da medula óssea. O índice de prognostico internacional do linfoma de células do manto (MIPI) simplificado (que inclui idade, pontuação ECOG e contagem basal de lactato desidrogenase e glóbulos brancos) foi intermediário em 43,5% e alto em 16,9% dos pacientes.
Com base na avaliação do investigador, 92% dos pacientes que responderam ao tratamento obtiveram uma resposta no segundo ciclo, com uma média de tempo de resposta documentada de 1,9 meses. A DR média não foi alcançada em média de 15,2 meses de acompanhamento após o início do tratamento. Setenta e dois por cento (72%) dos pacientes apresentaram uma resposta contínua aos 12 meses pela estimativa de Kaplan-Meier.
A TRG foi consistente entre todos os subgrupos pré-especificados, incluindo idade, nível do desempenho, estágio da doença e número de terapias anteriores.
Linfocitose
Após início do tratamento com CALQUENCE, houve um aumento temporário da contagem de linfócitos (definido como contagem absoluta de linfócitos ≥ 50% do valor basal, e uma avaliação posterior basal ≥ 5x109) em 31,15% dos pacientes do estudo LY-004. O tempo médio para início da linfocitose foi de 1,1 semana, e a sua duração média foi de 6,7 semanas.
Referências Bibliográficas
Barf et. al -Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J Pharmacol Exp Ther. 2017 Nov;363(2):240-252. doi: 10.1124/jpet.117.242909. Epub 2017 Sep 7
Byrd et al -Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med. 2016 Jan 28;374(4):323-32. doi: 10.1056/NEJMoa1509981. Epub 2015 Dec 7.
Wang et al -Acalabrutinib in relapsed or refractory mantle cell lymphoma (ACE-LY-004): a single-arm, multicentre, phase 2 trial. Lancet. 2017 Dec 11. pii: S0140-6736(17)33108-2. doi: 10.1016/S01406736(17)33108-2.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O acalabrutinibe é um inibidor seletivo de BTK. BTK é uma molécula de sinalização do receptor de antígenos das células B (BCR) e das vias dos receptores das citocinas. A sinalização pela BTK resulta em sobrevivência e proliferação das células B, e é necessária para a adesão celular, transporte e quimiotaxia.
O acalabrutinibe e o seu metabolito ativo, ACP-5862, formam uma ligação covalente com um resíduo de cisteína no sitio ativo da BTK, levando à inativação irreversível de BTK (IC50 ≤ 5 nM), com interações mínimas fora do alvo. Em uma triagem de 380 quinases de tipo selvagem de mamíferos, as únicas interações adicionais de quinases em concentrações clinicamente relevantes de acalabrutinibe e ACP-5862 foram com BMX e ERBB4, com potência de 3 a 4 vezes menor do que com a BTK.
Em estudos não clínicos, acalabrutinibe inibiu a ativação mediada por BTK das proteínas CD86 e CD69 de desregulação, inibiu a proliferação e sobrevivência de células B malignas e teve atividade mínima em outras células imunes (células T e células NK).
Efeitos Farmacodinâmicos
Em pacientes com neoplasias das células B submetidos a doses de 100 mg de acalabrutinibe duas vezes ao dia, manteve-se a ocupação média de BTK no estado de equilíbrio de ≥ 95% no sangue periférico por mais de 12 horas, resultando em inativação de BTK ao longo do intervalo de dosagem recomendado.
Eletrofisiologia Cardíaca
Em um estudo QT dedicado, com dose 4 vezes maior que a dose máxima recomendada, concluiu-se que CALQUENCE não prolonga o intervalo QT/QTc a nenhuma extensão clinicamente relevante (ou seja, não superior ou igual a 10 ms).
Propriedades Farmacocinéticas
A farmacocinética de acalabrutinibe foi estudada em indivíduos saudáveis e em pacientes com neoplasia das células B. O acalabrutinibe exibe uma farmacocinética quase linear em um intervalo de dose de 75 a 250 mg e apresenta proporcionalidade à dose. Na dose recomendada de 100 mg duas vezes ao dia, a área sob a curva (ASC) de concentração do medicamento no plasma diária foi de 1111 ng•h/ml e a concentração plasmática máxima (Cmáx) de acalabrutinibe foi de 323 ng/ml.
Absorção
O tempo médio para o pico das concentrações plasmáticas de acalabrutinibe (Tmáx) foi de 0,75 horas. A biodisponibilidade absoluta de CALQUENCE foi de 25%.
Distribuição
A ligação reversível de acalabrutinibe à proteína plasmática humana foi de 97,5% A proporção média in vitro de sangue-plasma foi de 0,7. O volume médio de distribuição (Vss) no estado de equilíbrio foi aproximadamente 34 L.
Biotransformação/Metabolismo
In vitro, o acalabrutinibe é predominantemente metabolizado pela enzima CYP3A e, em menor grau, pela conjugação de glutationa e pela hidrólise de amida. O ACP-5862 foi identificado como o principal metabólito no plasma com exposição geométrica média (ASC) de aproximadamente 2 a 3 vezes maior que a exposição ao acalabrutinibe. Em relação à inibição da BTK, o ACP-5862 é aproximadamente 50% menos potente que o acalabrutinibe.
In vitro, o acalabrutinibe é um inibidor fraco de CYP3A4/5, CYP2C8 e CYP2C9, mas não inibe CYP1A2, CYP2B6, CYP2C19 e CYP2D6.
O metabólito ativo (ACP-5862) é um inibidor fraco de CYP2C8, CYP2C9 e CYP2C19, mas não inibe in vitro CYP1A2, CYP2B6, CYP2D6 ou CYP3A4/5. O acalabrutinibe é um indutor fraco de CYP1A2, CYP2B6 e CYP3A4; o metabólito ativo (ACP-5862) induz fracamente o CYP3A4.
Interações com proteínas de transporte
O acalabrutinibe in vitro não é um substrato de transportadores de absorção renal OAT1, OAT3 e OCT2, ou transportadores hepáticos. O ATP1B1 e OATP1B3. O acalabrutinibe não inibe OAT1, OAT3, OCT2, OATP1B1 e OATP1B3 em concentrações clinicamente relevantes.
Efeitos de acalabrutinibe em P-gp e BCRP
In vitro, o acalabrutinibe é um substrato da glicoproteína P (P-gp) e da proteína de resistência ao câncer de mama BCRP, mas não inibe P-gp. O acalabrutinibe pode inibir os substratos BCRP intestinais (ver seção 6. INTERAÇÕES MEDICAMENTOSAS).
Eliminação
Após uma única dose oral de 100 mg de acalabrutinibe, a meia-vida de eliminação terminal média (t1/2) de acalabrutinibe foi 0,9 horas (intervalo: 0,6 a 2,8 horas). A t1/2 do seu metabolito ativo, ACP-5862, foi 6,9 horas.
A média da depuração oral aparente (CL/F) de acalabrutinibe foi de 159 L/h, com farmacocinética similar entre pacientes e indivíduos saudáveis, baseado na análise da farmacocinética populacional.
Após a administração de uma dose única de 100 mg de [14C]-acalabrutinibe radiomarcado em indivíduos saudáveis, 84% da dose foi recuperada nas fezes e 12% da dose foi recuperada na urina, com menos de 1% da dose excretada como acalabrutinibe inalterado.
Populações especiais
Com base na análise farmacocinética populacional, idade, sexo, etnia (caucasiana, afro-americana), e peso corporal não tiveram efeitos clinicamente significativos na farmacocinética de acalabrutinibe.
Insuficiência renal
Acalabrutinibe sofre uma eliminação renal mínima. Não foi realizado estudo farmacocinético em pacientes com insuficiência renal. Com base na análise farmacocinética populacional, nenhuma diferença farmacocinética clinicamente relevante foi observada em 282 pacientes com insuficiência renal leve (TFGe entre 60 e 89 mL/min/1,73m2 estimado por MDRD e 86 pacientes com insuficiência renal moderada (TFGe entre 30 e 59 mL/min/1,73m2) em comparação a 226 pacientes com função renal normal (TFGe maior ou igual a 90 mL/min/1,73m2). A farmacocinética do acalabrutinibe é desconhecida em pacientes com insuficiência renal grave (TFGe inferior a 29 mL/min/1,73m2) ou insuficiência renal que requer diálise. Pacientes com níveis de creatinina superiores a 2,5 vezes o limite superior normal institucional não foram incluídos nos ensaios clínicos (ver seção 8. POSOLOGIA E MODO DE USAR).
Insuficiência hepática
O acalabrutinibe é metabolizado no fígado. Em um estudo de insuficiência hepática, em comparação com indivíduos com função hepática normal (n = 6), a exposição ao acalabrutinibe (ASC) foi aumentada em 1,9 vezes e 1,5 vezes em indivíduos com insuficiência hepática leve (n = 6) (Child-Pugh A) e moderada (n = 6) (Child-Pugh B), respectivamente. Com base em uma análise farmacocinética populacional, não foi observada qualquer diferença clinicamente relevante nas exposições de acalabrutinibe entre indivíduos com insuficiência hepática leve (n = 41) ou moderada (n = 3) (bilirrubina total entre 1,5 a 3 vezes o LSN e qualquer AST) quando comparados a indivíduos com função hepática normal (n = 527) (bilirrubina total e AST dentro do LSN). A farmacocinética do acalabrutinibe não foi avaliada em pacientes com insuficiência hepática grave (Child-Pugh C ou bilirrubina total entre 3 a 10 vezes ULN e qualquer AST) (ver seção 8. POSOLOGIA E MODO DE USAR).
Dados de segurança pré-clínica
Carcinogenicidade
Estudos de carcinogenicidade não foram realizados com acalabrutinibe.
Genotoxicidade / Mutagenicidade
O acalabrutinibe não foi mutagênico no ensaio de mutação reversa bacteriana, no ensaio in vitro de aberração cromossômica, ou em um ensaio in vivo de micronúcleo de medula óssea de rato.
Toxicidade de dose repetida
A administração oral diária de acalabrutinibe por até 6 meses em ratos, e por até 9 meses em cães, foi tolerada em níveis de exposição que excedem as exposições terapêuticas humanas na dose recomendada (4 vezes em ratos, 14 vezes em cães, com base na ASC).
Em ratos, observaram-se efeitos renais incluindo degeneração tubular em exposições 7 vezes superiores ou mais à dose humana recomendada. Os efeitos renais foram reversíveis com recuperação completa em ratos expostos a níveis 7 vezes a dose humana recomendada, e com recuperação parcial em ratos com exposições maiores (12 vezes ou mais).
Em ratos, foram observados achados hepáticos reversíveis em resposta à dose, incluindo necrose hepatocitária individual, após exposição de 7 vezes superior ou mais à dose humana recomendada.
Em ratos, foram observadas toxicidades cardíacas (hemorragia miocárdica, inflamação, necrose) em exposições equivalentes a 12 vezes ou mais a dose humana recomendada, e foram associadas à morte prematura. A reversão dos problemas cardíacos não pôde ser avaliada, já que esses problemas apenas foram observados em doses acima da dose máxima tolerada. Em exposições que representam 7 vezes a dose humana recomendada, não foram observadas toxicidades cardíacas.
Toxicologia Reprodutiva
Não foram observados efeitos sobre a fertilidade em ratos machos ou fêmeas nas exposições 18 ou 16 vezes maiores que a exposição da ASC humana na dose recomendada, respectivamente.
Em um estudo combinado sobre fertilidade e desenvolvimento embriofetal em ratas fêmeas, acalabrutinibe foi administrado oralmente em doses de até 200 mg/kg/dia, com início 14 dias antes do acasalamento até o dia 17 de gestação. Não foram observados efeitos no desenvolvimento embriofetal e na sobrevivência do feto. A ASC das ratas prenhas que administraram a dose de 200 mg/kg/dia foi aproximadamente 16 vezes maior que a ASC em pacientes que administraram a dose recomendada de 100 mg duas vezes por dia. Foi confirmada a presença do acalabrutinibe e seu metabólito ativo no plasma fetal de ratos.
Em um estudo embriofetal em coelhas prenhas, acalabrutinibe foi administrado por via oral em doses de até 200 mg/kg/dia durante o período da organogênese (dias gestacionais 6 a 8). Acalabrutinibe não produziu toxicidade materna nem evidência de teratogenicidade ou comprometimento no desenvolvimento, crescimento ou sobrevivência fetal em doses de 50 mg/kg/dia (aproximadamente equivalente à exposição da ASC humana na dose recomendada). Foram observadas diminuição do peso corporal fetal e atraso na ossificação em níveis de exposição que produziram toxicidade materna (doses ≥ 100 mg/kg/dia), que foram aproximadamente 4 vezes superiores aos níveis de exposição humana na dose recomendada.
4. CONTRAINDICAÇÕES
Não há contraindicações estabelecidas para utilização de CALQUENCE.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hemorragia
Eventos hemorrágicos graves, incluindo eventos fatais, foram relatados em pacientes com neoplasias hematológicas (n = 610) tratados com CALQUENCE em monoterapia. Os eventos hemorrágicos de grau 3 ou superiores, incluindo gastrointestinais, intracranianos e epitaxiais, foram relatados em 2% dos pacientes. Em ACE-LY-004 (n = 124), um paciente (0,8%) apresentou evento hemorrágico de grau 3.
O mecanismo para eventos hemorrágicos não é bem compreendido. Os pacientes que recebem terapias antiplaquetárias ou anticoagulantes podem estar em maior risco de hemorragia e devem ser monitorados quanto a sinais de hemorragia. O risco benefício de descontinuar CALQUENCE durante pelo menos 3 dias antes e depois da cirurgia deve ser considerado.
No geral, ocorreram eventos hemorrágicos em aproximadamente 50% dos pacientes com neoplasias hematológicas, incluindo hematomas e petéquias de qualquer grau.
Infecções
Infecções graves (bacterianas, virais ou fúngicas), incluindo eventos fatais e infecções oportunistas, foram relatados em pacientes com neoplasias hematológicas (n = 610) tratados com CALQUENCE em monoterapia. As infecções de grau 3 ou superiores ocorreram em 16,2% desses pacientes. A infecção de grau 3 ou 4 mais frequentemente relatada foi a pneumonia. Ocorreram infecções devido à reativação do vírus da hepatite B (HBV) e leucoencefalopatia multifocal progressiva (LMP).
Monitorar os pacientes quanto a sinais e sintomas de infecção e trata-los conforme medicamente apropriado.
Considerar a profilaxia em pacientes com risco aumentado de desenvolver infecções.
Citopenias
As citopenias de grau 3 ou 4 emergentes do tratamento, incluindo neutropenia (22,1%), anemia (10,7%) e trombocitopenia (7,5%) com base em medidas laboratoriais, ocorreram em pacientes com neoplasias hematológicas (n = 610) tratados com CALQUENCE em monoterapia. Consulte a Tabela 4 para frequências de eventos no ACE-LY-004.
Monitorar os hemogramas completos conforme medicamente apropriado.
Novas Neoplasias Malignas Primárias
Novas neoplasias malignas primárias, incluindo carcinomas não cutâneos foram relatadas em 9,5% dos pacientes com neoplasias hematológicas (n = 610) tratados com CALQUENCE em monoterapia, sendo o mais frequente o câncer de pele, relatado em 5,9% dos pacientes. Aconselhar proteção contra a exposição solar.
Fibrilação Atrial
Em pacientes com neoplasias hematológicas (n = 610) tratados com CALQUENCE em monoterapia, foram relatadas fibrilação atrial / palpitação de grau 1 ou 2 em 1,3% dos pacientes, e de grau 3 em 1% dos pacientes. Monitorar a fibrilação atrial e a palpitação e manejar conforme apropriado.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas:
CALQUENCE tem insignificante ou nenhuma influência sobre a capacidade de dirigir veículos e operar máquinas. No entanto, durante o tratamento com acalabrutinibe, fadiga e tonturas foram relatadas, e os pacientes que apresentarem esses sintomas devem ter cautela ao dirigir ou operar máquinas.
Uso durante a gravidez e lactação
Categoria de risco na gravidez: C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião dentista.
Gravidez
CALQUENCE não deve ser usado durante a gravidez, e mulheres em idade fértil devem ser aconselhadas a evitar a gravidez durante o tratamento com acalabrutinibe. Com base em resultados de estudos em animais, pode haver risco para o feto decorrente da exposição à acalabrutinibe durante a gravidez. Não há dados clínicos suficientes sobre o uso de CALQUENCE em mulheres grávidas para informar um risco de defeitos congênitos e abortos espontâneos associado ao medicamento. A administração de acalabrutinibe às coelhas prenhas em exposições 4 vezes a ASC da dose recomendada em humanos foi associada à redução do crescimento fetal (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Lactação
Não se sabe se acalabrutinibe é excretado no leite humano. Não existem dados sobre os efeitos de acalabrutinibe no lactente amamentado ou sobre a produção de leite. O acalabrutinibe e seu metabolito ativo estavam presentes no leite de ratos em lactação. O risco para o lactente não pode ser excluído. As mães que amamentam são aconselhadas a não amamentar durante o tratamento com CALQUENCE e durante 2 dias após receber a última dose.
Fertilidade
Não há dados disponíveis sobre o efeito de CALQUENCE na fertilidade humana. Em um estudo não clínico de acalabrutinibe em ratos machos e fêmeas, não foram observados efeitos adversos nos parâmetros de fertilidade (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
6. INTERAÇÕES MEDICAMENTOSAS
Substâncias ativas que podem aumentar as concentrações plasmáticas de acalabrutinibe
Inibidores da CYP3A
A administração concomitante com um potente inibidor da CYP3A (200 mg itraconazol uma vez por dia durante 5 dias), aumentou a Cmáx e a ASC de acalabrutinibe em 3,7 vezes e 5,1 vezes em indivíduos saudáveis (n = 17), respectivamente. Ao considerar tanto o acalabrutinibe quanto o seu metabolito ativo, simulações farmacocinéticas baseadas em fisiologias (PBPK) com inibidores potentes, moderados e fracos da CYP3A, não apresentaram alterações significativas na ASC total de componentes ativos.
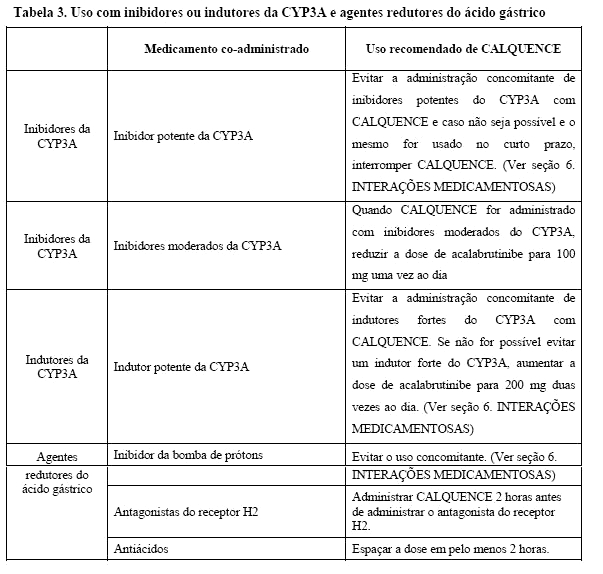
Evitar a administração concomitante de inibidores potentes do CYP3A com CALQUENCE, e caso não seja possível e o mesmo for usado no curto prazo, interromper CALQUENCE. Quando CALQUENCE for administrado com inibidores moderados do CYP3A, reduzir a dose de acalabrutinibe para 100 mg uma vez ao dia (Ver seção 8. POSOLOGIA). Os pacientes em tratamento com inibidores potentes da CYP3A (por exemplo, cetoconazol, conivaptan, claritromicina, indinavir, itraconazol, ritonavir, telaprevir, posaconazol, voriconazol) com CALQUENCE devem ser monitorados mais de perto quanto a reações adversas.
Substâncias ativas que podem diminuir as concentrações plasmáticas de acalabrutinibe
Indutores da CYP3A
A administração concomitante de um potente indutor da CYP3A (600 mg rifampicina uma vez por dia durante 9 dias) diminuiu a Cmáx e a ASC de acalabrutinibe em 68% e 77% em indivíduos saudáveis (n = 24), respectivamente.
Ao considerar tanto o acalabrutinibe quanto o seu metabolito ativo, simulações de PBPK com indutores potentes da CYP3A (carbamazepina e rifampicina) mostraram uma diminuição de 21-51% na ASC total dos componentes ativos. As simulações com um indutor moderado da CYP3A (efavirenz) mostraram uma redução de 25% na ASC total dos componentes ativos.
Evite a administração concomitante de indutores fortes do CYP3A (como fenitoína, rifampicina, carbamazepina) com CALQUENCE, e caso não seja possível, aumentar a dose de acalabrutinibe para 200 mg duas vezes ao dia (Ver seção 8. POSOLOGIA). Evite a erva de São João, que pode diminuir imprevisivelmente as concentrações plasmáticas do acalabrutinibe.
Medicamentos Redutores de Ácido Gástrico
A solubilidade do acalabrutinibe diminui com o aumento do pH. A administração concomitante do acalabrutinibe com um antiácido (1 g de carbonato de cálcio) diminuiu a ASC do acalabrutinibe em 53% em indivíduos saudáveis. A administração concomitante com um inibidor da bomba de prótons (40 mg de omeprazol durante 5 dias), diminuiu a ASC de acalabrutinibe em 43%.
Se o tratamento com um agente redutor de ácido gástrico for necessário, considere o uso de um antagonista do receptor H2 (por exemplo, ranitidina ou famotidina) ou um antiácido (por exemplo, carbonato de cálcio). Para uso com antiácidos, espace a dosagem em pelo menos 2 horas. Para os antagonistas do receptor H2, tome CALQUENCE 2 horas antes de tomar o antagonista do receptor H2.
Evite a administração concomitante com inibidores da bomba de prótons. Devido ao efeito duradouro dos inibidores da bomba de prótons, a separação de doses com inibidores da bomba de prótons pode não eliminar a interação com CALQUENCE.
Substâncias ativas cujas concentrações plasmáticas podem ser alteradas por CALQUENCE
Substratos de CYP3A
Com base em dados in vitro e no modelo de PBPK, não é esperada interação clinicamente significativa com substratos de CYP em concentrações clinicamente relevantes (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Efeitos de Acalabrutinibe em Sistemas Transportadores de Medicamentos
O acalabrutinibe pode aumentar a exposição a substratos de BCRP administrados concomitantemente (por exemplo, metotrexato) pela inibição do BCRP intestinal (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Efeito de alimentos sobre acalabrutinibe
Em indivíduos saudáveis, a administração de uma única dose de 75 mg de acalabrutinibe (0,75 vezes a dose única recomendada) com uma refeição rica em gordura e em calorias (aproximadamente 918 calorias, 59 gramas de carboidrato, 59 gramas de gordura e 39 gramas de proteína) não afetou a ASC média em comparação com a mesma dosagem em condições de jejum. A Cmáx resultante diminuiu 73% e Tmáx foi adiado por 1-2 horas.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
CALQUENCE deve ser conservado em temperatura ambiente (15°C a 30°C).
O prazo de validade de CALQUENCE é 24 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspecto físico
CALQUENCE 100 mg: cápsulas duras com tampa azul e corpo amarelo, com a sigla "ACA 100 mg" impressa em tinta preta.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Modo de usar
CALQUENCE deve ser engolido inteiro com água aproximadamente na mesma hora todos os dias. CALQUENCE pode ser tomado com ou sem alimento. A cápsula não deve ser mastigada, dissolvida ou aberta.
Este medicamento não deve ser partido, aberto ou mastigado.
Posologia
A dose recomendada de CALQUENCE é de 100 mg duas vezes ao dia (equivalente à dose diária total de 200 mg).
As doses devem ser separadas por aproximadamente 12 horas.
O tratamento com CALQUENCE deve ser iniciado e supervisionado por um médico experiente no uso de terapias oncológicas.
O tratamento com CALQUENCE deve continuar até progressão da doença ou toxicidade inaceitável.
Esquecimento de dose
Se um paciente esquecer de tomar uma dose de CALQUENCE por mais de 3 horas, instrua-o a tomar a próxima dose em seu horário habitual. Não se deve tomar cápsulas adicionais de CALQUENCE para compensar a dose perdida.
Ajustes da dose
Reações Adversas
As modificações da dose recomendada de CALQUENCE para reações adversas de grau ≥ 3 são fornecidas na Tabela 2.
Interromper temporariamente CALQUENCE para tratar uma reação adversa não hematológica de grau ≥ 3 relacionada com o tratamento, trombocitopenia de grau 3 com hemorragia significativa, trombocitopenia de grau 4 ou neutropenia de grau 4 com duração superior a 7 dias. Após a resolução da reação adversa para grau 1 ou para o valor da linha basal (recuperação), reinicie CALQUENCE conforme recomendado na Tabela 2.
Populações especiais
População pediátrica
A segurança e a eficácia de CALQUENCE em crianças e adolescentes de até 18 anos de idade ainda não foram estabelecidas.
Pacientes idosos (≥ 65 anos)
Não é necessário qualquer ajuste de dose com base na idade (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS)
Insuficiência Renal
Não é recomendado ajuste da dose em pacientes com insuficiência renal de leve a moderada (TFGe maior ou igual a 30 ml/min/1,73m2 como estimado pelo MDRD (modificação da dieta na equação da doença renal)). A farmacocinética e a segurança da CALQUENCE em pacientes com insuficiência renal grave (TFGe inferior a 29 ml/min/1,73m2) ou doença renal em fase terminal não foram estudadas (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Insuficiência Hepática
Não é recomendado ajuste da dose em pacientes com insuficiência hepática leve ou moderada (Child-Pugh A, Child-Pugh B ou bilirrubina total entre 1,5-3 vezes o limite superior do normal [LSN] e qualquer AST). A farmacocinética e a segurança da CALQUENCE em pacientes com insuficiência hepática grave (Child-Pugh C ou bilirrubina total 3-10 vezes o LSN e qualquer AST) não foram estudadas (ver seção 3. CARACTERÍSTICAS FARMACOLÓGICAS).
9. REAÇÕES ADVERSAS
Resumo Geral de Reações Adversas ao Medicamento
O perfil geral de segurança do acalabrutinibe baseia-se em dados de 610 pacientes com neoplasias hematológicas que receberam monoterapia com acalabrutinibe em doses que variavam de 100 mg duas vezes ao dia a 200 mg duas vezes ao dia. O LCM foi estudado em 124 pacientes a uma dose de 100 mg duas vezes ao dia.
As reações adversas ao medicamento (RAMs) mais comuns (≥ 20%) de qualquer grau relatadas em pacientes que receberam acalabrutinibe foram dor de cabeça, hematoma, diarreia, náusea e erupção cutânea. A maioria das reações adversas reportadas foi de grau 1 ou 2.
As reações adversas de grau ≥ 3 mais frequentemente relatadas (≥ 5%) foram neutropenia (9,3%), anemia (7%) e pneumonia (5,7%).
As reduções de dose e as interrupções decorrentes de eventos adversos foram relatadas em 2,5% e 33,4% dos pacientes, respectivamente. Descontinuações decorrentes de eventos adversos foram relatadas em 6,1% dos pacientes. A intensidade mediana da dose foi de 98,5%.
As seguintes reações adversas graves são discutidas com maiores detalhes na seção 5. ADVERTÊNCIAS E PRECAUÇÕES: hemorragias, infecções, citopenias, novas neoplasias malignas primárias e fibrilação atrial/ palpitação.
Lista tabulada das reações adversas
As seguintes reações adversas ao medicamento (RAMs) foram identificadas em estudos clínicos com pacientes que receberam CALQUENCE em monoterapia como tratamento para neoplasias hematológicas. A duração mediana do tratamento com acalabrutinibe no conjunto de dados agrupados foi de 14,2 meses.
As reações adversas a medicamentos são listadas de acordo com a classe de sistemas de órgãos (SOC) no MedDRA. Dentro de cada classe de órgãos do sistema, as reações adversas ao medicamento são classificadas por frequência, com as reações mais frequentes em primeiro lugar. Dentro de cada grupo de frequência, as reações adversas ao medicamento são apresentadas por ordem decrescente de gravidade. Além disso, a categoria de frequência correspondente para cada RAM é baseada na convenção CIOMS III e definida como: muito comum (≥ 1/10); comum ( > 1/100 a < 1/10); incomum (≥ 1/1.000 a < 1/100); rara (≥ 1/10.000 a < 1/1.000); muito rara ( < 1/10.000); não conhecida (não pode ser estimada a partir dos dados disponíveis).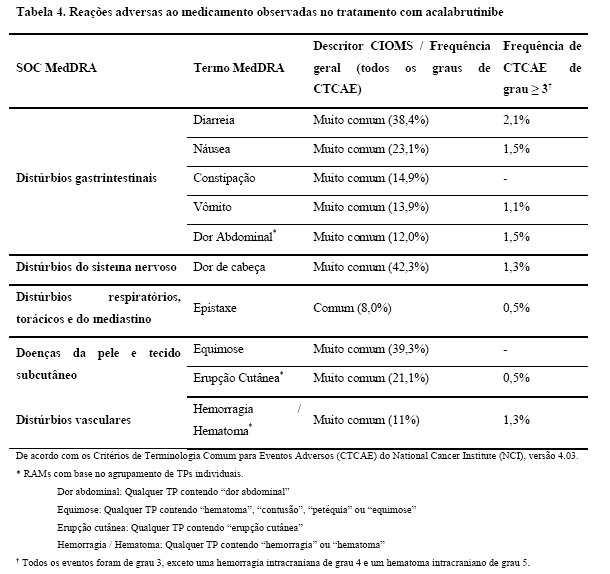

Estudo LY-004
Os dados de segurança descritos abaixo refletem a exposição de 124 pacientes, previamente tratados para LCM, ao CALQUENCE (100 mg duas vezes ao dia) no estudo LY-004 (vide sessão 2. RESULTADOS DE EFICÁCIA). O tempo médio de duração do tratamento com CALQUENCE foi 16,6 meses (variação de 0,1 a 26,6 meses). Um total de 91 pacientes (73,4%) foram tratados com CALQUENCE por ≥ 6 meses, e 74 pacientes (59,7%) foram tratados por ≥ 1 ano.
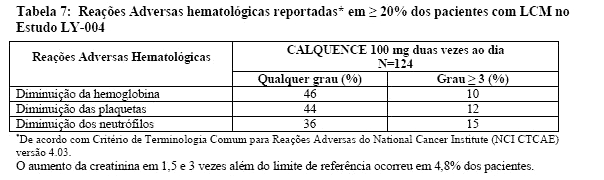
As reações adversas mais comuns de qualquer grau (≥ 20%) foram anemia, trombocitopenia, dor de cabeça, neutropenia, diarreia, fadiga, mialgia e contusões. As reações adversas não-hematológicas de severidade Grau 1 mais comuns foram: dor de cabeça (25%), diarreia (16%), fadiga (20%), mialgia (15%) e contusões (19%). A reação adversa não-hematológica de Grau ≥3 mais comum (reportadas em pelo menos 2% dos pacientes) foi diarreia.
A redução da dose ou descontinuação do tratamento devido a qualquer reação adversa foi reportada em 1,6% e 6,5% dos pacientes, respectivamente.
As Tabelas 6 e 7 apresentam a frequência de reações adversas e suas categorias observadas nos pacientes com LCM tratados com CALQUENCE.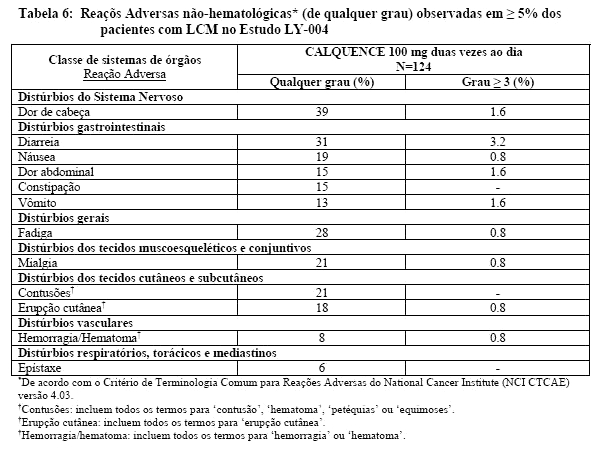
Populações Especiais
Idosos
Oitenta (64,5%) dos 124 pacientes com LCM em estudos clínicos de CALQUENCE tinham 65 anos de idade ou mais, e 32 pacientes (25,8%) tinham 75 anos ou mais. Não se observaram diferenças clinicamente relevantes na segurança ou eficácia entre pacientes com ≥ 65 anos de idade e pacientes mais jovens.
Atenção: Este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária -NOTIVISA, disponível em http://www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
10. SUPERDOSE
Não há tratamento específico para a superdose de acalabrutinibe, e os sintomas da superdose não foram estabelecidos. No caso de uma superdose, os pacientes devem ser acompanhados de perto quanto a sinais ou sintomas de reações adversas e um tratamento sintomático apropriado deve ser instituído.
Em caso de intoxicação ligue para 0800 722 6001, se você precisar de mais orientações.
Dizeres legais.
MS - 1.1618.0269.001-0
VENDA SOB PRESCRIÇÃO MÉDICA
Esta bula foi aprovada pela ANVISA em 03/01/2019.