BRAFTOVI
PFIZER
encorafenibe
Antineoplásico.
Apresentações.
Braftovi® 50 mg em blister contendo 28 cápsulas duras (7 blisters com 4 cápsulas cada).
Braftovi® 75 mg em blister contendo 42 cápsulas duras (7 blisters com6 cápsulas cada).
VIA DE ADMINISTRAÇÃO: USO ORAL
USO ADULTO
CUIDADO: AGENTE FOTOTÓXICO
Composição.
Cada cápsula dura de Braftovi® 50 mg contém 50 mg de encorafenibe
Cada cápsula dura de Braftovi® 75 mg contém 75 mg de encorafenibe.
Excipientes: conteúdo da cápsula: copovidona, poloxâmero, celulose microcristalina, ácido succínico, crospovidona, dióxido de silício, estearato de magnésio. Cápsula: gelatina, dióxido de titânio, óxido de ferro vermelho, óxido de ferro amarelo, óxido de ferro preto. Tinta de impressão: goma-laca, óxido de ferro preto, propilenoglicol.
Informações técnicas.
1. INDICAÇÕES
Braftovi® (encorafenibe) é indicado:
- em associação com Mektovi® (binimetinibe) para o tratamento de pacientes adultos com melanoma irressecável ou metastático com mutação BRAF V600 (vide îtem 5. Advertências e Precauções).
- em associação com cetuximabe, para o tratamento de pacientes adultos com câncer colorretal metastático (CCRm) com mutação BRAF V600E, que receberam terapia sistêmica prévia (vide item 5. Advertências e Precauções).
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
Melanoma Irressecável ou Metastático com mutação BRAF V600
A segurança e a eficácia do encorafenibe em combinação com o binimetinibe foram avaliadas em um estudo Fase III, randomizado (1:1:1), ativo-controlado, aberto, multicêntrico de 2 partes, em pacientes com melanoma irressecável ou metastático com mutação BRAF V600E ou V600K (Estudo CMEK162B2301), detectada por um teste para mutação BRAF. Os pacientes possuíam melanoma primário cutâneo ou primário desconhecido confirmado histologicamente, porém aqueles que apresentavam melanoma uveal ou mucoso foram excluídos. Os pacientes foram autorizados a receber terapia prévia adjuvante e uma linha prévia de imunoterapia para doença irresecável localmente avançada ou metastática. O tratamento prévio com inibidores de BRAF/MEK não foi permitido.
Estudo CMEK162B2301, parte 1
Na parte 1, os pacientes do estudo foram randomizados para receber a combinação de encorafenibe 450 mg via oral diariamente e binimetinibe 45 mg via oral duas vezes ao dia (Combo 450, n=192), encorafenibe 300 mg via oral diariamente (Enco 300, n=194), ou vemurafenibe 960 mg via oral duas vezes ao dia (Vem, n=191). O tratamento se manteve contínuo até a progressão da doença ou toxicidade inaceitável. A randomização foi estratificada pela classificação do Comitê Conjunto Americano para Estadiamento do Câncer (AJCC, do inglês American Joint Committee on Cancer) (IIIB, IIIC, IVM1a ou IVM1b, vs IVM1c), escala de desempenho ECOG, do inglês Eastern Cooperative Oncology Group (0 versus 1) e imunoterapia prévia para doença irresecável ou metastática (sim versus não).
O desfecho primário de eficácia foi a sobrevida livre de progressão (PFS, do inglês progression free survival) do Combo 450 em comparação com o vemurafenibe, conforme avaliado por um comitê de revisão independente cego (BIRC, do inglês blinded independent review committee). A PFS avaliada pelos investigadores (avaliação do investigador) foi uma análise de apoio. Um desfecho secundário adicional incluiu a PFS do Combo 450 em comparação com o Enco 300. Outras comparações secundárias de eficácia entre o Combo 450 e o vemurafenibe ou o Enco 300 incluíram a sobrevida global (OS, do inglês overall response rate), taxa de resposta objetiva (ORR, do inglês overall response rate), duração da resposta (DoR, do inglês duration of response) e taxa de controle de doença (DCR, do inglês, disease control rate) avaliadas pelo BIRC e pela avaliação do investigador.
A mediana de idade dos pacientes foi de 56 anos (variação de 20-89 anos), 58% eram do sexo masculino, 90% eram caucasianos e 72% tinham status de desempenho (ECOG) 0 no basal. A maioria dos pacientes tinha doença metastática (95%) e estágio IVM1c (64%); 27% apresentavam desidrogenase láctica (DHL) elevada, 45% tinham pelo menos 3 órgãos envolvidos no basal e 3,5% tinham metástases cerebrais. Vinte e sete pacientes (5%) receberam previamente inibidores de ponto de verificação (anti-PD1/PDL1 ou ipilimumabe) (8 pacientes no grupo Combo 450 (4%), 7 no braço do vemurafenibe (4%); 12 no braço do Enco 300mg (6%), incluindo 22 pacientes no estado metastático (6 pacientes no braço do Combo 450, 5 pacientes no braço vemurafenibe, 11 no braço Enco 300) e 5 pacientes cenário da adjuvância (2 no braço do Combo 450, 2 no braço vemurafenibe, 1 paciente no braço Enco 300).
A mediana de duração de exposição foi de 11,7 meses em pacientes tratados com Combo 450, 7,1 meses no braço Enco 300 e 6,2 meses em pacientes tratados com vemurafenibe. A mediana da intensidade da dose relativa (RDI do inglês, median relative dose intensity) para o Combo 450 foi de 100% para o encorafenibe e 99,6% para o binimetinibe; 86,2% para o Enco 300 e 94,5% para o vemurafenibe.
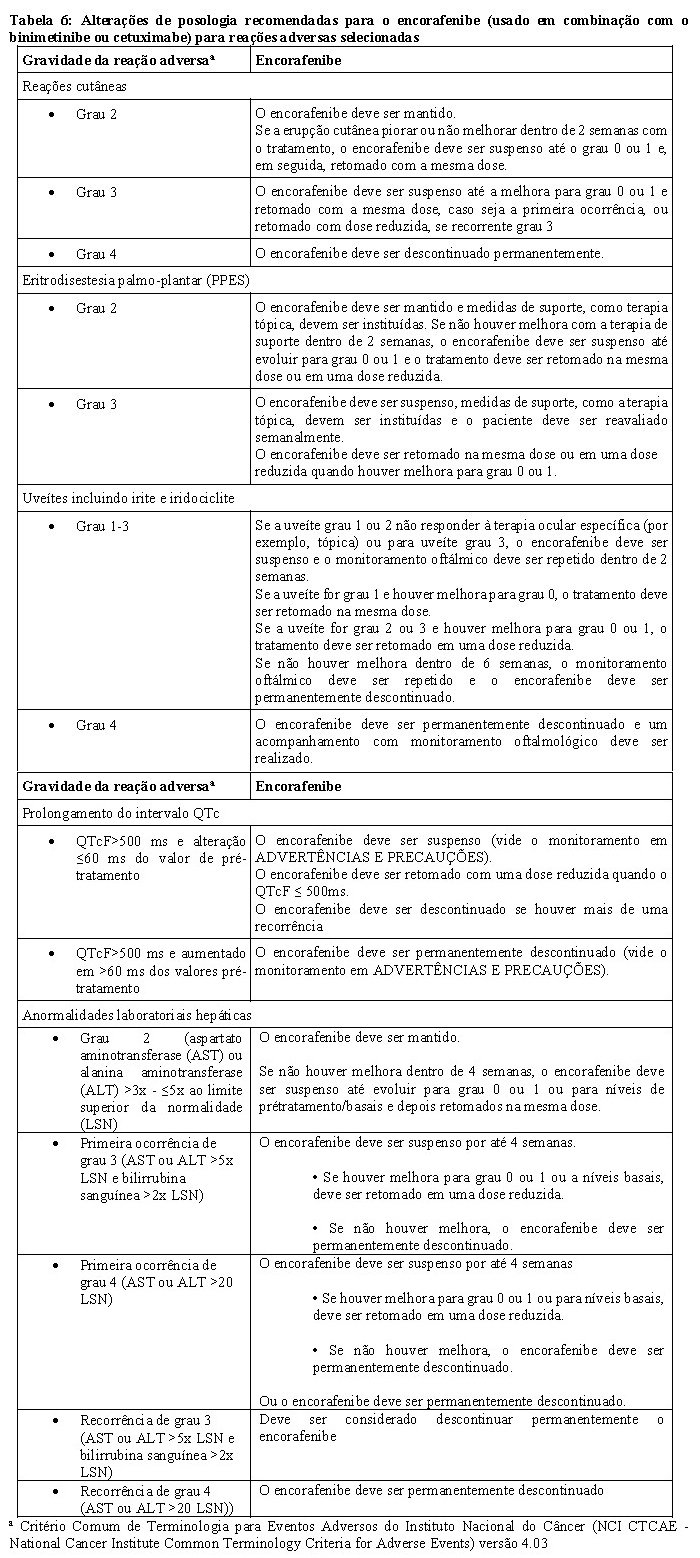
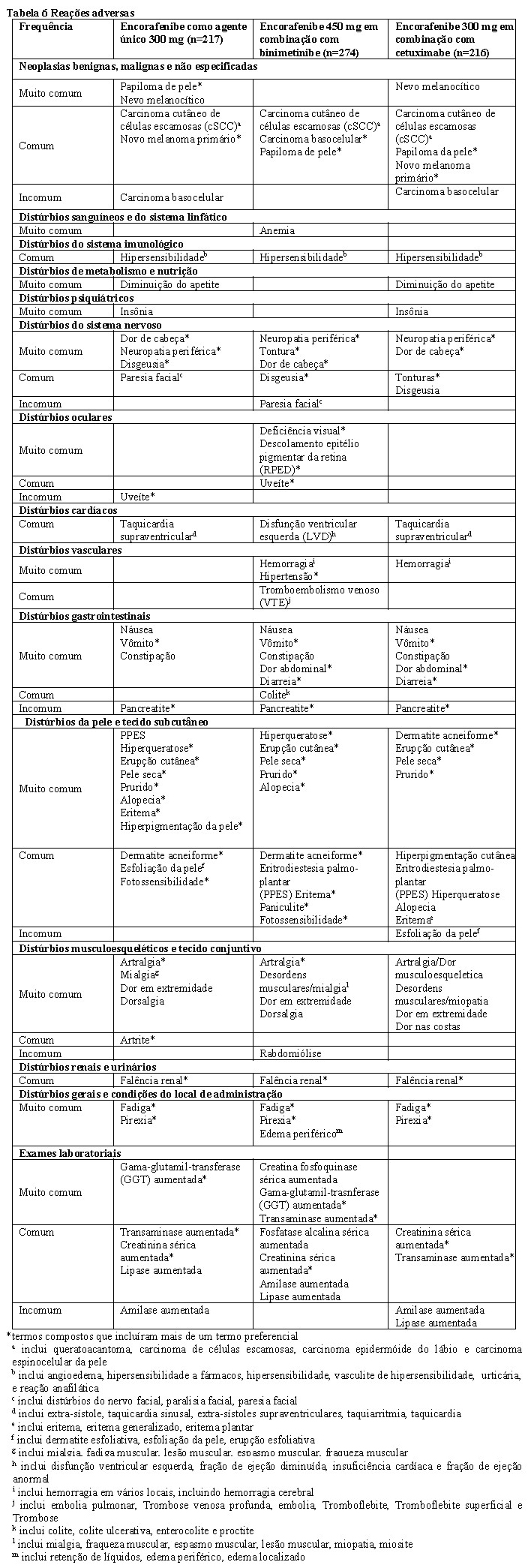
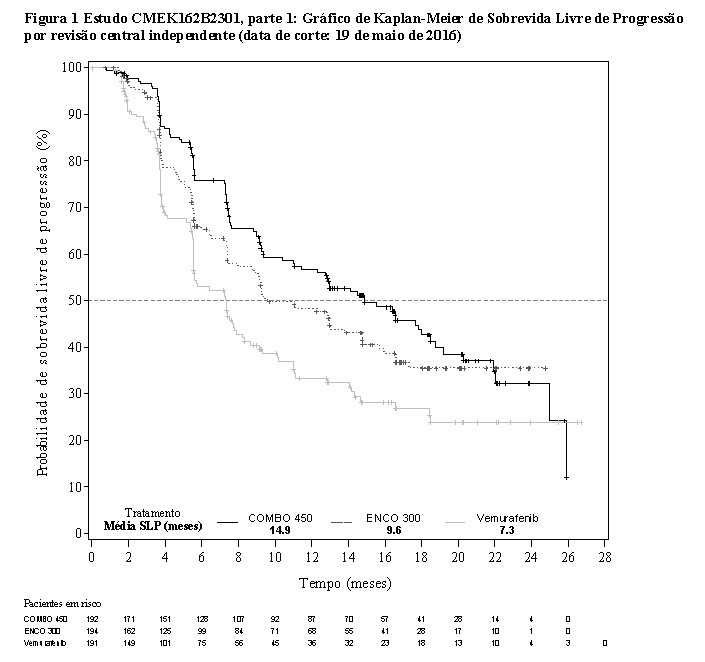
A parte 1 do estudo CMEK162B2301 demonstrou uma melhora estatisticamente significativa na PFS nos pacientes tratados com Combo 450 em comparação com os pacientes tratados com vemurafenibe. A Tabela 1 e a Figura 1 resumem a PFS e outros resultados de eficácia baseados na revisão central dos dados por um comitê cego independente de radiologia.
Os resultados de eficácia baseados na avaliação do investigador foram consistentes com a avaliação central independente (BIRC). Análises não estratificadas de subgrupos demonstraram pontos de estimativas a favor do Combo 450, incluindo DHL basal, status de desempenho ECOG e estágio por AJCC.
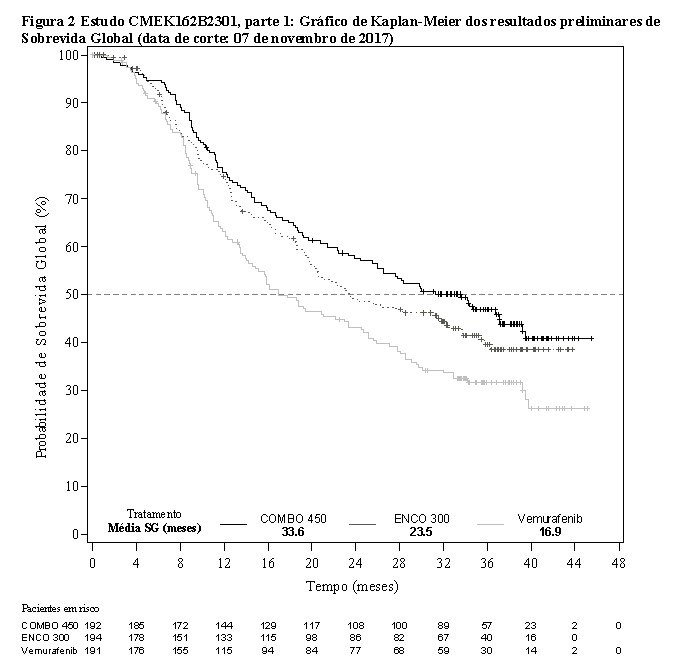
A análise preliminar da OS do Estudo CMEK162B2301 parte 1, (data de corte 07 de novembro de 2017) demonstrou aumento estatisticamente significante na OS para o Combo 450 em comparação com o vemurafenibe (vide Tabela 2 e Figura 2).
Uma proporção similar de pacientes em cada grupo recebeu tratamento subsequente com inibidores de ponto de verificação, principalmente pembrolizumabe, nivolumabe e ipilimumabe (34,4% do braço do Combo 450, 36,1% do braço do encorafenibe, 39,8% do braço do vemurafenibe).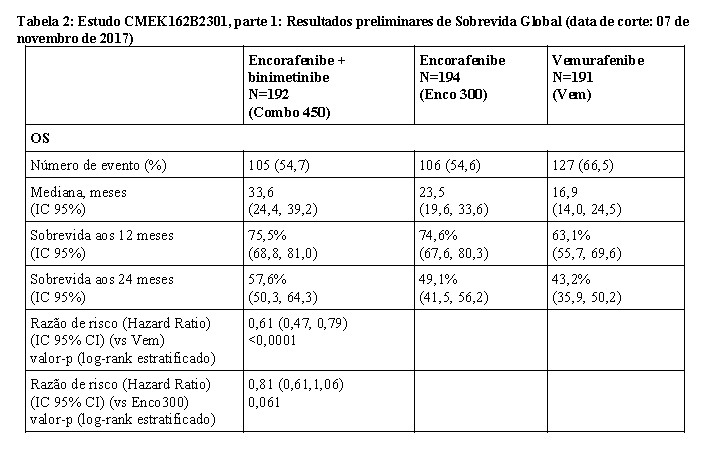
Qualidade de Vida (QoL, do inglês quality of life)) (data de corte: 19 de maio de 2016)
Para explorar as medidas de qualidade vida relacionadas à saúde, status funcional, sintomas do melanoma, reações adversas relacionadas ao tratamento e os resultados relatados pelos pacientes (PRO, do inglês patient response outcomes), foram utilizados a Avaliação Funcional para Terapia de Câncer - Melanoma (FACT-M - Functional Assessment of Cancer Therapy Melanoma), o Questionário de Qualidade de Vida da Organização Europeia para Pesquisa e Tratamento do Câncer (EORTC QLQ-C30 - European Organisation for Research and Treatment of Cancer's core quality of life questionnaire) e o exame EuroQoL-5dimensões- 5níveis (EQ-5D-5L). Uma deterioração definitiva de 10% no FACT-M e no EORTC QLQ-C30 foi significativamente retardada nos pacientes tratados com Combo450 em relação a outros tratamentos. A mediana do tempo para a deterioração definitiva de 10% na escala FACT-M não foi atingida no braço do Combo 450 e foi de 22,1 meses (IC 95%: 15.2, NE) no braço do vemurafenibe com um HR para a diferença de 0.46 (IC 95%: 0.29, 0.72). A análise do tempo para deterioração definitiva de 10% na escala EORTC QLQ-C30 forneceu resultados semelhantes.
Os pacientes que receberam o Combo 450 não relataram mudança ou ligeira melhora na variação média em relação à pontuação basal da escala EQ-5D-5L em todas as consultas, enquanto os que receberam vemurafenibe ou encorafenibe referiram piora em todas as visitas (com diferenças estatisticamente significativas). Uma avaliação de mudança ao longo do tempo na pontuação acarretou a mesma tendência para o EORTC QLQ-C30 e em todas as visitas para o FACT-M.
Estudo CMEK162B2301, parte 2
A parte 2 do estudo CMEK162B2301 foi designada para avaliar a contribuição do binimetinibe para a combinação de encorafenibe e binimetinibe.
A PFS para o encorafenibe 300 mg via oral diariamente utilizada em combinação com o binimetinibe 45 mg via oral duas vezes ao dia (Combo 300, n=258) foi comparada com a PFS para o Enco 300 (n=280, incluindo 194 pacientes da parte 1 e 86 pacientes da parte 2). A inclusão na parte 2 iniciou após todos os pacientes da parte 1 serem randomizados.
Dados preliminares da parte 2, na data de corte de 09 de novembro de 2016, demonstraram a contribuição do binimetinibe com estimativa de mediana de PFS de 12,9 meses (IC 95%: 10.1, 14.0) para o Combo 300 em comparação com 9,2 meses (IC 95%: 7.4, 11.0) para o Enco 300 (partes 1 e 2) por revisão central independente (BIRC). Resultados similares foram observados pela avaliação do investigador.
A ORR confirmada por BIRC foi de 65,9% (IC 95%: 59.8, 71.1) para o Combo 300 e 50,4% (IC 95%: 44.3, 56.4) para o Enco 300 (partes 1 e 2). A mediana de DoR para respostas confirmadas pelo BIRC foi de 12,7 meses [IC 95%: 9.3, 15.1] para o Combo 300 e 12,9 meses [IC 95%: 8.9, 15.5] para o Enco 300. A mediana da duração do tratamento foi mais longa para o Combo 300 versus Enco 300, 52,1 semanas versus 31,5 semanas.
Eletrofisiologia Cardíaca
Na análise de segurança de estudos agrupados, a incidência de novo prolongamento de QTc > 500ms foi de 0,7% (2/268) no grupo encorafenibe 450mg mais binimetinibe e de 2,5% (5/203) no grupo encorafenibe como agente único. O prolongamento de Qtc > 60ms comparado aos valores do pré-tratamento foi observado em 4,9% (13/268) dos pacientes no grupo encorafenibe mais binimetinibe e em 3,4% (7/204) no grupo encorafenibe como agente único.
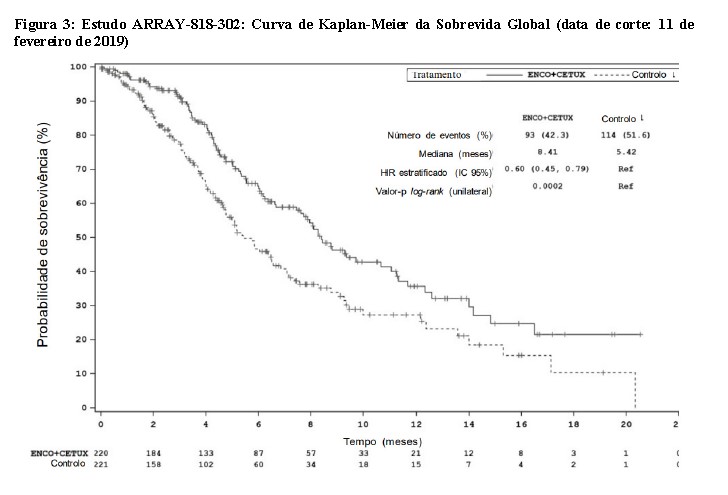
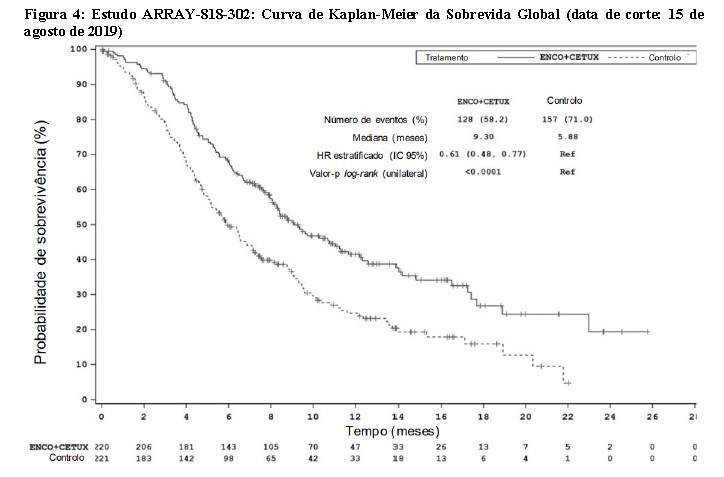
Câncer Coltroetal Metastático com Mutação BRAF V600E - Estudo ARRAY-818-302
Encorafenibe em associação com cetuximabe foi avaliado num ensaio multicêntrico, randomizado, aberto e controlado com comparador ativo (ARRAY 818-302, BEACON CRC). Os pacientes elegíveis deveriam apresentar câncer colorretal metastático com mutação BRAF V600E que tivessem progredido após 1 ou 2 regimes prévios. Os pacientes incluídos foram elegíveis para receber cetuximabe conforme localmente aprovado em relação ao status mutacional do gene RAS. Foi proibida a utilização prévia de inibidores RAF, inibidores MEK ou inibidores EGFR. A randomização foi estratificada pelo status de desempenho (ECOG), utilização prévia de irinotecano e formulação de cetuximabe.
Um total de 665 pacientes (1:1:1) foram randomizados para receber encorafenibe, 300 mg, por via oral, uma vez ao dia, em associação a cetuximabe na dose aprovada em bula (n=220), no grupo dupleto, ou encorafenibe, 300 mg, por via oral, uma vez ao dia, em associação com binimetinibe, 45 mg, por via oral, duas vezes ao dia e cetuximabe na dose aprovada em bula (n=224), no grupo tripleto ou irinotecano mais cetuximabe ou irinotecano/5-fluorouracilo/ácido folínico (FOLFIRI) com cetuximabe, n= 221), no grupo controle. O tratamento foi mantido até à progressão da doença ou toxicidade inaceitável.
Os desfechos de eficácia avaliados incluíram a sobrevivência global (OS) e a taxa de resposta global (ORR) avaliadas por um BIRC, comparando encorafenibe 300 mg em associação a cetuximabe versus o grupo controle. As outras medidas de eficácia encontram-se resumidas na Tabela 3 abaixo.
A idade mediana dos pacientes foi de 61 anos (intervalo 26-91), 47% eram do sexo masculino e 83% caucasianos. Cinquenta e um porcento dos pacientes apresentavam um status de desempenho ECOG na linha basal de 0, 51% e haviam recebido irinotecano previamente; 46,8% dos pacientes tinham pelo menos 3 órgãos com envolvimento tumoral no basal.
A duração mediana da exposição foi de 3,2 meses nos pacientes tratados com encorafenibe 300 mg em associação com cetuximabe e 1,4 meses nos pacientes tratados com irinotecano/cetuximabe ou FOLFIRI/cetuximabe (braço controle). Nos pacientes tratados com a associação de encorafenibe 300 mg e cetuximabe, a mediana da intensidade da dose relativa (RDI) foi de 98% para encorafenibe e de 93,5% para cetuximabe. No braço controle, a mediana da RDI foi de 85,4% para cetuximabe, 75,7% para irinotecano e no subgrupo de pacientes que receberam ácido folínico e 5-FU, a mediana da RDI foi de 75,2% e 75%, respetivamente.
O grupo dupleto de encorafenibe 300 mg em associação com cetuximabe demonstrou uma melhora estatisticamente significativa da OS, da ORR e da PFS em comparação ao controle. Os resultados de eficácia encontram-se resumidos na Tabela 3 e nas Figuras 3 e 4.
Os resultados de eficácia com base na avaliação do investigador foram consistentes com a avaliação do comitê central independente.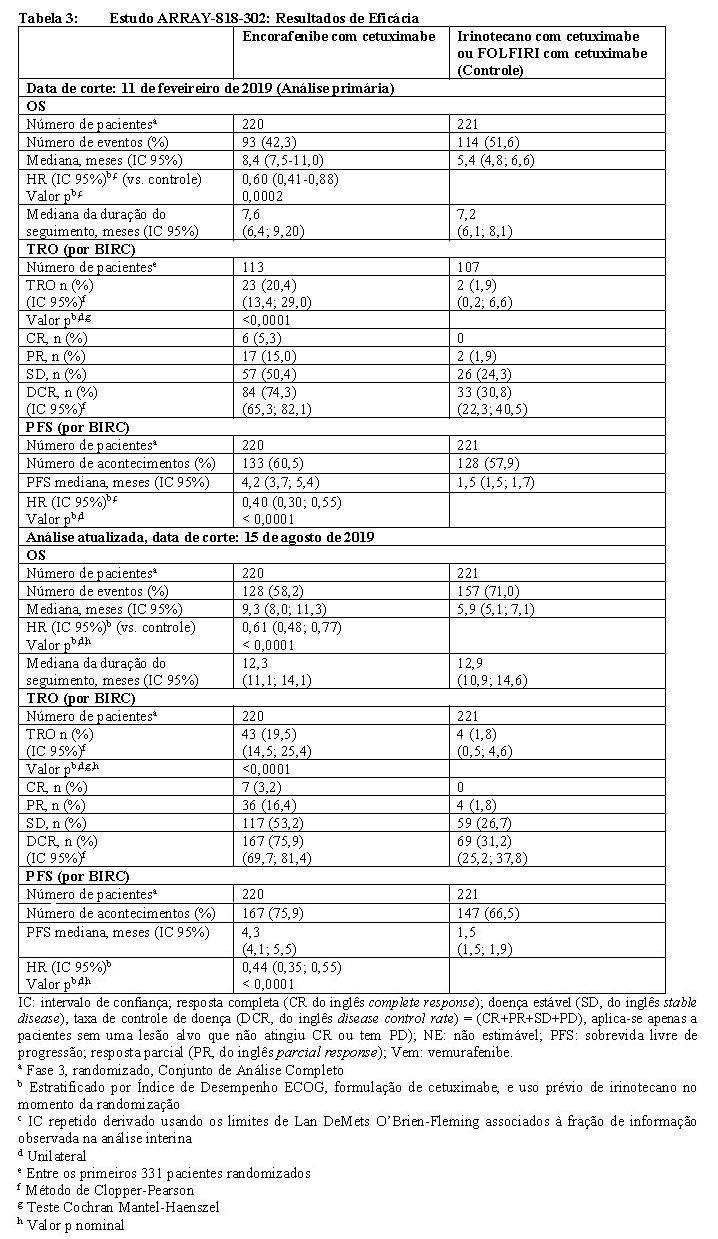
Eletrofisiologia Cardíaca
Na análise de segurança do conjunto de segurança de Fase 3 (ARRAY-818-302) na indicação CCRm, a incidência de novo prolongamento do QTcF > 500 ms foi de 3,2% (7/216) e o prolongamento QTcF > 60 ms em comparação com os valores pré-tratamento foi observado em 8,8% (19/216) dos pacientes do braço encorafenibe + cetuximabe (vide item 8. Posologia e Modo de usar e item 5. Advertências e Precauções).
População pediátrica
A Agência Europeia de Medicamentos diferiu a obrigação de apresentação dos resultados dos estudos com encorafenibe em um ou mais subgrupos da população pediátrica no melanoma (vide item 8. Posologia e Modo de usar para informação sobre utilização pediátrica).
A Agência Europeia de Medicamentos dispensou a obrigação de apresentação dos resultados dos estudos com encorafenibe em todos os sub-grupos da população pediátrica no carcinoma colorretal (vide item 8. Posologia e Modo de usar para informação sobre utilização pediátrica).
Referências Bibliográticas
1. Dummer R, Ascierto PA, Gogas HJ, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2018;19(5):603-615. doi:10.1016/S1470-2045(18)30142-6;
2. Dummer R, Ascierto PA, Gogas HJ et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2018 Oct;19(10):1315-1327.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: agente antineoplásico, inibidor de cinase de proteína, código ATC: L01XE46
Mecanismo de ação
O encorafenibe é uma pequena molécula, inibidora potente e altamente seletiva da cinase RAF por competição com o ATP.
A metade da concentração inibitória máxima (CI50) do encorafenibe contra as enzimas BRAF V600E, BRAF e CRAF foi determinada como sendo 0.35, 0.47 e 0.30 nM, respectivamente. A meia-vida da dissociação do encorafenibe foi > 30 horas e resultou em inibição prolongada de pERK. O encorafenibe suprime a via RAF/MEK/ERK em células tumorais que expressam várias formas mutadas de cinase BRAF (V600E, D e K). Especificamente, o encorafenibe inibe in vitro e in vivo o crescimento de células de melanoma com mutação BRAF V600E, D e K. O encorafenibe não inibe a sinalização RAF/MEK/ERK em células que expressam BRAF do tipo selvagem.
Combinação com binimetinibe
O encorafenibe e o binimetinibe (um inibidor de MEK, vide item 3. Características Farmacológicas da bula do Mektovi®) inibem a via MAPK, resultando em alta atividade anti-tumoral.
Adicionalmente, a combinação de encorafenibe e binimetinibe preveniu o aparecimento de resistência in vivo nos xenoenxertos de melanoma humano com mutação BRAF V600E.
Associação com cetuximabe
Um dos principais mecanismos de resistência de pacientes com CCRm com mutação BRAF, em relação aos inibidores RAF, tem sido a identificação da reativação do EGFR com desvio da transdução de sinal via BRAF. As associações de um inibidor BRAF, por exemplo, encorafenibe, e agentes dirigidos ao EGFR, por exemplo, cetuximabe, têm demonstrado uma melhora da eficácia anti-tumoral em modelos não clínicos.
Propriedades farmacocinéticas
A farmacocinética do encorafenibe foi estudada em indivíduos saudáveis e em pacientes com tumores sólidos, incluindo melanoma avançado e irressecável ou metastático, portador de mutação BRAF V600E ou K e em pacientes adultos com câncer colorretal metastático com uma mutação BRAF V600E. A farmacocinética do encorafenibe demonstrou ter dose aproximada linear após doses únicas e múltiplas. Após a repetição de dose uma vez ao dia, as condições de estabilidade foram atingidas em 15 dias. A taxa de acumulação de, aproximadamente, 0,5 é, provavelmente, devida à indução automática do CYP3A4. A variabilidade inter- indivíduos (CV%) da aera sobre a curva (AUC, do inglês área under curve) varia entre 12,3% e 68,9%.
Absorção
Após a administração oral, o encorafenibe é rapidamente absorvido com um Tmax médio de 1,5 a 2 horas. Em seguida a uma dose oral única de 100 mg [14C] de encorafenibe em indivíduos saudáveis, pelo menos 86% da dose de encorafenibe é absorvida. A administração de uma dose única de 100 mg de encorafenibe com uma refeição rica em gordura e elevado teor calórico diminuiu a Cmax em 36%, enquanto a AUC permaneceu inalterada. Um estudo de interação medicamentosa em indivíduos saudáveis indicou que a extensão da exposição do encorafenibe não foi alterada na presença de um agente que altere o pH gástrico(rabeprazol).
Distribuição
O encorafenibe é moderadamente (86,1%) ligado a proteínas plasmáticas humanas in vitro. Após uma dose oral única de 100 mg [14C] de encorafenibe em indivíduos saudáveis, a média do desvio padrão (SD, do inglês standard deviation) da relação entre a concentração sanguínea e a plasmática é de 0,58 (0,02) e a média (CV%) do volume aparente de distribuição (Vz/F) do encorafenibe é de 226 L (32,7%).
Biotransformação
O metabolismo é a principal via de depuração do encorafenibe (aproximadamente 88% da dose radioativa recuperada), após uma dose oral única de 100 mg [14C]. A reação de biotransformação predominante do encorafenibe foi a N-desalquilação. Outras vias metabólicas envolvem hidroxilação, hidrólise carbamato, glucuronidação indireta e formação de glicose conjugada.
Eliminação
Após uma dose oral única de 100 mg [14C] de encorafenibe em indivíduos saudáveis, a radioatividade foi eliminada igualmente tanto nas fezes como na urina (média de 47,2%). Na urina, 1,8% da radioatividade foi eliminada com encorafenibe. A depuração aparente (CL/F) média (CV%) do encorafenibe foi de 27,9 L/h (9,15%). A mediana (variação) da meia-vida terminal do encorafenibe foi de 6,32h (3,74 a 8,09h).
Interações medicamentosas
Efeito da enzima CYP no encorafenibe
O encorafenibe é metabolizado pela CYP3A4, CYP2C19 e CYP2D6. In vitro, previa-se que a CYP3A4 fosse a principal enzima que contribui para a depuração oxidativa total do encorafenibe em microssomas hepáticos humanos (~83,3%), seguido por CYP2C19 e CYP2D6 (~16,0% e 0,71%, respectivamente).
Efeito do encorafenibe nos substratos de CYP
Experimentos in vitro indicam que o encorafenibe é um inibidor reversível relativamente potente de UGT1A1, CYP2B6, CYP2C9 e CYP3A4/5, bem como um inibidor tempo dependente de CYP3A4. O encorafenibe induziu CYP1A2, CYP2B6, CYP2C9 e CYP3A4 em hepatócitos primários humanos. Simulações de 450 mg de encorafenibe coadministrado com sonda dos substratos para CYP2B6, CYP1A2, CYP2C9, CYP2C19 e CYP2D6 nos dias 1 e 15 indicaram que não são esperadas interações clinicamente relevantes. Para a coadministração com substratos de CYP3A4 e UGT1A1, que são submetidos à extração intestinal, espera-se uma interação menor a moderada. Embora o binimetinibe seja um substrato de UGT1A1, ele não é submetido à extração intestinal e, portanto, não é esperado nenhuma interação medicamentosa com o encorafenibe.
Efeito dos transportadores no encorafenibe
Constatou-se que o encorafenibe é um substrato de transportadores de glicoproteína P (P-gp). É improvável que a inibição da P-gp resulte em um aumento clinicamente relevante nas concentrações de encorafenibe, uma vez que este exibe uma elevada permeabilidade intrínseca. O envolvimento de diversas famílias de transportadores de captação (OCT1, OATP1B1, OATP1B3 e OATPB1) foi investigado in vitro utilizando inibidores de transportadores relevantes. Os dados sugerem que os transportadores de captação hepática não estão envolvidos na distribuição do encorafenibe nos hepatócitos humanos primários.
Efeito do encorafenibe nos transportadores
A administração repetida de encorafenibe 450 mg uma vez ao dia e binimetinibe 45 mg duas vezes ao dia com uma dose única de rosuvastatina (um substrato OATP1B1, OATP1B3 e BCRP) aumentou a Cmax da rosuvastatina em 2,7 vezes e a AUC em 1,6 vezes, indicando uma inibição leve de OATP1B1, OATP1B3 e/ou transportadores BCRP.
In vitro, o encorafenibe inibiu o transportador hepático OCT1, porém é improvável que seja um inibidor eficaz clinicamente. Com base em estudos in vitro, existe o potencial do encorafenibe inibir os transportadores renais OCT2, OAT1, OAT3 em concentrações clínicas. Ademais, o encorafenibe pode inibir o P-gp no intestino nas concentrações clínicas esperadas.
Populações especiais
População pediátrica
A segurança e eficácia do encorafenibe ainda não foram estabelecidas em crianças e adolescentes. Não há dados disponíveis.
Idade
Com base em uma análise farmacocinética populacional, verificou-se que a idade é uma covariável no volume de distribuição do encorafenibe, porém com alta variabilidade. Dada à pequena magnitude dessas alterações e a alta variabilidade, é pouco provável que sejam clinicamente relevantes, e nenhum ajuste de dose é necessário para pacientes idosos.
Gênero
Baseado em uma análise farmacocinética populacional, o gênero não foi considerado um modelo relevante de covariável na depuração ou no volume de distribuição. Como resultado, nenhuma alteração importante na exposição do encorafenibe é esperada com base no gênero.
Peso corporal
Com base em uma análise farmacocinética populacional, o peso corporal foi considerado um modelo relevante de covariável na depuração e no volume de distribuição. No entanto, devido à pequena magnitude da alteração na depuração e a alta variabilidade no volume de distribuição previsto no modelo, é improvável que o peso tenha uma influência clinicamente relevante na exposição do encorafenibe.
Etnia
Não há dados suficientes para avaliar potenciais diferenças na exposição do encorafenibe devido à raça ou etnia.
Insuficiência hepática
Os resultados de um estudo clínico específico indicam uma exposição total ao encorafenibe, 25% mais elevada em pacientes com insuficiência hepática leve (Classe A de Child-Pugh) em comparação com indivíduos com função hepática normal. Isso se traduz em um aumento de 55% do encorafenibe não ligado.
A farmacocinética do encorafenibe não foi clinicamente avaliada em pacientes com insuficiência hepática moderada (Classe B de Child-Pugh) ou grave (Classe C de Child-Pugh). Como o encorafenibe é principalmente metabolizado e eliminado por via hepática, com base no modelo PBPK, os pacientes com insuficiência hepática moderada a grave podem apresentar maiores aumentos na exposição do que aqueles com insuficiência leve. Não há como fazer qualquer recomendação posológica em pacientes com insuficiência hepática moderada ou grave (vide item 3. Características Farmacológicas e item 5. Advertências e Precauções).
Insuficiência renal
O encorafenibe sofre mínima eliminação renal. Nenhum estudo clínico formal foi realizado para avaliar o efeito da insuficiência renal na farmacocinética do encorafenibe.
Em uma análise farmacocinética populacional, não se observou uma tendência clara na CL/F do encorafenibe nos pacientes com insuficiência renal leve (eGFR 60 a 90 mL/min/1.73m2) ou moderada (eGFR 30 a 59 mL/min/1.73m2) em comparação com indivíduos com função renal normal (eGFR≥90 mL/min/1.73m2). Foi prevista uma pequena diminuição na CL/F (≤5%) para pacientes com insuficiência renal leve e moderada, o que, provavelmente, não é clinicamente relevante. A farmacocinética do encorafenibe não foi estudada em pacientes com insuficiência renal grave.
Dados de segurança pré-clínica
Nos estudos de toxicidade em ratos, com duração de 4 semanas e 13 semanas, observou-se sinais clínicos, redução do peso corporal, redução dos epidídimos e do peso da próstata, além de achados microscópicos nos testículos, epidídimos, estômago e pele. A reversibilidade parcial desses achados foi observada após um período de recuperação de quatro semanas.
Adicionalmente, no estudo de 13 semanas observaram-se alterações patológicas reversíveis em doses ≥100mg/kg/d. Nenhum nível sem efeitos adversos observáveis (NOEL, do inglês no observed effect level) pode ser estabelecido para o estudo de 4 semanas. O NOEL para o estudo de 13 semanas foi mais de 10 vezes a exposição terapêutica humana.
No estudo de toxicidade em macacos, com duração de 4 semanas e 13 semanas, observaram-se episódios isolados/esporádicos de emese e diarreia, bem como lesões oftálmicas, ligeiramente acima das exposições terapêuticas humanas. As lesões oftálmicas eram parcialmente reversíveis e consistiam em uma separação ou descolamento na retina entre as hastes externas e a camada de cones e o epitélio pigmentar da retina na mácula central na fóvea. Essa observação foi semelhante à descrita em humanos como corioretinopatia serosa central ou retinopatia serosa central.
O encorafenibe não é genotóxico.
Não foram conduzidos estudos de fertilidade com o encorafenibe. Nos estudos de toxicologia em ratos de 13 semanas, o tratamento com encorafenibe a 6 mg/kg/d (nível de dose mais de 5 vezes a exposição humana na dose terapêutica) resultou em diminuição do peso dos testículos e epidídimo com degeneração tubular e oligospermia. No estudo de 13 semanas, a reversibilidade parcial foi observada no nível de dose mais alto (60 mg/kg/d).
O estudo de desenvolvimento embrionário-fetal em ratos indicou que o encorafenibe induziu toxicidade fetal com pesos fetais mais baixos e atraso no desenvolvimento esquelético.
O mesmo tipo de estudo em coelhos indicou que o encorafenibe induziu toxicidade fetal com menores pesos fetais e alterações transitórias no desenvolvimento esquelético. Observou-se dilatação do arco aórtico em alguns fetos.
O encorafenibe foi fototóxico em um Teste de Captura de Vermelho Neutro 3T3 in vitro. O encorafenibe não foi um sensibilizante no ensaio de sensibilização em camundongo in vivo. Coletivamente, esses dados indicam que o encorafenibe tem risco de potencial fototóxico e risco mínimo de sensibilização em doses terapêuticas em pacientes.
4. CONTRAINDICAÇÕES
Hipersensibilidade à substância ativa ou a qualquer um dos excipientes mencionados na composição.
5. ADVERTÊNCIAS E PRECAUÇÕES
Encorafenibe deve ser administrado em associação com binimetinibe (para pacientes com melanoma irressecável ou metastático com mutação BRAF V600) ou em associação com cetuximabe (para pacientes com câncer colorretal metastático com mutação BRAF V600E). Para informação adicional sobre advertências e precauções associadas ao tratamento com binimetinibe ou cetuximabe, vide item 5. Advertências e Precauções da bula do Mektovi® (binimetinibe) ou do cetuximabe.
Teste de mutação BRAF
Antes de tomar encorafenibe, os pacientes devem ter confirmação de melanoma metastático ou irressecável com mutação BRAF V600 ou câncer colorretal metastático com mutação BRAF V600E utilizando um teste validado. A eficácia e segurança do encorafenibe foram estabelecidas apenas em pacientes com melanomas com mutações BRAF V600E e V600K ou tumores colorretal que expressam mutação BRAF V600E. O encorafenibe não deve ser utilizado em pacientes com melanoma BRAF tipo selvagem. O encorafenibe não deve ser usado em pacientes com melanoma maligno BRAF tipo selvagem ou câncer colorretal BRAF tipo selvagem.
Encorafenibe em combinação com binimetinibe em pacientes que progrediram com um inibidor BRAF
Há uma limitação de dados para uso da combinação de encorafenibe e binimetinibe em pacientes que progrediram após a utilização prévia de inibidor de BRAF para o tratamento de melanoma irressecável ou metastático com mutação BRAF V600. Esses dados mostram que a eficácia da combinação é menor nestes pacientes.
Encorafenibe em combinação com binimetinibe em pacientes com metástases cerebrais
Os dados de eficácia da combinação de encorafenibe e binimetinibe em pacientes com melanoma com mutação BRAF V600 e metástases cerebrais são limitados. (vide item 2. Resultados de Eficácia).
Disfunção ventricular esquerda (LVD, do inglês left ventricular dysfunction)
A LVD definida como reduções sintomáticas ou assintomáticas na fração de ejeção foi relatada quando o encorafenibe é utilizado em combinação com o binimetinibe. Recomenda-se que a fração de ejeção do ventrículo esquerdo (LVEF, do inglês left ventricular ejection fraction) seja avaliada por ecocardiograma ou ventriculografia radioisotópica (MUGA, do inglês multi-gated acquisition) antes do início do encorafenibe e binimetinibe, um mês após o início e depois trimestralmente, aproximadamente, ou com mais frequência, conforme indicação clínica durante o tratamento. Se, durante o tratamento, a LVD ocorrer, vide item 8. Posologia e Modo de usar da bula do Mektovi®.
A segurança do encorafenibe em combinação com o binimetinibe não foi estabelecida em pacientes com LVEF basal abaixo de 50% ou abaixo dos limites institucionais inferiores do normal. Portanto, nesses pacientes, o binimetinibe deve ser utilizado com cautela e para qualquer disfunção ventricular esquerda sintomática, LVEF grau 3-4 ou redução absoluta da LVEF basal ≥10%, o binimetinibe e o encorafenibe devem ser descontinuados e a LVEF deve ser avaliada a cada 2 semanas até a recuperação.
Hemorragia
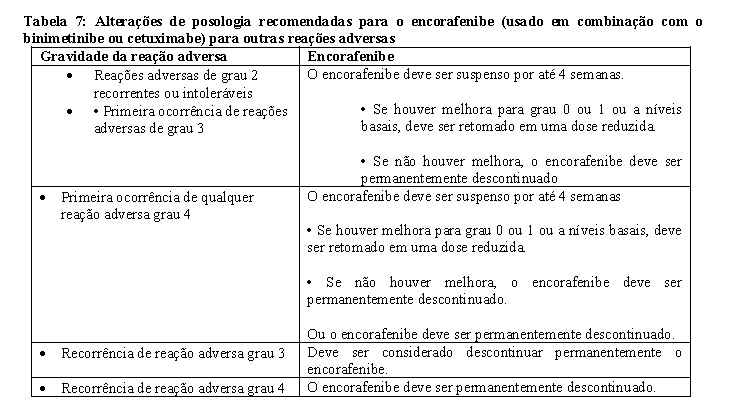
Hemorragias, incluindo os principais eventos hemorrágicos, podem ocorrer com o encorafenibe (vide item 9. Reações Adversas). O risco de hemorragia pode aumentar com o uso concomitante de anticoagulante e terapia antiplaquetária. A ocorrência de eventos hemorrágicos grau 3 deve ser controlada com interrupção da dose ou descontinuação do tratamento (vide Tabela 5 em Posologia e Modo de usar) e conforme clinicamente indicado.
Toxicidades oculares
As toxicidades oculares, incluindo uveíte, irite e iridociclite, podem ocorrer quando o encorafenibe é administrado. O descolamento do epitélio pigmentar da retina (DEPR) também foi relatado em pacientes tratados com encorafenibe em combinação com binimetinibe (vide item 9. Reações Adversas).
Os pacientes devem ser avaliados em cada visita para sintomas novos ou agravamento de comprometimento visual. Se forem identificados novos sintomas ou agravamento de comprometimento visual, incluindo visão central diminuída, visão turva ou perda de visão, recomenda-se um exame oftalmológico imediato.
Se ocorrer uveíte, incluindo iridociclite e irite, durante o tratamento, vide item 9. Reações Adversas.
Se, durante o tratamento, o paciente desenvolver distrofia epitelial pigmentar da retina (RPED, do inglês retinal pigment epithelial dystrophy) ou oclusão da veia central da retina (RVO, do inglês retinal vein occlusion), vide item 8. Posologia e Modo de usar da bula do Mektovi® para orientação.
Prolongamento do intervalo QT
O prolongamento do intervalo QT foi observado em pacientes tratados com inibidores de BRAF. Um estudo minucioso do intervalo QT para se avaliar o potencial prolongamento do intervalo QT associado ao encorafenibe não foi realizado.
No geral, os resultados sugerem que o encorafenibe como agente único tem o potencial de causar leves aumentos na frequência cardíaca. Através de estudos combinados de encorafenibe e binimetinibe na posologia recomendada e em um estudo do encorafenibe como agente único, os resultados sugerem que o encorafenibe tem o potencial de resultar em pequenos aumentos no intervalo QTc (vide item 2. Resultados de Eficácia).
Não existem dados suficientes para excluir um prolongamento do intervalo QT dependente da exposição clinicamente significativo. Devido ao potencial risco de prolongamento do intervalo QT, recomenda-se que as anomalias dos eletrólitos séricos, incluindo magnésio e potássio, sejam corrigidas e os fatores de risco para o prolongamento do intervalo QT (por exemplo, insuficiência cardíaca congestiva, bradiarritmias) controlados antes do início do tratamento e durante o mesmo.
Recomenda-se que o eletrocardiograma (ECG) seja avaliado antes do início do encorafenibe, um mês após o início e depois trimestralmente, aproximadamente, ou com maior frequência, conforme clinicamente indicado durante o tratamento. A ocorrência do prolongamento do intervalo QTc pode ser controlada com a diminuição da dose, interrupção ou descontinuação com a correção de eletrólitos anormais e controle dos fatores de risco (vide item 8. Posologia e Modo de usar).
Novos tumores primários
Novos tumores primários, cutâneos e não cutâneos, foram observados em pacientes tratados com inibidores de BRAF e podem ocorrer quando o encorafenibe é administrado (vide item 9. Reações Adversas.
Tumores cutâneos
Tumores cutâneos, como o carcinoma de células escamosas cutâneo (cSCC), incluindo queratoacantoma, foram observados em pacientes tratados com inibidores de BRAF, incluindo o encorafenibe.
Um novo melanoma primário foi observado em pacientes tratados com inibidores de BRAF, incluindo o encorafenibe (vide item 9. Reações Adversas).
Avaliações dermatológicas devemser realizadas previamente ao início do tratamento com o encorafenibe em combinação com o binimetinibe, bimestralmente durante o tratamento e por até 6 meses após a descontinuação da combinação. Lesões cutâneas suspeitas devem ser tratadas com excisão dermatológica e avaliação dermatológica.
Os pacientes devem ser instruídos a informar imediatamente seus médicos caso novas lesões de pele se desenvolvam. O encorafenibe deve ser continuado sem qualquer modificação na posologia.
Tumores não cutâneos
Com base em seu mecanismo de ação, o encorafenibe pode promover tumores associados à ativação de RAS por meio de mutações ou outros mecanismos. Os pacientes em tratamento com encorafenibe devem ser submetidos a um exame de cabeça e pescoço, tomografia computadorizada (TC) de tórax/abdome, exames anais e pélvicos (para mulheres) e contagem completa de células sanguíneas previamente ao início, durante e ao final do tratamento, conforme clinicamente apropriado. Deve-se considerar descontinuar permanentemente o encorafenibe em pacientes que desenvolvam tumores não cutâneos positivos para a mutação RAS. Os benefícios e riscos devem ser cuidadosamente considerados antes da administração de encorafenibe para pacientes com um câncer prévio ou concomitante associado à mutação RAS.
Anormalidades laboratoriais hepáticas
Anormalidades laboratoriais hepáticas, incluindo elevações de alanina aminotransferase (ALT) e aspartato aminotransferase (AST), foram observadas com o encorafenibe (vide item 9. Reações Adversas). Os valores laboratoriais hepáticos devem ser monitorados previamente ao início do encorafenibe e binimetinibe e, pelo menos, mensalmente durante os 6 primeiros meses de tratamento, depois conforme clinicamente indicado. Alterações laboratoriais hepáticas devem ser tratadas com interrupção da dose, redução ou descontinuação do tratamento (vide item 8. Posologia e Modo de usar).
Insuficiência hepática
Como o encorafenibe é principalmente metabolizado e eliminado por via hepática, pacientes com insuficiência hepática leve a grave podem ter aumento da exposição ao encorafenibe ao longo do intervalo de exposição à variabilidade individual (vide item 3. Características Farmacológicas).
Na ausência de dados clínicos, o encorafenibe não é recomendado em pacientes com insuficiência hepática moderada ou grave.
A administração do encorafenibe deve ser realizada com cautela na dose de 300 mg uma vez ao dia em pacientes com insuficiência hepática leve (vide item 8. Posologia e Modo de usar).
Recomenda-se monitoramento mais atento das toxicidades relacionadas ao encorafenibe em pacientes com insuficiência hepática leve, incluindo exame clínico e testes de função hepática, com avaliação dos ECGs, conforme clinicamente apropriado durante o tratamento.
Insuficiência renal
Não existem dados disponíveis em pacientes com insuficiência renal grave (vide item 3. Características Farmacológicas e item 8. Posologia e Modo de usar).
O encorafenibe deve ser utilizado com cautela em pacientes com insuficiência renal grave. A elevação de creatinina tem sido comumente relatada com o encorafenibe como agente único ou em combinação com o binimetinibe ou cetuximabe. Os casos observados de falência renal, incluindo lesão renal aguda e insuficiência renal, foram geralmente associados a vômitos e desidratação. Outros fatores contribuintes incluem diabetes e hipertensão. A creatinina sanguínea deve ser monitorada conforme clinicamente indicado e a elevação da creatinina controlada com alteração de dose ou descontinuação (vide Tabela 5 em Posologia e Modo de usar). Os pacientes devem ser orientados quanto a ingestão adequada de líquidos durante o tratamento.
Efeitos de outros medicamentos sobre o encorafenibe
O uso concomitante de inibidores fortes de CYP3A durante o tratamento com encorafenibe deve ser evitado. Se for necessário o uso concomitante com um inibidor forte de CYP3A, os pacientes devem ser cuidadosamente monitorados por segurança (vide item 5. Advertências e Precauções).
Deve-se ter cuidado caso um inibidor moderado de CYP3A for coadministrado com o encorafenibe.
Sensibilidade à luz solar
Apesar do risco de fototoxicidade ser mínimo com encorafenibe, é recomendado o uso de roupas adequadas para se proteger do sol e a aplicação de filtro solar antes de sair ao ar livre.
Mulheres em idade fértil / Contracepção em mulheres
As mulheres em idade fértil devem utilizar método de contracepção eficaz durante o tratamento com encorafenibe e durante, pelo menos, um mês após a última dose. O encorafenibe pode diminuir a eficácia de contraceptivos hormonais (vide item 5. Advertências e Precauções). Assim sendo, pacientes do sexo feminino que utilizam métodos contraceptivos hormonais são aconselhadas a utilizar um método adicional ou alternativo, como um método de barreira (por exemplo, preservativo) durante o tratamento com encorafenibe e durante, pelo menos, um mês após a última dose.
Fertilidade
Não existem dados a respeito dos efeitos do encorafenibe sobre a fertilidade em humanos. Com b