BRILINTA
SERVIER
ticagrelor
Antiagregante plaquetário. Antitrombótico.
Apresentações.
Comprimidos revestidos de 90 mg em embalagens com 20 ou 60 comprimidos.
VIA ORAL
USO ADULTO ACIMA DE 18 ANOS
Composição.
Cada comprimido revestido contém 90 mg de ticagrelor. Excipientes: manitol, fosfato de cálcio dibásico, amidoglicolato de sódio, hiprolose, estearato de magnésio, hipromelose, dióxido de titânio, talco, macrogol e óxido de ferro amarelo.
Informações técnicas.
1. INDICAÇÕES
BRILINTA é indicado para a prevenção de eventos trombóticos (morte cardiovascular [CV], infarto do miocárdio [IM] e acidente vascular cerebral [AVC]) em pacientes com Síndrome Coronariana Aguda (SCA) (angina instável, infarto agudo do miocárdio sem elevação do segmento ST [IAMSST] ou infarto agudo do miocárdio com elevação do segmento ST [IAMCST]), incluindo pacientes tratados clinicamente, e aqueles que são tratados com intervenção coronária percutânea (ICP) ou cirurgia de revascularização do miocárdio (RM).
2. RESULTADOS DE EFICÁCIA
A evidência clínica para a eficácia de BRILINTA é procedente do estudo PLATO (PLATelet Inhibition and Patient Outcomes), um estudo comparativo de BRILINTA e clopidogrel, ambos administrados em combinação com ácido acetilsalicílico e outras terapias padrão.
O estudo PLATO foi um estudo randomizado, duplo-cego, de grupos paralelos, fase III, com 18.624 pacientes, que avaliou eficácia e segurança de BRILINTA comparado com clopidogrel para prevenção de eventos vasculares em pacientes com Síndrome Coronariana Aguda (angina instável, infarto agudo do miocárdio sem elevação do segmento ST [IAMSST] ou infarto agudo do miocárdio com elevação do segmento ST [IAMCST]).
O estudo foi composto de pacientes que se apresentaram no prazo de 24 horas do início do episódio mais recente da dor no peito ou sintomas relacionados. Os pacientes foram randomizados para receber clopidogrel (75 mg uma vez ao dia, com uma dose de ataque inicial de 300 mg se a terapia com tienopiridina não houvesse sido administrada anteriormente. Uma dose de ataque adicional de 300 mg foi permitida, a critério do investigador), ou uma dose de ataque de 180 mg de BRILINTA seguido por uma dose de manutenção de 90 mg de BRILINTA duas vezes ao dia. Os pacientes poderiam ser controlados clinicamente, tratados com intervenção coronária percutânea (ICP) ou cirurgia de revascularização do miocárdio (RM).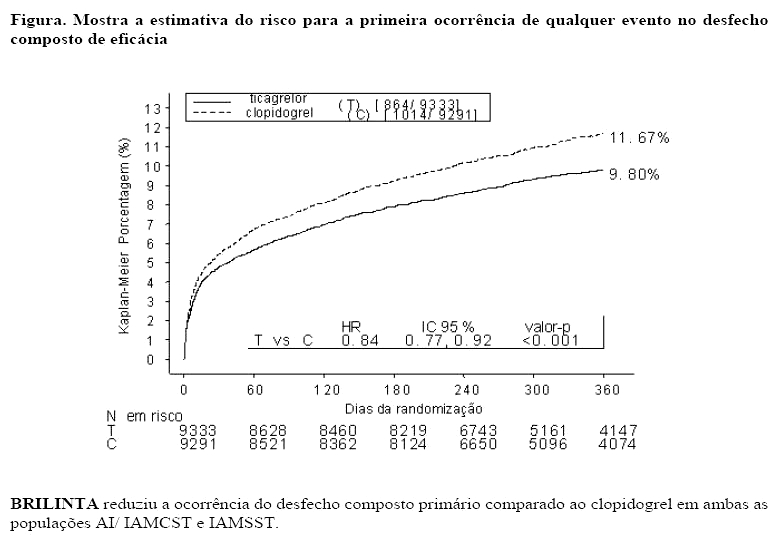
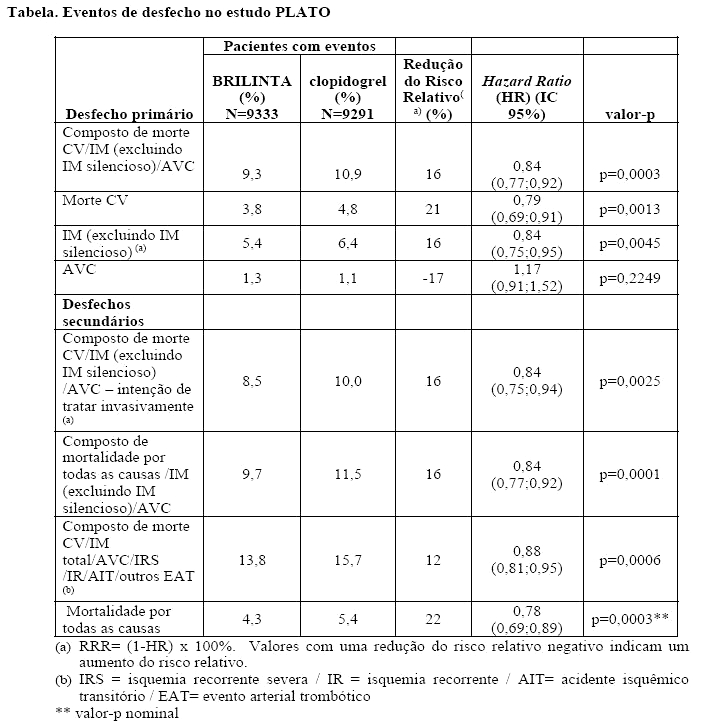
BRILINTA é superior ao clopidogrel na prevenção de eventos trombóticos (RRR 16%; RRA 1,9%; NNT =54) no desfecho composto de eficácia (morte CV, IM e AVC) em 12 meses. A diferença nos tratamentos foi determinada pela morte cardiovascular e infarto do miocárdio sem diferença nos acidentes vasculares cerebrais. BRILINTA demonstrou uma redução do risco relativo estatisticamente significativo de 16% (RRA 1,1%) para IM e uma redução do risco relativo de 21% (RRA 1,1%) para morte CV. Tratar 91 pacientes com BRILINTA ao invés de clopidogrel prevenirá 1 morte CV.
BRILINTA demonstrou superioridade em relação ao clopidogrel na prevenção do desfecho composto (morte CV, IM ou AVC). Os resultados foram precoces (redução de risco absoluto [RRA] de 0,6% e Redução do Risco Relativo [RRR] de 12% em 30 dias), com efeitos observados no tratamento mantidos durante o período de 12 meses, resultando em uma RRA de 1,9% ao ano com RRR de 16%. Isto sugere que o tratamento é apropriado por pelo menos 12 meses (ver item "Posologia e modo de usar").
No estudo PLATO um grande número de comparações de subgrupos foram conduzidas do desfecho de eficácia primário para avaliar a robustez e consistência do benefício global. O efeito do tratamento de BRILINTA em comparação ao clopidogrel parece consistente entre os múltiplos subgrupos de pacientes pelas características demográficas, incluindo peso, sexo, antecedentes clínicos, terapia concomitante e pelo diagnóstico final do evento (IAMSST, IAMCST e AI).
Uma fraca, mas significativa interação do tratamento foi observada por região em que o HR para o desfecho primário favorece BRILINTA no resto do mundo, mas favorece o clopidogrel na América do Norte, que representou aproximadamente 10% do total da população estudada (valor-p da interação = 0,045).
Essa aparente interação de tratamento por região observada no PLATO pode plausivelmente ser atribuída ao acaso, pelo menos em parte. Análises adicionais sugerem que a eficácia de BRILINTA em relação ao clopidogrel está associada à dose de ácido acetilsalicílico durante a terapia de manutenção. Os dados mostram uma maior eficácia de ticagrelor em relação ao clopidogrel, quando utilizados em associação com uma dose baixa de ácido acetilsalicílico (75-150 mg). A eficácia relativa de ticagrelor versus clopidogrel, quando utilizado com altas doses de ácido acetilsalicílico ( > 300 mg) é menos evidente. Baseado nessas observações da relação entre a dose de manutenção do ácido acetilsalicílico e a eficácia relativa de ticagrelor em comparação ao clopidogrel, é recomendado que BRILINTA seja utilizado com uma dose baixa de ácido acetilsalicílico de 75-150 mg (ver itens "Posologia e modo de usar" e "Advertências e precauções").
Os benefícios associados com BRILINTA também foram independentes do uso de outras terapias cardiovasculares indicadas na fase aguda e de longo-prazo, incluindo heparina, heparina de baixo peso molecular (HBPM), inibidores GpIIb/IIIa por via intravenosa, medicamentos hipolipemiantes, betabloqueadores, inibidores da enzima conversora da angiotensina (ECA), antagonistas dos receptores da angiotensina II e inibidores da bomba de prótons (ver item "Interações medicamentosas").
BRILINTA demonstrou uma redução do risco relativo (RRR) estatisticamente significativa no desfecho composto de morte cardiovascular (CV), infarto do miocárdio (IM) e acidente vascular cerebral (AVC) em pacientes com SCA com intenção de tratamento invasivo (RRR 16%; RRA 1,7%; p = 0,0025). Em uma análise exploratória, BRILINTA demonstrou uma redução do risco relativo do desfecho composto primário em pacientes com SCA com intenção de tratamento clínico (RRR 15%; RRA 2,3%; p nominal = 0,0444). Consistente com o desfecho primário do estudo, o efeito nesses dois grupos foi determinado pela morte CV e IM, sem efeito em AVC. Em pacientes recebendo stents houve numericamente menos trombose definitiva de stent entre pacientes tratados com ticagrelor comparado com o clopidogrel (73 versus 107; RRR 32%; RRA 0,6%; p nominal = 0,0123).
BRILINTA demonstrou uma RRR estatisticamente significativa de 16% (RRA 2,1%) para o composto de mortalidade por todas as causas, IM e AVC comparado com o clopidogrel.
O desfecho secundário final (mortalidade por todas as causas) foi avaliado. BRILINTA demonstrou uma RRR de 22% de mortalidade por todas as causas comparado com o clopidogrel com um nível de significância de p = 0,0003 e uma RRA de 1,4%.
Subestudo de Holter
Para estudar a ocorrência de pausas ventriculares e outros episódios arrítmicos durante o estudo PLATO, investigadores realizaram monitoramento de Holter em um subconjunto de cerca de 3.000 pacientes, dos quais aproximadamente 2.000 tinham gravações tanto na fase aguda da SCA quanto um mês depois. A principal variável de interesse foi a ocorrência de pausas ventriculares ≥ 3 segundos. Mais pacientes tiveram pausas ventriculares com BRILINTA (6,0%) do que com o clopidogrel (3,5%) na fase aguda; e 2,2% e 1,6%, respectivamente, um mês depois. Mais pacientes tiveram pausas ventriculares com BRILINTA que com clopidogrel, entretanto, não houve consequências clínicas adversas associadas a esta diferença (incluindo inserções de marcapasso) nesta população de pacientes.
O ticagrelor é ativo oralmente. Diferente do clopidogrel, ele não requer a atividade enzimática da CYP450 para inibir a agregação plaquetária. Polimorfismos no gene codificador para a enzima 2C19 da CYP450 podem impactar a eficácia de clopidogrel. Polimorfismo no gene codificador para o transportador (ABCB1) da glicoproteína-P pode impactar na eficácia de ambos, clopidogrel e ticagrelor.
No estudo PLATO, amostras genéticas de 10.285 pacientes foram analisadas para determinação do genótipo do CYP2C19 e loco ABCB1. Foram analisadas associações de grupos de genótipo em relação aos resultados de eficácia e segurança do estudo PLATO.
· A superioridade de BRILINTA em relação ao clopidogrel não é significativamente afetada em pacientes genótipo CYP2C19.
· BRILINTA reduz eventos CV maiores comparado ao clopidogrel independentemente do genótipo CYP2C19.
· As taxas de eventos para BRILINTA não variaram com o genótipo CYP2C19.
· No grupo tratado com clopidogrel, portadores do alelo da CYP2C19 de perda da função tiveram um aumento das taxas de eventos de desfecho primário em comparação aos não-portadores.
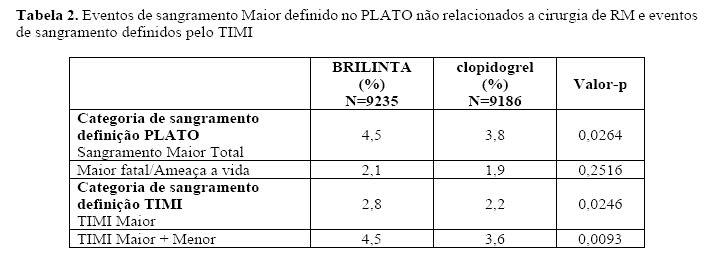
· Assim como no estudo PLATO, o sangramento maior total não diferiu entre BRILINTA e clopidogrel independentemente do genótipo CYP2C19, embora os pacientes com um ou mais alelos de ganho de função (GOF) tenham tido taxas mais altas de sangramento maior com clopidogrel.
· Assim como no estudo PLATO global, o sangramento não relacionado a procedimento cirúrgico de RM aorta-coronariana aumentou com BRILINTA em relação ao clopidogrel em paciente com alelo da CYP2C19 de perda de função.
· Sangramento não relacionado a procedimento cirúrgico de RM aorta-coronariana foi similar entre BRILINTA e clopidogrel em pacientes sem o alelo de perda de função.
Desfechos de eficácia e segurança combinados
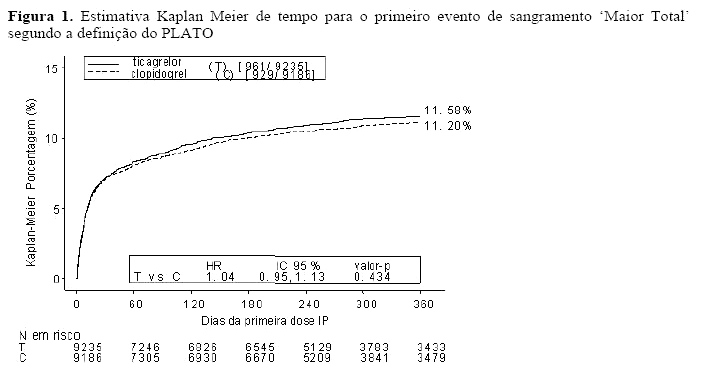
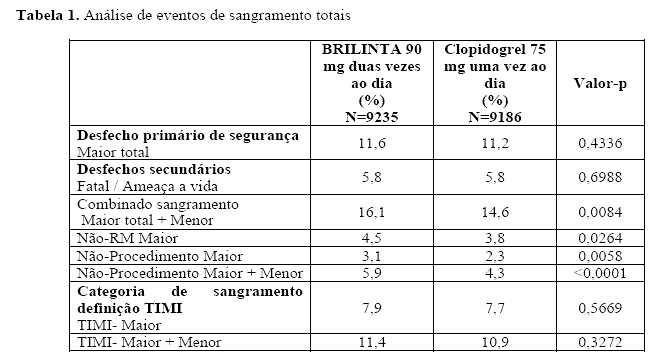
Desfechos de eficácia e segurança combinados (morte CV, IM, AVC, ou sangramento "maior total" segundo a definição do PLATO) sustentam o benefício clínico do ticagrelor comparado com o clopidogrel (RRR 8%; RRA 1,4%; HR 0,92; p = 0,0257) por mais de 12 meses após os eventos de SCA.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades Farmacodinâmicas
- Mecanismo de ação
BRILINTA contém ticagrelor um membro da classe química ciclopentiltriazolopirimidinas (CPTP), que é antagonista seletivo oral, de ação direta e de ligação reversível ao receptor P2Y12 que previne a ativação e agregação plaquetária mediada por adenosina difosfato (ADP) P2Y12 dependente. O ticagrelor não previne a ligação do ADP, mas quando ligado ao receptor P2Y12 previne a transdução de sinal ADP induzida. Como as plaquetas participam na iniciação e/ou evolução de complicações trombóticas da doença arterosclerótica, a inibição da função plaquetária tem demonstrado redução do risco de eventos cardiovasculares como morte, infarto do miocárdio ou acidente vascular cerebral.
O ticagrelor possui um mecanismo de ação adicional, aumentando os níveis de adenosina endógena pela inibição do transportador equilibrativo 1 de nucleosídeo (ENT-1). A adenosina é formada localmente nos pontos de hipóxia e dano tecidual, através da degradação da adenosina tri- e di-fosfato (ATP e ADP) liberada. Como a degradação da adenosina é essencialmente restrita ao espaço intracelular, a inibição do ENT-1 pelo ticagrelor prolonga a meia-vida da adenosina e, portanto, aumenta a sua concentração extracelular local promovendo aumento localizado das respostas à adenosina. O ticagrelor não possui efeito significativo direto nos receptores de adenosina (A1, A2A, A2B, A3) e não é metabolizado à adenosina. Tem sido documentado que a adenosina possui um número de efeitos que incluem: vasodilatação, cardioproteção, inibição da agregação plaquetária, modulação da inflamação e indução de dispneia, o que pode contribuir para o perfil clínico do ticagrelor.
Efeitos farmacodinâmicos:
- Início da Ação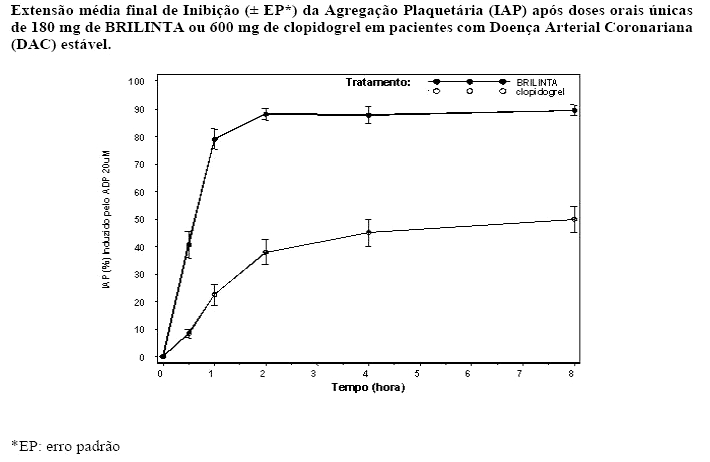
Em pacientes com DAC estável em terapia com o ácido acetilsalicílico, BRILINTA demonstra um rápido início de efeito farmacológico, como demonstrado pela média de IAP para BRILINTA em 0,5 horas após dose de ataque de 180 mg em torno de 41%, com o efeito IAP máximo de 87,9% a 89,6% por 2-4 horas pós-dose. 90% dos pacientes tiveram um alcance final de IAP > 70% por 2 horas pós-dose. O alto efeito da IAP de BRILINTA entre 87% -89% foi mantido entre 2-8 horas.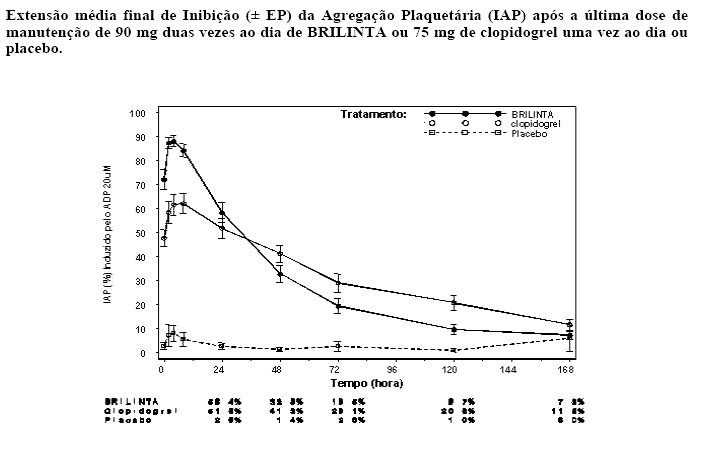
- Reversão de Efeito
Após o declínio das concentrações de BRILINTA e de seu metabólito ativo a um nível inferior ao requerido para saturação do receptor, a IAP diminui gradualmente com o declínio das concentrações plasmáticas. Uma vez que BRILINTA se liga reversivelmente, a recuperação da função plaquetária não depende da reposição de plaquetas. BRILINTA tem uma taxa de reversão mais rápida da IAP em comparação com o clopidogrel, conforme determinado pela inclinação de reversão de 4-72 horas após a última dose (ver item "Advertências e precauções").
A extensão média final da IAP medida após a última dose de BRILINTA é aproximadamente 20-30% maior para BRILINTA comparado com o clopidogrel. Entretanto, por 24 horas pós-dose, a % da IAP é similar entre BRILINTA e clopidogrel, e é menor para BRILINTA a partir de 72 horas em até 7 dias comparado com o clopidogrel. A % média da IAP para BRILINTA em 72 horas (Dia 3) após última dose foi comparável ao clopidogrel no Dia 5, e a % da IAP para BRILINTA no Dia 5 foi similar ao clopidogrel no Dia 7, que não é estatisticamente diferente do placebo.
- Respondedores ao BRILINTA
A IAP induzida por BRILINTA tem menor variabilidade nos picos de concentrações plasmáticas de BRILINTA e seu metabólito ativo observados com a dose de 90 mg duas vezes ao dia em comparação ao clopidogrel. Pacientes com doença arterial coronariana estável, predeterminados a terem menor resposta IAP ao clopidogrel (não-respondedores), e que receberam uma dose concomitante de ácido acetilsalicílico, exibiram maior média de resposta IAP após administração de BRILINTA comparado ao clopidogrel. Em não-respondedores ao clopidogrel, a resposta observada de IAP ao BRILINTA foi maior e mais consistente. O tratamento de BRILINTA resultou em IAP consistentemente mais elevada em comparação com o clopidogrel, e isso foi aparente após a dose para ambos os respondedores e não-respondedores.
- Dados de troca
A troca de clopidogrel para BRILINTA resulta em um aumento absoluto da IAP de 26,4% e a troca de BRILINTA para clopidogrel resulta em uma diminuição absoluta da IAP de 24,5%. Os pacientes podem ser transferidos de clopidogrel para BRILINTA sem a interrupção do efeito antiplaquetário.
- Mecanismo da Adenosina (ENT-1)
O ticagrelor aumenta a concentração plasmática de adenosina em pacientes SCA e tem demonstrado que amplia inúmeras respostas fisiológicas à adenosina. A adenosina é um vasodilatador; ticagrelor demonstrou que amplia o aumento do fluxo sanguíneo coronário induzido por adenosina em voluntários saudáveis e em pacientes SCA. Adenosina é um inibidor plaquetário endógeno; ticagrelor demonstrou que aumenta a inibição da agregação plaquetária mediada por adenosina em adição à inibição plaquetária decorrente do seu antagonismo ao P2Y12. A adenosina está associada ao efeito cardioprotetor de précondicionamento; em um modelo em ratos com lesão de reperfusão, ticagrelor demonstrou redução do tamanho do infarto através do mecanismo mediado por adenosina. A adenosina também induz dispneia; ticagrelor demonstrou ampliação da dispneia adenosina-induzida em voluntários saudáveis. Desta maneira, a dispneia observada em alguns pacientes que utilizam ticagrelor pode ser parcialmente ou completamente mediada por adenosina.
Propriedades Farmacocinéticas
- Geral
O ticagrelor demonstra farmacocinética linear, e a exposição ao BRILINTA e ao metabólito ativo (ARC124910XX) são aproximadamente proporcionais à dose.
- Absorção
A absorção de BRILINTA é rápida, com uma tmax mediana de aproximadamente 1,5 horas. A formação do principal metabólito circulante AR-C124910XX (também ativo) de BRILINTA é rápida, com uma Tmax mediana de aproximadamente 2,5 horas. A Cmax e a AUC de BRILINTA e do metabólito ativo aumentaram de uma maneira aproximadamente proporcional à dose por toda faixa de doses estudadas (301260 mg).
A biodisponibilidade média absoluta de BRILINTA foi estimada em 36% (faixa de 25,4% a 64,0%). A ingestão de uma refeição rica em gordura não teve efeito sobre a Cmax de BRILINTA ou a AUC do metabólito ativo, mas resultou em um aumento de 21% na AUC de BRILINTA e uma diminuição de 22% na Cmax do metabólito ativo. Estas pequenas alterações são consideradas de mínima relevância clínica, portanto, BRILINTA pode ser administrado com ou sem alimentos.
- Distribuição
O volume de distribuição de BRILINTA no estado de equilíbrio é 87,5 L. BRILINTA e o metabólito ativo são extensivamente ligados às proteínas plasmáticas humanas ( > 99,0%).
- Metabolismo
A CYP3A é a principal enzima responsável pelo metabolismo de BRILINTA e a formação do metabólito ativo e suas interações com outros substratos da CYP3A variam da ativação até a inibição. BRILINTA e o metabólito ativo são fracos inibidores da glicoproteína-P.
O principal metabólito de BRILINTA é o AR-C124910XX, que também é ativo como avaliado in vitro pela ligação ao receptor de ADP P2Y12 das plaquetas. A exposição sistêmica ao metabólito ativo é aproximadamente 30-40% do obtido por BRILINTA.
- Excreção
A principal via de eliminação de BRILINTA é por metabolização hepática. Quando BRILINTA marcado radioativamente é administrado, a recuperação média da radioatividade é de aproximadamente 84% (57,8% nas fezes, 26,5% na urina). Recuperações de BRILINTA e do metabólito ativo na urina foram menor que 1% da dose. A primeira via de eliminação do metabólito ativo é principalmente através da secreção biliar. A t1/2 média foi aproximadamente 6,9 horas (faixa 4,5-12,8 horas) para BRILINTA e 8,6 horas (faixa 6,5-12,8 horas) para o metabólito ativo.
- Populações especiais
Idosos: exposições maiores ao BRILINTA (aproximadamente 60% para Cmax e para AUC) e ao metabólito ativo (aproximadamente 50% para Cmax e para AUC) foram observadas em indivíduos idosos (≥ 65 anos) em comparação com indivíduos mais jovens. Estas diferenças não são consideradas clinicamente significativas (ver item "Posologia e modo de usar").
Pediátrico: BRILINTA não foi avaliado em uma população pediátrica (ver item "Posologia e modo de usar").
Sexo: exposições maiores ao BRILINTA (aproximadamente 52% e 37% para Cmax e AUC, respectivamente) e ao metabólito ativo (aproximadamente 50% para Cmax e para AUC) foram observadas em mulheres em relação aos homens. Estas diferenças não são consideradas clinicamente significativas.
Insuficiência renal: a exposição ao BRILINTA foi aproximadamente 20% menor e a exposição ao metabólito ativo foi aproximadamente 17% maior em pacientes com insuficiência renal grave comparado a indivíduos com função renal normal. O efeito de IAP de BRILINTA foi similar entre os dois grupos, entretanto, houve maior variabilidade observada na resposta individual em pacientes com insuficiência renal grave.
Em pacientes com doença renal terminal em hemodiálise, a AUC e a Cmax de BRILINTA 90 mg administradas em um dia sem diálise foram 38% e 51% superiores, respectivamente, em comparação com indivíduos com função renal normal. Um aumento similar na exposição foi observado quando o BRILINTA foi administrado imediatamente antes da diálise, demostrando que BRILINTA não é dialisável. A exposição do metabolito ativo aumentou em menor grau. O efeito da IAP do BRILINTA foi independente da diálise em pacientes com doença renal terminal e semelhante a indivíduos com função renal normal.
Não é necessário ajuste de dose em pacientes com insuficiência renal.
Insuficiência hepática: a Cmax e a AUC para BRILINTA foram 12% e 23% maiores em pacientes com insuficiência hepática leve em comparação com indivíduos saudáveis, respectivamente, entretanto, o efeito de IAP de BRILINTA foi similar entre os dois grupos. Não é necessário ajuste de dose em pacientes com insuficiência hepática leve. BRILINTA não foi estudado em pacientes com insuficiência hepática moderada ou grave (ver item "Posologia e modo de usar").
Raça: pacientes de origem asiática têm uma biodisponibilidade média 39% mais alta em comparação com pacientes caucasianos. Pacientes autoidentificados como negros tiveram uma biodisponibilidade 18% menor de BRILINTA comparados a pacientes caucasianos. Em estudos de farmacologia clínica, a exposição (Cmax e AUC) de BRILINTA em indivíduos japoneses foi aproximadamente 40% (20% após o ajuste para o peso corporal) maior comparada com a de caucasianos.
Dados de segurança pré-clínica
Dados pré-clínicos para o ticagrelor e o principal metabólito não demonstraram risco inaceitável para efeitos adversos para humanos baseado em estudos convencionais de farmacologia de segurança, toxicidade de dose única e repetida e potencial genotóxico.
Reações adversas não observadas em estudos clínicos, mas observadas em animais com níveis de exposição similar ou superior aos níveis de exposição clínica e com possível relevância para o uso clínico foram: GI (gastrointestinais) e irritação gastrointestinal.
Não foram observados tumores relacionados ao composto em um estudo de 2 anos com camundongos em doses orais até 250 mg/kg/dia ( > 18 vezes a exposição terapêutica humana). Não houve aumento nos tumores em ratos machos em doses orais até 120 mg/kg/dia ( > 15 vezes a exposição terapêutica humana). Houve um aumento de adenocarcinomas uterinos e adenomas hepatocelulares mais adenocarcinomas e uma redução nos adenomas hipofisários e fibroadenomas mamários em ratas expostas somente a altas doses ( > 25 vezes a exposição terapêutica humana). Não foi observada alteração na incidência de tumores em doses de 60 mg/kg/dia (diferença > 8 vezes para a dose terapêutica humana.). Os tumores uterinos observados apenas em ratos foram achados ser o resultado de um efeito endócrino não-genotóxico do desequilíbrio hormonal presente em ratos que receberam altas doses de ticagrelor. Os tumores benignos do fígado são considerados secundários a resposta pelo fígado para a carga metabólica localizada no fígado a partir de doses elevadas de ticagrelor.
O ticagrelor foi testado em uma faixa de testes in vitro e in vivo e não foi mostrado ser genotóxico.
O ticagrelor não demonstrou ter efeito na fertilidade de ratas fêmeas em doses orais até 200 mg/kg por dia (aproximadamente 20 vezes a exposição terapêutica humana) e não teve efeito sobre a fertilidade de ratos machos em doses até 180 mg/kg/dia (15,7 vezes a exposição terapêutica humana).
O ticagrelor não teve efeito no desenvolvimento fetal em doses orais até 100 mg/kg por dia em ratos (5,1 vezes a exposição terapêutica humana) e até 42 mg/kg/dia em coelhos (equivalente à exposição terapêutica humana). O ticagrelor não teve efeitos no parto ou no desenvolvimento pós-natal em ratos com doses até 60 mg/kg/dia (4,6 vezes a exposição terapêutica humana).
4. CONTRAINDICAÇÕES
BRILINTA é contraindicado a pacientes com hipersensibilidade ao ticagrelor ou a qualquer componente da fórmula.
Este medicamento é contraindicado a pacientes com sangramento patológico ativo, com antecedente de hemorragia intracraniana e/ou com insuficiência hepática grave.
5. ADVERTÊNCIAS E PRECAUÇÕES
Risco de sangramento
Assim como com outros agentes antiplaquetários, o uso de BRILINTA em pacientes com conhecido risco aumentado de sangramento deve ser balanceado em relação ao benefício em termos de prevenção de eventos trombóticos. Se clinicamente indicado, BRILINTA deve ser utilizado com cuidado nos seguintes grupos de pacientes:
As seguintes considerações devem ser seguidas:
· Pacientes com propensão a sangrar (por exemplo, devido a um trauma recente, cirurgia recente, sangramento gastrointestinal ativo ou recente, ou insuficiência hepática moderada). O uso de BRILINTA é contraindicado em pacientes com sangramento patológico ativo e em pacientes com antecedente de hemorragia intracraniana e insuficiência hepática grave (ver item "Contraindicações").
· Pacientes com administração concomitante de medicamentos que podem aumentar o risco de sangramento (por exemplo, anti-inflamatórios não-esteroidais (AINEs), anticoagulantes orais e/ou fibrinolíticos dentro de 24 horas da dose de BRILINTA).
Transfusão de plaquetas não reverteu o efeito antiplaquetário de BRILINTA em voluntários saudáveis e é improvável que exista benefícios em pacientes com sangramento. Uma vez que a coadministração de BRILINTA com desmopressina não diminuiu o tempo de sangramento padrão, é improvável que a desmopressina seja efetiva no manuseio clínico do sangramento.
Terapia antifibrinolítica (ácido aminocapróico ou ácido tranexâmico) e/ou fator VIIa recombinante pode aumentar a hemostasia. BRILINTA pode ser retomado após a causa de sangramento ter sido identificada e controlada.
Cirurgia
Se um paciente necessita de cirurgia, os médicos devem considerar o perfil clínico de cada paciente, bem como os benefícios e riscos da terapia antiplaquetária continuada determinando quando a interrupção do tratamento de BRILINTA deve ocorrer.
Devido à ligação reversível de BRILINTA, a restauração da agregação plaquetária ocorre mais rapidamente com BRILINTA comparado com o clopidogrel. No estudo OFFSET, a Inibição da Agregação Plaquetária (IAP) média para BRILINTA em 72 horas pós-dose foi comparável à IAP média para o clopidogrel em 120 horas pós-dose. A reversão do efeito mais rápida pode predizer uma redução do risco de complicações hemorrágicas, como por exemplo, em situações nas quais a terapia antiplaquetária deve ser temporariamente interrompida devido a cirurgia ou trauma.
Nos pacientes do estudo PLATO que se submeteram a RM, BRILINTA apresentou uma taxa similar de sangramentos maior em comparação ao clopidogrel em todos os dias da terapia com exceção do Dia 1 onde BRILINTA teve a maior taxa de sangramento.
Se um paciente for submetido a cirurgia eletiva e efeito antiplaquetário não é desejado, BRILINTA deve ser interrompido 5 dias antes da cirurgia. (ver item "Propriedades farmacodinâmicas").
Pacientes com insuficiência hepática moderada
É aconselhada cautela em pacientes com insuficiência hepática moderada, pois não há estudos com BRILINTA nesses pacientes. BRILINTA é contraindicado em pacientes com insuficiência hepática grave (ver item "Contraindicações").
Pacientes com risco de bradiarritmia
A monitorização por Holter ECG demonstrou, na maioria das vezes, uma frequência aumentada de pausas ventriculares assintomáticas durante o tratamento com ticagrelor em comparação com o clopidogrel. Eventos bradiarrítimicos foram relatados no cenário pós-comercialização. Nos estudos de fase 3 para avaliação da segurança e eficácia de BRILINTA, os eventos bradiarrítimicos foram relatados em uma frequência similar para o ticagrelor e comparadores (placebo, clopidogrel e ácido acetilsalicílico). Pacientes com um risco aumentado de eventos bradicárdicos (por exemplo, pacientes sem marcapasso que tinham síndrome do nó sinoatrial, bloqueio atrioventricular de 2° ou 3° grau ou síncope relacionada à bradicardia) foram excluídos dos estudos pivotais de BRILINTA. Portanto, devido à experiência clínica limitada nestes pacientes, recomenda-se precaução (ver também item "Propriedades farmacodinâmicas").
Púrpura Trombocitopênica Trombótica (PTT)
A púrpura trombocitopênica trombótica foi reportada muito raramente com o uso de BRILINTA. PTT é uma condição séria e requer tratamento imediato.
Interferência com testes laboratoriais
Testes de função plaquetária para diagnosticar trombocitopenia induzida pela heparina (TIH)
Resultados falso-negativos no teste da função plaquetária para trombocitopenia induzida por heparina (TIH) foram relatados em pacientes que receberam ticagrelor. Isso está relacionado à inibição do receptor P2Y12 nas plaquetas de doador saudável no teste com soro/plasma do paciente com ticagrelor. A informação sobre o tratamento concomitante com ticagrelor é necessária para interpretação dos testes de função plaquetária da TIH.
Antes de considerar a descontinuação do ticagrelor, o risco/benefício de continuar o tratamento deve ser avaliado, considerando tanto o estado protrombótico da TIH quanto o risco aumentado de hemorragia com tratamento concomitante com anticoagulante e ticagrelor.
Dispneia
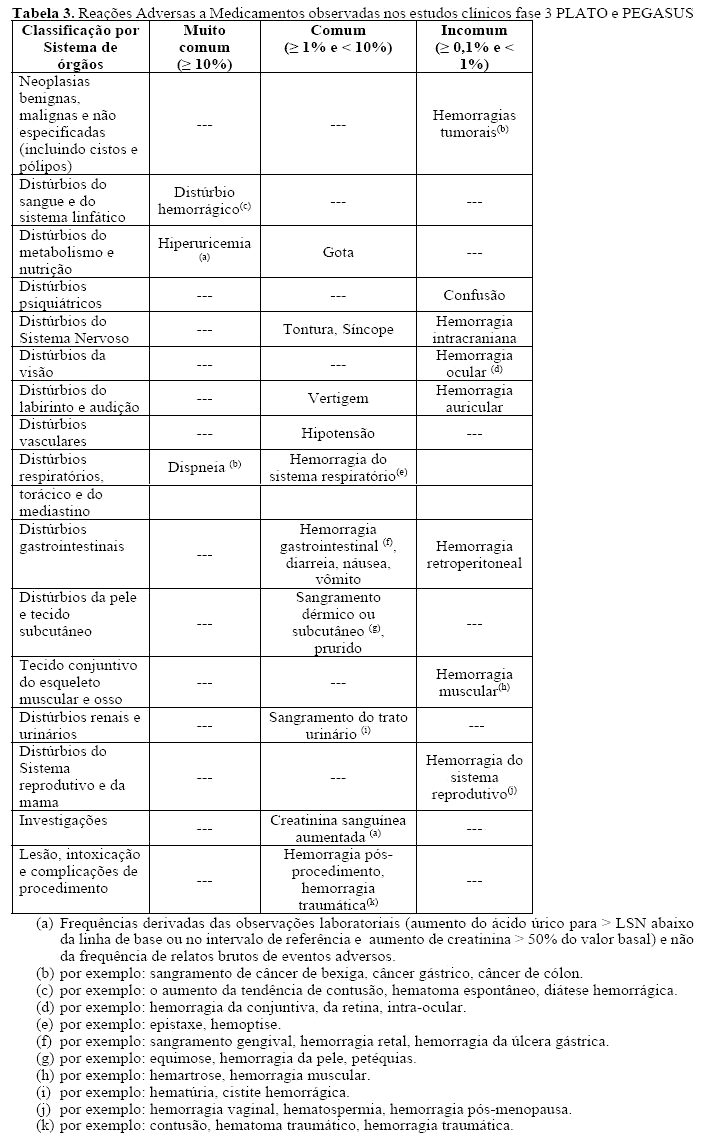
Dispneia, geralmente de leve a moderada intensidade e frequentemente de resolução espontânea sem a necessidade de descontinuação do tratamento, foi relatada em pacientes tratados com BRILINTA (aproximadamente 13,8%) (ver item "Reações adversas"). O mecanismo ainda não foi elucidado. Se o paciente relatar nova, prolongada ou piora da dispneia deve-se fazer uma investigação completa e se não tolerado, o tratamento com BRILINTA deve ser descontinuado.
Outros
Baseado na relação observada no estudo PLATO entre a dose de manutenção de ácido acetilsalicílico e a eficácia relativa do ticagrelor em comparação ao clopidogrel, a coadministração de ticagrelor com altas doses de ácido acetilsalicílico ( > 300 mg) não é recomendada (ver item "Propriedades farmacodinâmicas").
A coadministração de BRILINTA com potentes inibidores da CYP3A4 (por exemplo, cetoconazol, claritromicina, nefazodona, ritonavir e atazanavir) deve ser evitada visto que a coadministração pode levar a um aumento substancial de exposição ao BRILINTA (ver item "Interações medicamentosas").
Descontinuações
Os pacientes que requerem a descontinuação de BRILINTA estão em risco aumentado para eventos cardíacos ou AVC. A descontinuação prematura do tratamento deve ser evitada. Se BRILINTA tiver que ser temporariamente interrompido devido a evento(s) adverso(s), o tratamento deve ser reiniciado assim que os benefícios superarem os riscos do evento adverso ou quando o evento adverso for resolvido (ver item "Posologia e Modo de usar").
Efeito sobre a capacidade de dirigir veículos e operar máquinas
Não foram realizados estudos sobre os efeitos de BRILINTA sobre a capacidade de dirigir veículos e utilizar máquinas. BRILINTA não tem influência ou é insignificante, sobre a capacidade de dirigir veículos e utilizar máquinas. Durante o tratamento da Síndrome Coronariana Aguda, tontura e confusão foram relatadas. Portanto, pacientes que apresentarem estes sintomas devem ser cautelosos enquanto estiverem dirigindo ou utilizando máquinas.
Atenção: este medicamento contém manitol (126 mg/comprimido), portanto, deve ser usado com cautela e a critério médico em pacientes portadores de diabetes.
BRILINTA contém manitol, que pode ter um leve efeito laxativo.
Uso durante a gravidez e lactação
Categoria B
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica.
Não foi conduzido estudo clínico em mulheres grávidas ou lactantes.
Dados clínicos limitados sobre a exposição de BRILINTA durante a gravidez estão disponíveis.
Estudos em animais não indicam efeitos prejudiciais diretos com relação a gravidez, desenvolvimento embrionário/fetal, parto ou desenvolvimento pós-natal. O ticagrelor não teve efeito na fertilidade masculina ou feminina (ver item "Dados de segurança pré-clínica").
Como estudos de reprodução animal nem sempre são preditivos de uma resposta humana, o ticagrelor deve ser usado durante a gravidez somente se o benefício potencial para a mãe justificar qualquer risco potencial para o feto.
Não se sabe se este medicamento é excretado no leite humano. Estudos em ratos demonstraram que o ticagrelor e metabólitos ativos são excretados no leite. O uso de BRILINTA durante a amamentação não é recomendada.
6. INTERAÇÕES MEDICAMENTOSAS
Efeitos de outros medicamentos em BRILINTA
- Medicamentos metabolizados pela CYP3A4
Cetoconazol (potentes inibidores da CYP3A4): a coadministração de cetoconazol com ticagrelor aumentou a Cmax e AUC de ticagrelor igual a 2,4 vezes e 7,3 vezes, respectivamente. A Cmax e AUC do metabólito ativo foram reduzidas em 89% e 56%, respectivamente. Outros potentes inibidores da CYP3A4 (claritromicina, nefazodona, ritonavir e atazanavir), devem ter efeitos similares e não devem ser administrados concomitantemente com BRILINTA (ver item "Advertências e precauções").
Diltiazem (inibidores moderados da CYP3A4): a coadministração de ticagrelor e diltiazem aumentou a Cmax de ticagrelor em 69% e a AUC em 174% e diminuiu a Cmax do metabólito ativo em 38% e a AUC não foi alterada. Não houve efeito de ticagrelor nos níveis plasmáticos do diltiazem. Outros moderados inibidores da CYP3A4 (por exemplo, amprenavir, aprepitanto, eritromicina, fluconazol e verapamil) podem ser coadministrados com BRILINTA.
Rifampicina e outros indutores da CYP3A4: a coadministração de rifampicina com ticagrelor diminuiu a Cmax e AUC de ticagrelor em 73% e 86%, respectivamente. A Cmax do metabólito ativo foi inalterada e a AUC diminuiu em 46%, respectivamente. Outros indutores da CYP3A4 (por exemplo, fenitoína, carbamazepina e fenobarbital) devem diminuir a exposição ao ticagrelor e poderiam resultar em eficácia reduzida de BRILINTA.
Ciclosporina (GpP - glicoproteína P e inibidor CYP3A): a coadministração de ciclosporina (600 mg) com ticagrelor aumentou a Cmax e AUC de ticagrelor em 2,3 vezes e 2,8 vezes, respectivamente. A AUC do metabólito ativo aumentou 32% e a Cmax diminuiu 15% na presença da ciclosporina. Não houve efeito de ticagrelor nos níveis plasmáticos da ciclosporina.
Outros: Estudos de interação de farmacologia clínica demonstraram que a coadministração de ticagrelor com heparina, enoxaparina e ácido acetilsalicílico não têm qualquer efeito sobre os níveis plasmáticos de ticagrelor ou do metabólito ativo. A coadministração de ticagrelor e heparina não teve efeito sobre a heparina baseado nos testes de Tempo de Tromboplastina Parcial ativada (TTPa) e Tempo de Coagulação Ativado (TCA). A coadministração de ticagrelor e enoxaparina não teve efeito sobre a enoxaparina com base no teste de fator Xa.
Uma exposição menor e tardia aos inibidores P2Y12 por via oral, incluindo o ticagrelor e o seu metabolito ativo, foi reportada em pacientes tratados com morfina (aproximadamente 35% de redução no ticagrelor). Essa interação pode estar relacionada à redução da motilidade gastrintestinal, portanto, se aplica a outros opioides. A relevância clínica é desconhecida.
Efeitos de BRILINTA em outros medicamentos
- Medicamentos metabolizados pela CYP3A4
Sinvastatina: a coadministração de ticagrelor com sinvastatina aumentou a Cmax da sinvastatina em 81% e a AUC em 56% e aumentou a Cmax em 64% e a AUC em 52% da sinvastatina ácida, com alguns aumentos individuais iguais a 2 a 3 vezes. Consideração de significância clínica deve ser dada referente a magnitude e variação de alterações na exposição a sinvastatina em pacientes que requerem mais de 40 mg de sinvastatina. Não houve efeito da sinvastatina nos níveis plasmáticos de ticagrelor. BRILINTA pode ter efeito similar sobre a lovastatina, mas não é esperado ter um efeito clinicamente significativo sobre outras estatinas.
Atorvastatina: a coadministração de atorvastatina e ticagrelor aumentou a Cmax da atorvastatina ácida em 23% e a AUC em 36%. Aumentos similares na AUC e Cmax foram observados para todos os metabólitos da atorvastatina ácida. Estes aumentos não são considerados clinicamente significativos.
- Medicamentos metabolizados pela CYP2C9 - tolbutamida
A coadministração de ticagrelor com a tolbutamida não resultou em alteração dos níveis plasmáticos de cada fármaco, o que sugere que ticagrelor não é um inibidor da CYP2C9 e é improvável que altere o metabolismo de fármacos mediados pela CYP2C9 como a varfarina e a tolbutamida.
Anticoncepcionais orais: a coadministração de ticagrelor e levonorgestrel e etinilestradiol aumentou a exposição do etinilestradiol em aproximadamente 20%, mas não alterou a farmacocinética do levonorgestrel. Não é esperado efeito clinicamente relevante sobre a eficácia do contraceptivo oral quando levonorgestrel e etinilestradiol são coadministrados com BRILINTA.
Digoxina (substrato da GpP - glicoproteína P): a administração concomitante de ticagrelor aumentou a Cmax da digoxina em 75% e a AUC em 28%. Portanto, monitoramento laboratorial e/ou clínico adequado é recomendado quando da administração de medicamentos dependentes da GpP- glicoproteína P de índice terapêutico estreito como a digoxina concomitantemente com BRILINTA.
Outras terapias concomitantes: em estudos clínicos, BRILINTA foi geralmente administrado com ácido acetilsalicílico, heparina, heparina de baixo peso molecular, inibidores da GpIIb/IIIa por via intravenosa, inibidores da bomba de prótons, estatinas, betabloqueadores, inibidores da enzima conversora da angiotensina e bloqueadores dos receptores da angiotensina, conforme a necessidade para condições concomitantes. Esses estudos não apresentaram qualquer evidência de interações adversas clinicamente significativas.
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
BRILINTA deve ser mantido em temperatura ambiente (15°C a 30°C).
Esse medicamento tem validade de 36 meses a partir da data de fabricação.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Os comprimidos de BRILINTA são apresentados da seguinte maneira: comprimidos revestidos, redondos, biconvexos e de cor amarela, com a impressão 90T de um lado e liso do outro.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Modo de usar
BRILINTA deve ser administrado por via oral e pode ser ingerido com ou sem alimentos.
Os comprimidos de BRILINTA não devem ser partidos ou mastigados.
Posologia
O tratamento de BRILINTA deve ser iniciado com uma dose única de 180 mg (dois comprimidos de 90 mg) e então continuada com a dose de 90 mg duas vezes ao dia.
Os pacientes que estiverem utilizando BRILINTA devem também tomar ácido acetilsalicílico diariamente a menos que especificamente contraindicado. Após uma dose inicial de ácido acetilsalicílico, BRILINTA deve ser utilizado com uma dose de manutenção de 75-150 mg de ácido acetilsalicílico (ver item "Propriedades farmacodinâmicas").
Lapsos durante a terapia devem ser evitados. Se o paciente esquecer-se de tomar uma dose de BRILINTA deve tomar um comprimido de 90 mg (sua próxima dose) no horário programado.
Os