CABLIVI
SANOFI MEDLEY
caplacizumabe
Tratamento da PTTa.
Apresentações.
CABLIVI® (caplacizumabe) pó liofilizado para solução injetável: embalagem com 1 frasco-ampola com 10 mg de caplacizumabe e 1 seringa preenchida com 1 mL de água para injetáveis.
USO INTRAVENOSO (IV) E USO SUBCUTÂNEO
USO ADULTO
Composição.
Cada frasco-ampola de pó liofilizado branco contendo 10 mg de caplacizumabe. Excipientes (sacarose, ácido cítrico, citrato de sódio di-hidratado, polissorbato 80)
Cada seringa preenchida de diluente contém 1 mL de água para injetáveis.
Informações técnicas.
1. INDICAÇÃO
CABLIVI® é indicado no tratamento de adultos com um episódio de púrpura trombocitopênica trombótica adquirida (PTTa), em conjunto com troca plasmática e imunossupressão.
2. RESULTADOS DE EFICÁCIA
A eficácia e segurança de caplacizumabe em adultos com um episódio de púrpura trombocitopênica trombótica adquirida (PTTa) foram estabelecidas no estudo clínico de Fase III, HERCULES - contemplando um total de 145 pacientes.
HERCULES
HERCULES, um estudo fase III duplo-cego, controlado por placebo, no qual 145 pacientes ([com idade média de 45 anos (18 a 79 anos), sendo 69% do sexo feminino e 73% brancos]) com um episódio de PTTa foram randomizados na proporção de 1:1 para receber CABLIVI® (n=72) ou placebo (n=73) em combinação com troca plasmática e imunossupressão. As características da doença na linha de base eram típicas de púrpura trombocitopênica trombótica adquirida (PTTa). Os pacientes foram estratificados de acordo com a gravidade do envolvimento neurológico (pontuação da escala de coma de Glasgow ≤12 ou 13 a 15). Pacientes com sepse, infecção por E. coli 0157, síndrome hemolítica urêmica atípica, coagulação intravascular disseminada ou púrpura trombocitopênica trombótica congênita não foram elegíveis para a inscrição. Foi administrada aos pacientes uma única injeção intravenosa de caplacizumabe 10 mg ou placebo antes realização da primeira troca plasmática no estudo. Após esta etapa, foi administrada uma injeção subcutânea diária de caplacizumabe ou placebo após a finalização de cada troca plasmática pela duração do período diário de troca plasmática e por 30 dias depois. Se ao final desse período de tratamento houvesse evidência de atividade imunológica persistente subjacente à doença, como níveis de atividade ADAMTS13 suprimidos permanecerem presentes (ou seja, indicativo de um risco iminente de recorrência), o tratamento poderia ser estendido semanalmente por um período máximo de 4 semanas, juntamente com otimização de imunossupressão. Pacientes que apresentaram recorrência durante o tratamento medicamentoso do estudo foram transferidos para um estudo não-cego de caplacizumabe. Esses pacientes receberam novamente o tratamento pela duração da troca plasmática diária e por 30 dias depois. Se, no final deste período de tratamento, houve evidências de doença imunológica subjacente em andamento, o tratamento aberto com caplacizumabe poderá ser estendido semanalmente por um período máximo de 4 semanas, juntamente com a otimização da imunossupressão. Os pacientes tiveram acompanhamento durante 1 mês após a descontinuação do tratamento. Em casos de recorrência durante o período de acompanhamento (após todo o tratamento medicamentoso do estudo ter sido interrompido), não houve reinício do caplacizumabe e a recorrência deveria ser tratada de acordo com o padrão de atendimento. A duração médica do tratamento com caplacizumabe no período duplo cego do estudo foi de 35 dias. No HERCULES, os pacientes foram tratados até no máximo 65 dias. A eficácia do CABLIVI® em pacientes com PTTa foi estabelecida com base no tempo até a resposta da contagem de plaquetas (contagem de plaquetas ≥150.000 / mLseguida pela interrupção da troca plasmática diária em 5 dias). O tratamento com caplacizumabe resultou em uma redução estatisticamente significativa no tempo de resposta à contagem de plaquetas (p < 0,01). Os pacientes tratados com caplacizumabe apresentaram 1,55 vezes mais chances de obter resposta da contagem de plaquetas em um determinado momento, em comparação aos pacientes tratados com placebo.
O tratamento com caplacizumabe resultou em uma redução de 74% na porcentagem de pacientes com morte relacionada ao PTTa, recorrência de PTTa ou pelo menos um evento tromboembólico importante durante o tratamento medicamentoso do estudo (p < 0,0001 -consulte a Tabela 1).
A proporção de pacientes com recorrência de PTTa no período geral do estudo (ou seja, o período de tratamento medicamentoso mais os 28 dias de acompanhamento após a descontinuação do tratamento medicamentoso) foi 67% menor no grupo caplacizumabe (12,7%) em comparação ao grupo placebo (38,4%) (p < 0,001). Nos 6 pacientes do grupo caplacizumabe que apresentaram recorrência de PTTa durante o período de acompanhamento (ou seja, uma recaída), os níveis de atividade do ADAMTS13 foram < 10% no final do tratamento medicamentoso do estudo, indicando que a doença imunológica subjacente ainda estava ativa no momento em que o caplacizumabe foi interrompido. Nenhum paciente tratado com caplacizumabe apresentou doença refratária (definida como ausência de duplicação de plaquetas após 4 dias de tratamento padrão e LDH elevado), em comparação com três pacientes (4,2%) tratados com placebo. Observou-se uma tendência à normalização mais rápida dos marcadores de danos aos órgãos, lactato desidrogenase, troponina I cardíaca e creatinina sérica em pacientes tratados com caplacizumabe. O tratamento com caplacizumabe reduziu o número médio de dias de troca plasmática, o volume de plasma usado, o tempo médio de permanência na Unidade de Terapia Intensiva e o tempo médio de hospitalização durante o período de tratamento medicamentoso do estudo (ver Tabela 2).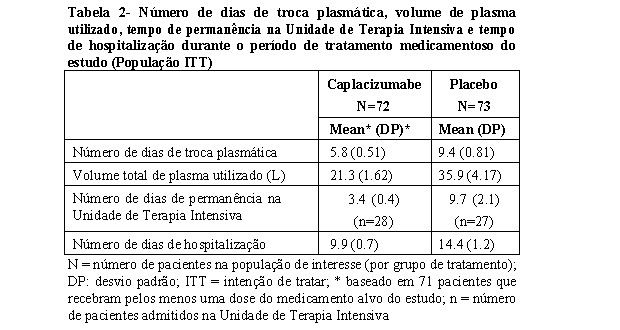
3. CARACTERÍSTICAS FARMACOLÓGICAS
Mecanismo de ação
O caplacizumabe é um nano anticorpo bivalente humanizado (fragmento de anticorpo) que consiste em dois blocos de construção humanizados idênticos (PMP12A2hum1), geneticamente ligados por um ligante de três alanina, visando o domínio A1 do fator de von Willebrand e inibindo a interação entre o fator de von Willebrand e as plaquetas. Como tal, o caplacizumabe evita a agregação plaquetária mediada por Fator de von Willebrand de alto peso molecular, característica da aTTP. Também afeta a disposição do fator de von Willebrand, levando a reduções transitórias dos níveis totais de antígeno do fator de von Willebrand e à redução concomitante dos níveis de fator VIII: C durante o tratamento.
Propriedades Farmacodinâmicas
Inibição alvo
O efeito farmacológico do caplacizumabe na inibição do alvo foi avaliado usando dois biomarcadores para a atividade do fator de von Willebrand; agregação plaquetária induzida por ristocetina (RIPA) e cofator de ristocetina (RICO). A inibição total da agregação plaquetária mediada pelo fator von Willebrand pelo caplacizumabe é indicada pelos níveis de atividade do RIPA e RICO que caem abaixo de 10% e 20%, respectivamente. A dose subcutânea de 10 mg em pacientes com PTTa provocou inibição total da agregação plaquetária mediada pelo fator von Willebrand, como evidenciado pelos níveis de atividade do RICO < 20% aproximadamente 4 horas após a dose e durante o período de tratamento. A atividade do RICO retornou aos valores basais dentro de 7 dias após a descontinuação do medicamento.
Disposição alvo
O efeito farmacológico do caplacizumabe na disposição alvo foi medido usando o antígeno do fator von Willebrand e a atividade de coagulação do fator VIII (fator VIII: C) como biomarcadores. Após a administração repetida de caplacizumabe, observou-se uma diminuição de 30-50% nos níveis de antígeno do fator de von Willebrand em estudos clínicos, atingindo um máximo em 1-2 dias após o tratamento. Como o fator de von Willebrand atua como portador do fator VIII, os níveis reduzidos de antígeno do fator de von Willebrand resultaram em uma redução semelhante nos níveis de fator VIII: C. O antígeno do fator de von Willebrand reduzido e os níveis de FVIII: C foram transitórios e retornaram à linha de base após a interrupção do tratamento.
Propriedades Farmacocinéticas
A farmacocinética do caplacizumabe foi avaliada em sujeitos saudáveis após uma única infusão intravenosa e após uma única e repetidas injeções subcutâneas. A farmacocinética em pacientes com PTTa foi avaliada após uma única e repetidas injeções intravenosas. CABLICI exibe uma farmacocinética não proporcional à dose, caracterizada pela disposição mediada pelo alvo.
Absorção
Após administração subcutânea, caplacizumabe é rapidamente e quase completamente absorvido (F estimado > 0.901) na circulação sistêmica. Em voluntários saudáveis com administração subcutânea de caplacizumabe uma vez ao dia, a concentração máxima foi observada em 6 - 7 horas após a administração e o estado estacionário
Distribuição
Após a absorção, caplacizumabe liga-se ao alvo e é distribuído aos órgãos bem perfundidos. Em pacientes com PTTa o volume central de distribuição estimado é de 6,33 L.
Metabolismo / Eliminação
Presume-se que o caplacizumabe ligado ao alvo seja catabolizado no fígado, enquanto que o caplacizumabe não ligado é eliminado por via renal. A farmacocinética do caplacizumabe depende da expressão do fator de von Willebrand alvo. Níveis mais altos de antígeno do fator von Willebrand, como em pacientes com PTTa, aumentam a fração do complexo droga-alvo retido na circulação. A meia-vida do caplacizumabe é, portanto, dependente da concentração e do nível alvo.
Anticorpos Antidrogas:
Não foram observadas diferenças clinicamente significativas na farmacocinética do caplacizumabe em pacientes com anticorpos antidrogas pré-existentes ou emergentes do tratamento.
Populações Especiais
A farmacocinética do caplacizumabe foi determinada utilizando uma análise farmacocinética da população em dados farmacocinéticos reunidos. As diferenças nas diferentes subpopulações foram investigadas. Nas populações estudadas, sexo e idade (18 a 79 anos), raça, grupo sanguíneo, tratamento concomitante (anticoagulantes, imunossupressores, antibióticos) e a classe de anticorpos antidrogas não afetaram a farmacocinética do caplacizumabe. Enquanto o peso corporal afetou os parâmetros de disposição, não se espera que o nível de exposição previsto resulte em efeitos farmacodinâmicos significativamente diferentes na faixa estudada de 50 a 150 kg. Não foram realizados estudos formais dos efeitos da insuficiência renal ou hepática na farmacocinética de CABLIVI®. Pacientes com insuficiência renal leve (CrCl: 60 a 90 mL / min), moderada (CrCl: 30 a 60 mL / min) ou grave (CrCl: 15 a 30 mL / min) foram incluídos na população PTTa estudada. No modelo PK / PD da população, o comprometimento renal grave foi associado a um aumento mínimo na exposição prevista (AUCss). Nos estudos clínicos de pacientes com PTTa, aqueles com insuficiência renal grave não apresentaram risco adicional de eventos adversos.
DADOS DE SEGURANÇA NÃO CLÍNICOS
Toxicologia Animal e / ou Farmacologia
Foram realizados estudos de toxicologia em porquinhos-da-índia e macacos cynomolgus com doses produzindo exposições respectivas de 50 e 24 vezes a exposição esperada (AUC) a partir da dose diária humana de 10 mg e a farmacologia de segurança foi conduzida como parte dos estudos de dose repetida.
Carcinogênese, mutagênese, comprometimento da fertilidade
Não foram realizados estudos para avaliar o potencial mutagênico do caplacizumabe, pois esses testes não são relevantes para os biológicos. Com base em uma avaliação de risco de carcinogenicidade, estudos dedicados não foram considerados necessários.
Estudos em animais dedicados avaliando os efeitos do caplacizumabe na fertilidade masculina e feminina não foram realizados. Em estudos de toxicidade de dose repetida em macacos cynomolgus, não foi observado impacto do caplacizumabe nos parâmetros de fertilidade em animais machos (tamanho testicular, função espermática, análise histopatológica de testículo e epidídimo) e fêmea (análise histopatológica de órgãos reprodutivos, citologia vaginal periódica).
4.CONTRAINDICAÇÕES
Este medicamento é contraindicado para pacientes com hipersensibilidade ao caplacizumabe ou a qualquer excipiente da formulação.
5.ADVERTÊNCIAS E PRECAUÇÕES
Este medicamento é contraindicado para pacientes com hipersensibilidade ao caplacizumabe ou a qualquer excipiente da formulação.
Sangramento
Caplacizumabe aumenta o risco de sangramento. Foram notificados casos de hemorragia grave, incluindo hemorragia fatal e com risco de vida em pacientes fazendo o uso de caplacizumabe, principalmente naqueles que utilizam agentes antiplaquetários ou anticoagulantes concomitantes. O caplacizumabe deve ser usado com cautela em pacientes com condições subjacentes que podem predispô-los a um risco maior de sangramento. Interrompa o uso de CABLIVI® se ocorrer sangramento clinicamente significativo. Se necessário, o fator de von Willebrand concentrado pode ser administrado para corrigir rapidamente a hemostasia. Se o CABLIVI® for reiniciado, monitore cuidadosamente os sinais de sangramento.
Uso concomitante de anticoagulantes orais ou altas doses de heparina
O risco de sangramento aumenta com o uso concomitante de CABLIVI® com medicamentos que afetam a hemostasia e a coagulação. O início ou a continuação do tratamento com anticoagulantes orais (por exemplo, antagonistas da vitamina K ou anticoagulantes orais diretos [DOAC], como inibidores de trombina ou inibidores do fator Xa) ou heparina em altas doses requer uma avaliação de benefício / risco e monitoramento clínico rigoroso.
Uso concomitante de agentes antiplaquetários e/ou heparina de baixo peso molecular (HBPM)
Embora nenhum risco aumentado de sangramento tenha sido observado em ensaios clínicos, o tratamento concomitante com agentes antiplaquetários e/ou HBPM requer uma avaliação de risco/benefício risco e monitoramento clínico rigoroso.
Pacientes com coagulopatias
Devido a um risco potencial aumentado de sangramento, o uso de caplacizumabe em pacientes com coagulopatias subjacentes (por exemplo, hemofilia, outras deficiências dos fatores de coagulação) deve ser acompanhado por monitoração clínica rigorosa.
Pacientes submetidos à cirurgia
Se um paciente for submetido a uma cirurgia eletiva, a um procedimento odontológico invasivo ou a outras intervenções invasivas, o paciente deve ser aconselhado a informar o médico ou dentista que está usando caplacizumabe e recomenda-se suspender o tratamento por pelo menos 7 dias antes da intervenção planejada. O paciente também deve notificar o médico que supervisiona o tratamento com caplacizumabe sobre o procedimento planejado. Depois que o risco de sangramento cirúrgico for resolvido e o CABLIVI® for retomado, monitore de perto os sinais de sangramento. Se for necessária cirurgia de emergência, recomenda-se o uso do concentrado de fator de von Willebrand para corrigir a hemostasia
Insuficiência hepática grave
Não foram realizados estudos formais com caplacizumabe em pacientes com insuficiência hepática grave e não há dados disponíveis sobre o uso de caplacizumabe nessas populações. O uso de CABLIVI® em pacientes com insuficiência hepática grave requer uma avaliação benefício / risco e um acompanhamento clínico rigoroso devido a um risco potencial aumentado de sangramento.
Pacientes em pediatria
A segurança e a eficácia não foram estabelecidas na população pediátrica.
Gravidez e aleitamento
Não há dados disponíveis sobre o uso de CABLIVI® em mulheres grávidas. Não é possível tirar conclusões sobre se o caplacizumabe é ou não seguro para uso durante a gravidez. O CABLIVI® deve ser utilizado durante a gravidez apenas se os benefícios potenciais para a mãe superarem os riscos potenciais, incluindo aqueles para o feto. Todos os pacientes que recebem CABLIVI®, incluindo mulheres grávidas, correm risco de sangrar. Monitore gestantes e recém-nascidos quanto a qualquer evidência de sangramento excessivo.
Não há dados disponíveis sobre a presença de CABLIVI® no leite humano, efeito sobre a produção de leite ou os efeitos sobre o lactente. Não é possível tirar conclusões sobre se o caplacizumabe é ou não seguro para uso durante a amamentação. CABLIVI® deve ser utilizado durante a amamentação apenas se os benefícios potenciais para a mãe superarem os riscos potenciais, incluindo os da criança amamentada.
Homens e mulheres com potencial reprodutivo
Os efeitos do caplacizumabe na fertilidade em humanos são desconhecidos. Estudos em animais dedicados avaliando os efeitos do caplacizumabe na fertilidade masculina e feminina não foram realizados. Em um estudo de toxicidade de dose repetida de 13 semanas em macacos cynomolgus, não foi observado impacto do caplacizumabe nos parâmetros de fertilidade masculina e feminina
Alterações na capacidade de dirigir veículos e operar máquinas
Não houve estudos para investigar o efeito do caplacizumabe no desempenho de direção ou na capacidade de operar máquinas. Não são previstos efeitos prejudiciais em tais atividades a partir da farmacologia do caplacizumabe. Com base no perfil de segurança clínica, não se espera que o caplacizumabe influencie a capacidade de dirigir ou usar máquinas.
Categoria de risco de gravidez: C. Este medicamento não deve ser usado por mulheres grávidas sem orientação médica.
Não há dados disponíveis sobre o uso a longo prazo de CABLIVI®.
Atenção diabéticos: este medicamento contém açúcar.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo relacionado a interação medicamentosa foi conduzido com caplacizumabe.
Nenhum estudo de interação avaliando o uso de caplacizumabe com anticoagulantes orais (como, por exemplo, antagonistas da vitamina K ou anticoagulantes orais diretos [DOAC] como inibidores da trombina ou inibidores do fator Xa) ou altas doses de heparina foram conduzidos (veja em ADVERTÊNCIAS E PRECAUÇÕES).
7.CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Este medicamento deve ser armazenado sob refrigeração (entre 2° e 8°C). Conservar na embalagem original para proteger da luz. Não congelar.
CABLIVI® pode ser armazenado a uma temperatura não superior a 30°C por um único período de até 2 meses, mas não além do prazo de validade. Não retorne o CABLIVI® ao armazenamento refrigerado após armazenamento em temperatura ambiente.
Após reconstituição
A estabilidade química e física em uso foi demonstrada por 4 horas a 2°C -8°C.
Do ponto de vista microbiológico, a menos que o método de reconstituição impeça o risco de contaminação microbiana, o produto deve ser utilizado imediatamente.
Prazo de validade: 48 meses após a data de fabricação (frasco fechado a 2°C -8°C)
Número do lote e datas de fabricação e validade: consulte a embalagem.
Não use o medicamento com prazo de validade vencido. Mantenha-o em sua embalagem original.
Características físicas e organolépticas
CABLIVI® é um pó liofilizado branco. O solvente é um líquido transparente e incolor
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças
8. POSOLOGIA E MODO DE USAR
O tratamento com CABLIVI® deve ser um complemento da troca plasmática e da terapia imunossupressora.
O tratamento com CABLIVI® deve ser iniciado e supervisionado por médicos com experiência no tratamento de pacientes com microangiopatias trombóticas.
O caplacizumabe deve ser administrado após o início da terapia de troca plasmática. A dose recomendada de CABLIVI® é a seguinte:
Primeira dose
Primeiro dia de tratamento: injeção intravenosa de 10 mg pelo menos 15 minutos antes da troca plasmática, seguida por injeção subcutânea de 10 mg após a conclusão da troca plasmática naquele dia.
Doses subsequentes
Administração subcutânea diária de 10 mg de caplacizumabe após a conclusão de cada troca plasmática durante o tratamento diário de troca plasmática.
Tratamento após período de troca plasmática
Injeção subcutânea diária de 10 mg de caplacizumabe por 30 dias após a interrupção do tratamento diário de troca plasmática. Se, no final deste período, houver evidência de doença imunológica não resolvida, recomenda-se otimizar o regime de imunossupressão e continuar a administração subcutânea diária de 10 mg de caplacizumabe até que os sinais da doença imunológica subjacente sejam resolvidos (por exemplo, normalização sustentada do nível de atividade do ADAMTS13).
No programa de desenvolvimento clínico, o caplacizumabe foi administrado diariamente por até 77 dias. Não existem dados disponíveis sobre repetição de tratamento com caplacizumabe. Recomenda-se suspender o tratamento se o paciente apresentar mais de duas recorrências de PTTa enquanto estiver usando CABLIVI®.
Dose perdida
A administração diária e a continuidade do tratamento são críticas. Contudo, se faltar uma dose de CABLIVI® durante o período de permuta plasmática, deve ser administrada o mais rapidamente possível. Se uma dose de CABLIVI® for perdida após o período de troca plasmática, ela poderá ser administrada dentro de 12 horas após o horário programado de administração. Após 12 horas, a dose esquecida deve ser pulada e a próxima dose diária administrada de acordo com o esquema posológico usual.
Descontinuação para cirurgia e outras intervenções
Reter o tratamento com CABLIVI® 7 dias antes da cirurgia eletiva, procedimentos odontológicos invasivos ou outras intervenções invasivas (consulte 5. ADVERTÊNCIA E PRECAUÇÕES).
Reconstituição e Administração
CABLIVI® deve ser preparado e reconstituído antes da administração. Instruções detalhadas sobre preparação e administração são fornecidas nas Instruções de Uso.
A primeira dose deve ser administrada por injeção intravenosa por um profissional de saúde. As doses subsequentes devem ser administradas por injeções subcutâneas no abdômen por um profissional de saúde.
Não injetar na área ao redor do umbigo e não usar o mesmo quadrante abdominal para injeções consecutivas.
Na ausência de estudos de compatibilidade, o CABLIVI® não deve ser misturado com outros medicamentos. Para administração intravenosa, se estiver usando uma linha intravenosa, a linha pode ser lavada com 0,9% de injeção de cloreto de sódio ou injeção de glicose a 5% (p / v).
Populações Especiais
Pacientes idosos
Os estudos clínicos de CABLIVI® não incluíram número suficiente de indivíduos com 65 anos ou mais para determinar se eles respondem diferentemente dos indivíduos mais jovens. Embora a experiência com o uso de caplacizumabe em idosos seja limitada, não há evidências que sugiram que o ajuste da dose ou precauções especiais sejam necessárias para pacientes idosos (consulte 3. CARACTERÍSTICAS FARMACOLÓGICAS).
Comprometimento renal (diminuição da função renal)
Não é necessário ajuste da dose em doentes com comprometimento renal.
Comprometimento hepático (diminuição da função hepática)
Não existem dados em doentes com comprometimento hepático, uma vez que não foram realizados estudos formais de caplacizumabe nestes doentes. Com base no metabolismo do caplacizumabe, não é necessário ajuste da dose em pacientes com insuficiência hepática (consulte 3. CARACTERÍSTICAS FARMACOLÓGICAS). Em caso de insuficiência hepática crônica, consulte ADVERTÊNCIAS E PRECAUÇÕES para considerações especiais em pacientes com insuficiência hepática grave.
9.REAÇÕES ADVERSAS
A seguinte classificação de frequência do CIOMS é usada, quando aplicável: Muito comum ≥ 10%; Comum ≥ 1 e < 10%; Pouco frequentes ≥ 0,1 e < 1%; Raro ≥ 0,01 e < 0,1%; Muito raro < 0,01%; Desconhecido (a frequência não pode ser calculada a partir dos dados disponíveis).
A segurança do CABLIVI® foi avaliada em dois estudos clínicos controlados por placebo (HERCULES, em que 71 pacientes receberam caplacizumabe; e TITAN, em que 35 pacientes receberam CABLIVI®). Os dados descritos abaixo e em ADVERTÊNCIAS E PRECAUÇÕES refletem a exposição ao CABLIVI® durante os períodos cegos de ambos os estudos, que incluem 106 pacientes com PTTa que receberam pelo menos uma dose, com idades entre 18 e 79 anos, dos quais 69% eram do sexo feminino e 73% eram brancos. A duração média do tratamento com CABLIVI® foi de 35 dias (intervalo de 177 dias).
As reações adversas mais frequentemente relatadas ( > 15%) foram epistaxe, dor de cabeça e sangramento gengival. Sete pacientes (7%) no grupo CABLIVI® experimentaram uma reação adversa que levou ao estudo da descontinuação do medicamento. Nenhuma das reações adversas que levaram à descontinuação foi observada em mais de 1% dos pacientes.
Entre os 106 pacientes tratados com CABLIVI® durante os estudos TITAN e HERCULES, os efeitos adversos hemorrágicos graves relatados em ≥2% dos pacientes incluíram epistaxe (4%). Um caso de hemorragia subaracnóidea foi observado em cada estudo, o que poderia estar relacionado à PTTa ou ao tratamento.
As reações adversas que ocorreram em ≥2% dos pacientes tratados com CABLIVI® e com mais frequência do que naqueles tratados com placebo nos dados agrupados dos dois estudos estão resumidas na Tabela 3: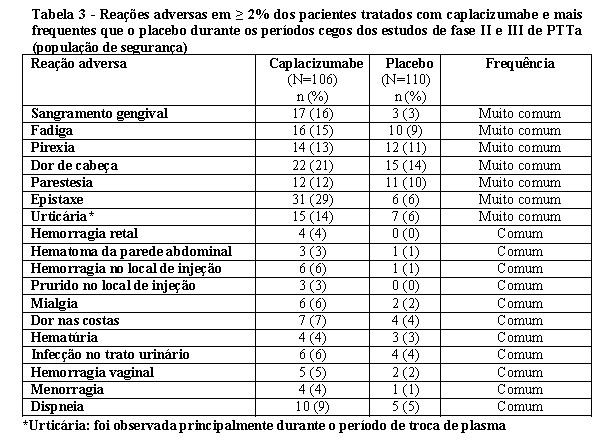
Eventos de Sangramento - Frequência comum (Comum ≥ 1 e < 10%):
Houve 2 notificações dos estudos de fase II e III de PTTa de hemorragia subaracnóidea e uma notificação de infarto cerebral hemorrágico, hemorragia ocular, hemoptise, hematêmese, melena e hemorragia gastrointestinal superior.
Imunogenicidade
Os anticorpos antidrogas emergentes do tratamento (TE ADA) foram detectados em 3,1% dos pacientes tratados com CABLIVI® no estudo HERCULES. TE ADA foram caracterizados como tendo potencial neutralizante. Não houve impacto na eficácia ou segurança clínica.
Reações adversas Pós-Comercialização
As seguintes reações adversas foram identificadas durante o uso pós-aprovação de CABLIVI®. Como essas reações são relatadas voluntariamente por uma população de tamanho incerto, nem sempre é possível estimar com segurança sua frequência ou estabelecer uma relação causal com a exposição ao caplacizumabe.
-Desordens gerais e condições no local de administração: Eritema no local da injeção. Frequência: Desconhecido.
-Desordens do Sistema Sanguíneo e Linfático: Sangramento grave, incluindo eventos fatais e com risco de vida. Frequência: Desconhecido.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema VigiMed, disponível no Portal da Anvisa.
10. SUPERDOSE
Em caso de sobredosagem, com base na ação farmacológica do caplacizumabe, existe o potencial de aumento do risco de sangramento.
Recomenda-se um monitoramento cuidadoso dos sinais e sintomas de sangramento. Se necessário, o uso do concentrado de fator de von Willebrand pode ser considerado para corrigir a hemostasia.
Em caso de intoxicação, ligue para 0800 722 6001 se precisar de mais orientações.
Dizeres legais.
VENDA SOB PRESCRIÇÃO MÉDICA
USO RESTRITO A HOSPITAIS
MS - 1.8326.0480
Esta bula foi aprovada pela Anvisa em 21/10/2021.