BLENREP
GLAXOSMITHKLINE
belantamabe mafodotina
Anticorpo monoclonal. Antineoplásico.
Apresentações.
Pó para solução para infusão.
BLENREP® é apresentado em embalagem com 1 frasco-ampola com 70 mg ou 100 mg de belantamabe mafodotina (50mg/mL após reconstituição).
USO INTRAVENOSO
USO ADULTO
Composição.
Cada frasco-ampola de 70mg contém belantamabe mafodotina 77mg*, excipientes*** q.s.p para 1 frasco-ampola
Cada frasco-ampola de 100mg contém belantamabe mafodotina110mg**, excipientes*** q.s.p para 1 frasco-ampola
* Cada frasco contém 77 mg de belantamabe mafodotina; a alegação do rótulo de 70 mg de belantamabe mafodotina é baseada no volume extraível de 1,4 mL após a reconstituição do pó liofilizado. O enchimento excessivo (0,14 mL) permite um volume extraível de 1,4 mL após a reconstituição com 1,4 mL de água para injetáveis estéril. Nenhum excesso está incluído.
**Cada frasco contém 110 mg de belantamabe mafodotina; a alegação do rótulo de 100 mg de belantamabe mafodotina é baseada no volume extraível de 2,0 mL após a reconstituição do pó liofilizado. O enchimento excessivo (0,2 mL) permite um volume extraível de 2,0 mL após a reconstituição com 2,0 mL de água para injetáveis estéril. Nenhum excesso está incluído.
excipientes***: citrato de sódio di-hidratado, ácido cítrico monohidratado, trealose di-hidratada, edetato dissódico di-hidratado, polissorbato 80 e água para injetáveis.
BLENREP® pode causar alterações geralmente reversíveis no epitélio da córnea, resultando em alterações na visão, incluindo redução da acuidade visual, e achados do exame da córnea (ver seção "Advertências e Precauções"). Realize exames oftalmológicos antes de cada uma das primeiras 6 doses e, posteriormente, conforme indicação clínica. Ajuste a dose ou suspenda o uso de BLENREP® até que ocorra melhora e retome o uso, ou interrompa-o permanentemente, com base na gravidade (ver seção "Posologia").
Informações técnicas.
1. INDICAÇÕES
BLENREP® é indicado para o tratamento de adultos com mieloma múltiplo:
• em combinação com bortezomibe e dexametasona em pacientes que receberam pelo menos uma terapia anterior; e ·
• em combinação com pomalidomida e dexametasona em pacientes que receberam pelo menos uma terapia anterior incluindo lenalidomida.
2. RESULTADOS DE EFICÁCIA
DREAMM-7: Combinação com bortezomibe e dexametasona
O DREAMM-7 foi um estudo de Fase III, aberto, multicêntrico, que avaliou o belantamabe mafodotina em combinação com bortezomibe e dexametasona (BVd) em comparação com daratumumabe, bortezomibe e dexametasona (DVd) em pacientes com mieloma múltiplo (MM) recidivado ou refratário.
Os pacientes elegíveis apresentavam um diagnóstico confirmado de MM, conforme definido pelos critérios do Grupo de Trabalho Internacional de Mieloma (IMWG), haviam sido tratados anteriormente com pelo menos 1 linha de terapia anterior para MM e deveriam ter apresentado progressão da doença documentada durante ou após a terapia mais recente.
Os pacientes foram randomizados em uma proporção de 1:1. No braço BVd (N = 243), os pacientes receberam belantamabe mafodotina 2,5 mg/kg por infusão intravenosa (IV) a cada 3 semanas no dia 1 de cada Ciclo; bortezomibe 1,3 mg/m2 (por via subcutânea) nos dias 1, 4, 8 e 11 dos Ciclos 1 a 8 (Ciclos de 21 dias); e dexametasona 20 mg (IV ou oral) no dia do tratamento com bortezomibe e no dia seguinte. No braço DVd (N = 251), os pacientes receberam daratumumabe 16 mg/kg (IV) em Ciclos de 21 dias: toda semana nos Ciclos 1 a 3, a cada 3 semanas nos Ciclos 4 a 8 e a cada 4 semanas para o Ciclo 9 em diante. Os cronogramas de dexametasona e bortezomibe foram os mesmos em ambos os braços nos primeiros 8 Ciclos. O tratamento continuou em ambos os braços até a progressão da doença, morte, toxicidade inaceitável, retirada do consentimento ou término do estudo.
No total, 494 pacientes foram avaliados quanto à eficácia no DREAMM-7. Os dados demográficos e as características basais foram semelhantes em ambos os braços. As características basais para o braço BVd (N = 243) foram: idade mediana: 65 anos (35% com idade entre 65 e 74 anos e 15% com 75 anos ou mais); 53% do sexo masculino, 47% do sexo feminino; 85% brancos, 12% asiáticos, 3% pretos; estágio R-ISS na triagem I (42%), II (53%), III (4%); 28% de alto risco citogenético, número mediano de 1 linha de terapia anterior; 5% com doença extramedular (EMD); e dos que receberam tratamento (n = 242), o status de desempenho do Eastern Cooperative Oncology Group (ECOG PS) foi 0 (50%), 1 (46%) ou 2 (4%). No braço BVd, 90% dos pacientes receberam terapia anterior com inibidores de proteassoma (bortezomibe, carfilzomibe, ixazomibe), 81% dos pacientes receberam terapia anterior com imunomoduladores (lenalidomida, talidomida, pomalidomida), 1% dos pacientes recebeu terapia anterior com daratumumabe e 67% dos pacientes receberam transplante autólogo de células-tronco (ASCT). Houve 9% de pacientes refratários à terapia com inibidores de proteassoma e 39% de pacientes refratários à terapia com imunomoduladores.
O desfecho primário foi a sobrevida livre de progressão (PFS), conforme avaliado por um Comitê de Revisão Independente (IRC) em caráter cego, com base nos critérios do IMWG para mieloma múltiplo.
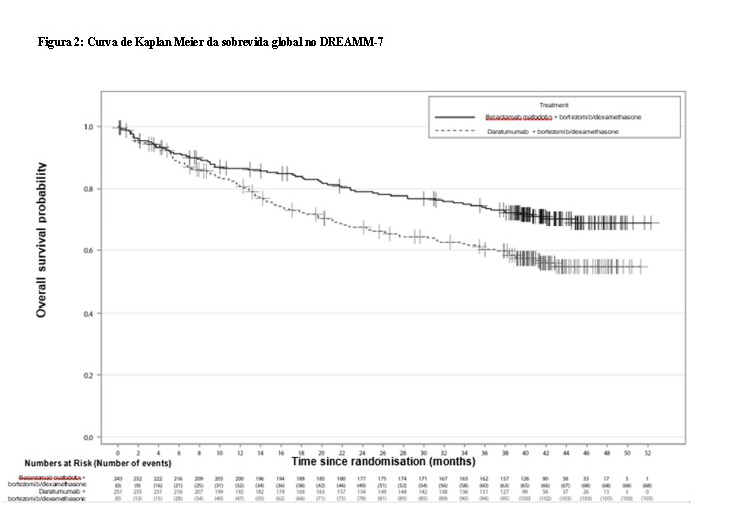
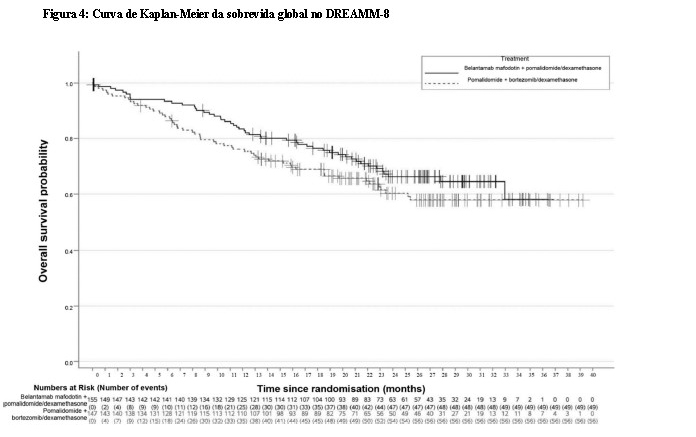
Os pacientes tratados com belantamabe mafodotina em combinação com bortezomibe e dexametasona apresentaram uma melhora estatisticamente significativa na PFS, sobrevida global (OS) e doença residual mínima (MRD) na população geral em comparação com daratumumabe, bortezomibe e dexametasona.
Os resultados de eficácia no momento da primeira análise interina (corte de dados em 2 de outubro de 2023, exceto OS, onde os dados são da segunda análise interina, corte de dados em 7 de outubro de 2024) estão apresentados na Tabela 1 e nas Figuras 1 e 2.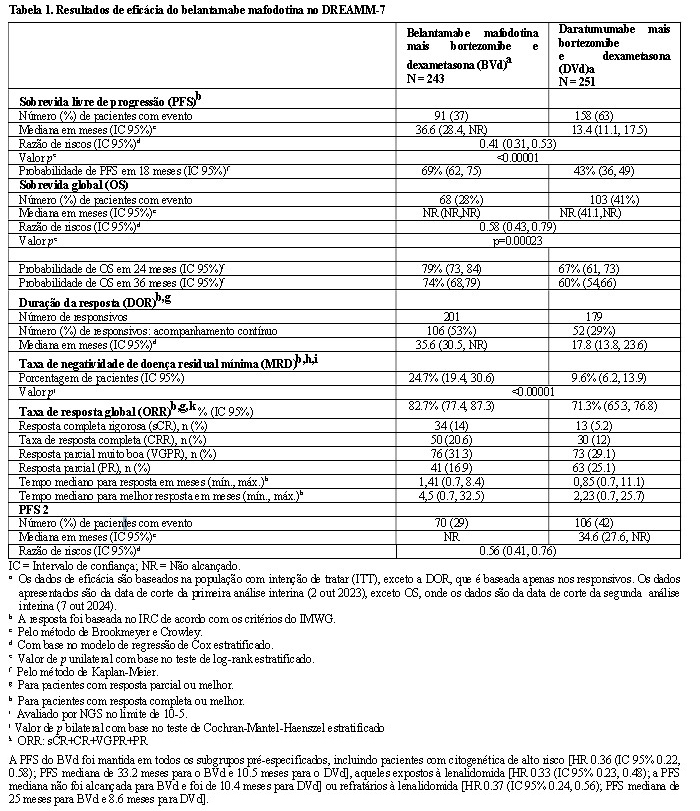
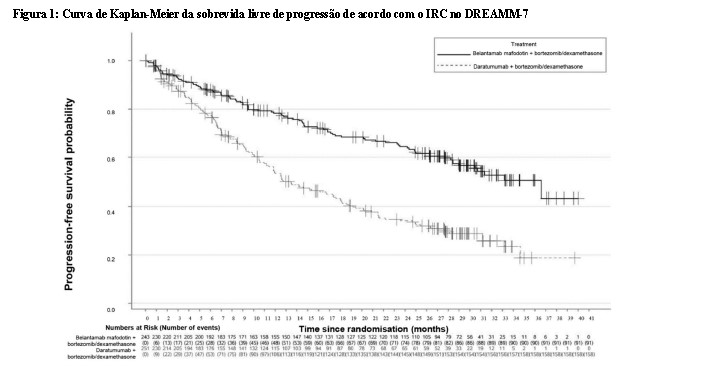
Durante o estudo, as modificações de dose recomendadas, que incluíram atrasos e reduções de dose, controlaram as reações adversas e permitiram que os pacientes continuassem o tratamento.
A dose média por paciente foi de 2.2 mg/kg (mediana: 2.1 mg/kg). A exposição ao belantamabe mafodotina observada durante o DREAMM-7 está apresentada na Tabela 2.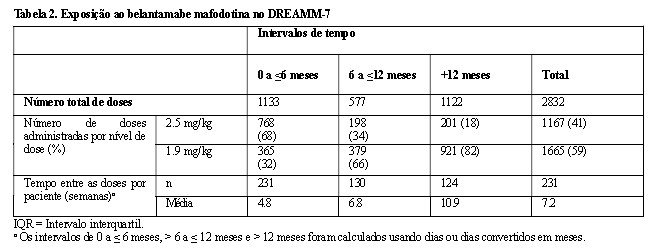
Os resultados relatados pelos pacientes (PROs) foram avaliados usando o Questionário de Qualidade de Vida (QLQ) C30 da Organização Europeia para Pesquisa e Tratamento do Câncer (EORTC) e o QLQ-IL52 da EORTC. Os pacientes que receberam BVd ou DVd mantiveram a qualidade de vida global ( < 10 pontos de alteração em relação ao período basal), conforme medido pelo domínio de estado de saúde global/QoL do QLQ-C30 do EORTC, e não houve diferenças entre os grupos de tratamento (≥ 10 pontos). Da mesma forma, não houve diferenças (≥ 10 pontos) entre os grupos de tratamento quanto ao desempenho da função, funcionamento físico, fadiga e sintomas da doença.
DREAMM-8: Combinação com pomalidomida e dexametasona
O DREAMM-8 foi um estudo de Fase III, aberto, multicêntrico, que avaliou o belantamabe mafodotina em combinação com pomalidomida e dexametasona (BPd) em comparação com pomalidomida, bortezomibe e dexametasona (PVd) em pacientes com mieloma múltiplo recidivado ou refratário.
Os pacientes elegíveis apresentavam um diagnóstico confirmado de mieloma múltiplo (MM), conforme definido pelos critérios do IMWG, haviam sido tratados anteriormente com pelo menos 1 linha de terapia anterior para MM, incluindo lenalidomida, e deveriam ter apresentado progressão da doença documentada durante ou após a terapia mais recente.
Os pacientes foram randomizados em uma proporção de 1:1 para receber BPd ou PVd, estratificados pelo número de linhas de tratamento anteriores, exposição anterior ao bortezomibe, tratamento anterior com anti-CD38 e status do Sistema Internacional de Estadiamento (ISS). No braço BPd (N = 155), os pacientes receberam belantamabe mafodotina 2,5 mg/kg (IV) uma vez no dia 1 no Ciclo 1 (Ciclo de 28 dias) seguido por belantamabe mafodotina 1,9 mg/kg (IV) a cada 4 semanas no dia 1 do Ciclo 2 em diante (Ciclos de 28 dias); pomalidomida 4 mg (via oral [PO]) administrada nos dias 1 a 21 e dexametasona 40 mg (PO) nos Dias 1, 8, 15 e 22 em todos os Ciclos (Ciclos de 28 dias).
No braço PVd (N = 147), a pomalidomida 4 mg (PO) foi administrada a cada 3 semanas nos dias 1 a 14 em todos os ciclos (ciclos de 21 dias); bortezomibe 1,3 mg/m2 foi administrado por via subcutânea nos dias 1, 4, 8 e 11 nos Ciclos 1 a 8, e nos dias 1 e 8 no Ciclo 9 em diante (Ciclos de 21 dias). Dexametasona 20 mg (PO) foi administrada no dia anterior e no dia seguinte ao bortezomibe. O nível de dose de dexametasona em cada braço foi reduzido pela metade em pacientes com 75 anos de idade ou mais. O tratamento em ambos os braços continuou até doença progressiva, toxicidade inaceitável, retirada do consentimento, início de outra terapia anticâncer, término do estudo ou morte.
No total, 302 pacientes com MM foram avaliados quanto à eficácia no DREAMM-8. Os dados demográficos e as características basais foram semelhantes em ambos os braços. As características basais para o braço BPd (N = 155) foram: idade mediana: 67 anos (46% com idade entre 65 e 74 anos e 12% com 75 anos ou mais); 64% do sexo masculino, 36% do sexo feminino; 86% brancos, 13% asiáticos, < 1% nativo do Havaí ou das ilhas do Pacífico, < 1% raça mista; estágio ISS na triagem I (60%), II (25%), III (14%); 34% de alto risco citogenético, número mediano de 1 linha de terapia anterior; 13% com EMD; e dos que receberam tratamento (n = 150), o ECOG PS foi 0 (53%), 1 (45%) ou 2 (3%). No braço BPd, 100% dos pacientes receberam terapia anterior com imunomoduladores (lenalidomida, talidomida), 90% dos pacientes receberam terapia anterior com inibidores de proteassoma (bortezomibe, carfilzomibe, ixazomibe), 25% dos pacientes receberam terapia anti-CD38 anterior (daratumumabe, isatuximabe) e 64% dos pacientes receberam ASCT anteriormente. Houve 82% de pacientes refratários à terapia com imunomoduladores, 26% de pacientes refratários à terapia com inibidores de proteassoma e 23% de pacientes refratários à terapia anti-CD38.
O desfecho primário foi a Sobrevida Livre de Progressão (PFS), conforme avaliado por um Comitê de Revisão Independente (IRC) cego, com base nos critérios do Grupo de Trabalho Internacional de Mieloma (IMWG) para mieloma múltiplo.
Os pacientes tratados com belantamabe mafodotina em combinação com pomalidomida e dexametasona apresentaram uma melhora estatisticamente significativa na PFS na população geral em comparação com pomalidomida, bortezomibe e dexametasona.
Os resultados de eficácia no momento da primeira análise parcial (corte de dados em 29 de janeiro de 2024) estão apresentados na Tabela 3 e nas Figuras 3 e 4.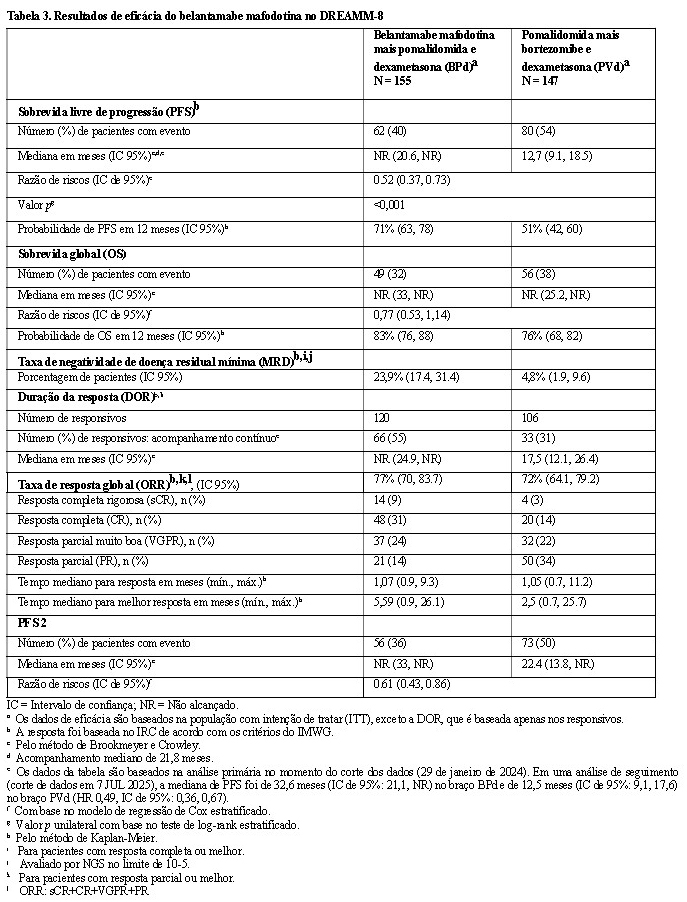
A PFS do BPd foi mantida em todos os subgrupos pré-especificados, incluindo pacientes com citogenética de alto risco, [HR 0,57 (IC 95% 0,34, 0,95); PFS mediana de 17,6 meses para BPd e 9,1 meses para PVd], aqueles refratários à lenalidomida [HR 0,45 (IC 95% 0,31, 0,65); PFS mediana de 24 meses para BPd e 9,2 meses para PVd] ou refratários aos agentes anti-CD38 [HR 0,65 (IC 95% 0,36, 1,18); PFS mediana de 11,5 meses para BPd e 6,4 meses para PVd].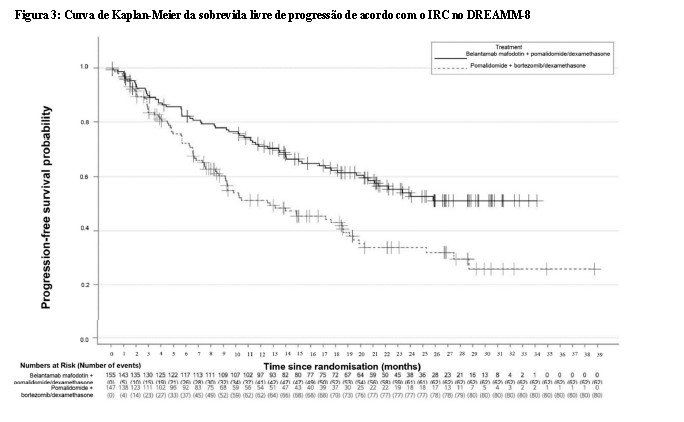
Durante o estudo, as modificações de dose recomendadas, que incluíram atrasos e reduções de dose, controlaram as reações adversas e permitiram que os pacientes continuassem o tratamento.
A dose média por paciente foi de 2 mg/kg (mediana: 2 mg/kg). A exposição ao belantamabe mafodotina observada durante o DREAMM-8 está apresentada na Tabela 4.
Os resultados relatados pelos pacientes (PROs) foram avaliados usando o Questionário de Qualidade de Vida (QLQ) C30 da Organização Europeia para Pesquisa e Tratamento do Câncer (EORTC) e o QLQ-IL52 da EORTC. Os pacientes que receberam BPd ou PVd mantiveram a qualidade de vida global ( < 10 pontos de alteração em relação ao período basal), conforme medido pelo domínio de estado de saúde global/QoL do QLQ-C30 do EORTC, e não houve diferenças entre os grupos de tratamento (≥ 10 pontos). Da mesma forma, não houve diferenças entre os grupos de tratamento quanto ao funcionamento físico, fadiga, desempenho da função e sintomas da doença (≥10 pontos), exceto nas semanas 117 e 137, em que foram observadas diferenças ≥10 a favor do BPd para o desempenho da função e os sintomas da doença, respectivamente.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Farmacodinâmica
Classificação ATC
L01FX15 belantamabe mafodotina
Mecanismo de ação
BLENREP® é um anticorpo monoclonal IgG1 kappa humanizado conjugado com um agente citotóxico, maleimido-caproil monometil auristatina F (mcMMAF). BLENREP® se liga ao BCMA da superfície celular e é rapidamente internalizado. Uma vez dentro da célula tumoral, o agente citotóxico (cys mcMMAF) é liberado, interrompendo a rede de microtúbulos, levando à parada do ciclo celular e à apoptose. O anticorpo também aumenta o recrutamento e a ativação de células imunológicas efetoras, matando as células tumorais por citotoxicidade celular dependente de anticorpo e fagocitose. A apoptose induzida pelo BLENREP® é acompanhada por marcadores de morte celular imunogênica, que podem contribuir para uma resposta imune adaptativa às células tumorais.
Propriedades farmacodinâmicas
Relação de resposta à exposição em terapias combinadas
Para as terapias combinadas BVd e BPd, uma maior exposição ao Ciclo 1 de BLENREP® foi associada a uma maior probabilidade de resposta [por ex., resposta parcial muito boa (VGPR+)] e maior incidência de algumas reações adversas de segurança (por ex., achados de exame da córnea de grau ≥2). Para a maior parte do intervalo de exposição ao Ciclo 1 de BLENREP®, a probabilidade de VGPR ou melhor foi maior do que a probabilidade de reações adversas oculares e desfechos relacionados à BCVA.
Eletrofisiologia Cardíaca
O belantamabe mafodotina ou o cys-mcMMAF não apresentaram prolongamento significativo do QTc ( > 10 ms) em doses de até 3,4 mg/kg uma vez a cada 3 semanas.
Imunogenicidade
A incidência de anticorpos (ADAs) anti-belantamabe mafodotina foi baixa em pacientes tratados com BLENREP® em terapias combinadas, sem impacto clínico observado na farmacocinética, segurança e eficácia.
Nos estudos pivotais de terapia combinada (DREAMM-7 e DREAMM-8) e no estudo de suporte à terapia combinada (DREAMM-6), 3% dos pacientes (15/515) apresentaram resultado positivo para ADAs decorrentes do tratamento. Dois pacientes apresentaram resultado positivo para anticorpos neutralizantes (NAb) anti-belantamabe mafodotina.
Propriedades Farmacocinéticas
Absorção
A concentração máxima de BLENREP® ocorreu no momento ou logo após o término da infusão, enquanto as concentrações de cys-mcMMAF atingiram o pico cerca de 24 horas após a administração.
As Tabelas 5 e 6 descrevem a farmacocinética do BLENREP® para doses de 2,5 mg/kg no Ciclo 1, Dia 1, ao final dos primeiros intervalos de 3 e 4 semanas.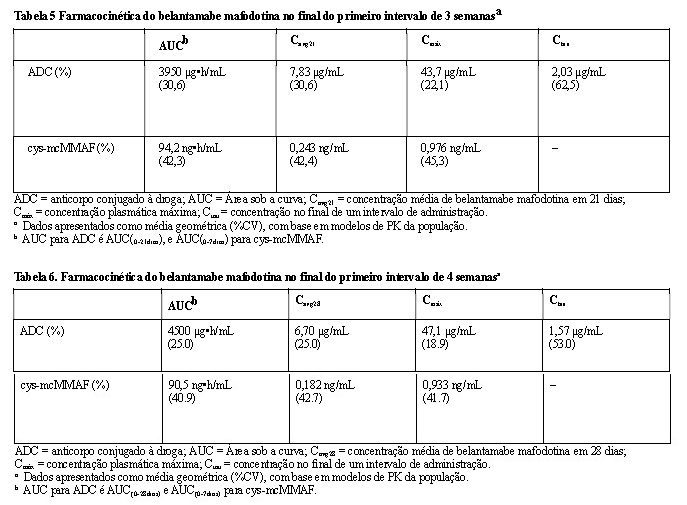
O acúmulo de belantamabe mafodotina (ADC) foi mínimo a moderado, conforme observado em estudos clínicos com um regime de administração a cada 3 semanas.
Distribuição
In vitro, o cys-mcMMAF apresentou baixa ligação à proteína (70% não ligado em uma concentração de 5 ng/mL) no plasma humano de maneira dependente da concentração. Com base na análise PK da população, a média geométrica (CV% geométrico) para o volume de distribuição em estado de equilíbrio do belantamabe mafodotina foi de 10,8 L (22%).
Metabolismo
Espera-se que a porção de anticorpo monoclonal do belantamabe mafodotina sofra proteólise em pequenos peptídeos e aminoácidos individuais por enzimas proteolíticas ubíquas. O Cys-mcMMAF teve depuração metabólica limitada em estudos de incubação da fração S9 hepática humana.
Interações medicamentosas
Estudos in vitro demonstraram que o cys-mcMMAF não é um inibidor, um indutor ou um substrato sensível das enzimas do citocromo P450, mas é um substrato do polipeptídeo transportador de ânions orgânicos (OATP)1B1 e OATP1B3, da proteína associada à resistência a múltiplos medicamentos (MRP)1, MRP2, MRP3, da bomba de exportação de sais biliares (BSEP) e um possível substrato da glicoproteína P (P-gp).
Efeito de outros medicamentos sobre o belantamabe mafodotina
Uma análise farmacocinética (PK) da população foi usada para avaliar a terapia combinada na PK do ADC belantamabe mafodotina e cysmcMMAF. As terapias combinadas com bortezomibe, lenalidomida, pomalidomida e/ou dexametasona não afetaram a PK do ADC e do cysmcMMAF.
Efeito do belantamabe mafodotina sobre outros medicamentos
Para terapias combinadas com lenalidomida, bortezomibe e pomalidomida, os perfis de PK foram avaliados em estudos clínicos e comparados com dados históricos. A PK observada para lenalidomida, bortezomibe e pomalidomida sugeriu a falta de impacto do belantamabe mafodotina na PK das terapias combinadas incluídas.
Eliminação
Com base na análise PK da população, a média geométrica (CV% geométrico) da CL sistêmica inicial do belantamabe mafodotina (ADC) foi de 0,901 L/dia (40%) e a meia-vida de eliminação foi de 13 dias (26%). Após o tratamento, o CL no estado de equilíbrio foi de 0,605 L/dia (43%) ou aproximadamente 33% menor do que o CL sistêmico inicial com uma meia vida de eliminação de 17 dias (31%). A fração de cys-mcMMAF excretada na urina não foi substancial (aproximadamente 18% da dose) após a dose do Ciclo 1, sem evidência de outros metabólitos relacionados ao MMAF.
Linearidade/não linearidade
O belantamabe mafodotina apresenta farmacocinética proporcional à dose ao longo da faixa de dose recomendada, com uma redução na depuração ao longo do tempo.
Carcinogênese/mutagênese
O belantamabe mafodotina foi genotóxico em um ensaio de triagem de micronúcleos in vitro em linfócitos humanos, consistente com o efeito farmacológico da interrupção de microtúbulos mediada por cys-mcMMAF, causando aneuploidia. Não foram realizados estudos de carcinogenicidade ou genotoxicidade definitiva com belantamabe mafodotina.
Toxicidade reprodutiva
Não foram realizados estudos em animais para avaliar os efeitos potenciais do belantamabe mafodotina na reprodução ou no desenvolvimento. O mecanismo de ação é matar as células que se dividem rapidamente, o que afetaria um embrião em desenvolvimento, que tem células que se dividem rapidamente. Há também um potencial risco de alterações hereditárias por meio de aneuploidia nas células germinativas femininas.
Efeitos nos órgãos reprodutivos masculinos e femininos foram observados em animais em doses de ≥10 mg/kg, o que é aproximadamente 4 vezes a exposição da dose clínica. Folículos não ovulatórios luteinizados foram observados nos ovários de ratas após 3 doses semanais. Os achados nos órgãos reprodutivos masculinos, que foram adversos e progrediram após a repetição da administração em ratos, incluíram degeneração/atrofia acentuada dos túbulos seminíferos que, em geral, não reverteram após a interrupção da administração.
Toxicologia e/ou farmacologia animal
Em estudos não clínicos, os principais achados adversos (relacionados diretamente à belantamabe mafodotina) no rato e no macaco, em exposições ≥1,2 vez a dose clínica recomendada de 2,5 mg/kg, foram enzimas hepáticas elevadas, algumas vezes associadas à necrose hepatocelular em ≥10 e ≥3 mg/kg, respectivamente, e aumentos nos macrófagos alveolares associados a material eosinofílico nos pulmões em ≥3 mg/kg (somente no rato). A maioria dos achados em animais estava relacionada ao conjugado de medicamentos citotóxicos; as alterações histopatológicas observadas nos testículos e pulmões não eram reversíveis em ratos.
Foi observada necrose de uma única célula no epitélio da córnea e/ou aumento das mitoses das células epiteliais da córnea em ratos e coelhos. Em coelhos, foi observada inflamação do estroma da córnea, correlacionada com opacidade superficial e vascularização. O belantamabe mafodotina foi absorvido pelas células de todo o corpo por um mecanismo não relacionado à expressão do receptor do BCMA na membrana celular.
Populações especiais de pacientes
Crianças
Não há dados farmacocinéticos disponíveis em pacientes pediátricos.
Idosos
Com base em uma população de pacientes com idades entre 32 e 89 anos, a idade não foi uma covariável significativa nas análises farmacocinéticas da população.
Insuficiência renal
Em pacientes com comprometimento renal grave (eGFR: 15 a 29 mL/min), a Cmáx de BLENREP® diminuiu 23% e a AUC(0-tau) diminuiu 16% em comparação com pacientes com comprometimento renal normal ou leve (eGFR ≥60 mL/min). Quanto ao cys-mcMMAF, a Cmáx e a AUC(0168h) diminuíram 56% e 44%, respectivamente, em comparação com pacientes com comprometimento renal normal ou leve. A função renal (eGFR: 12 a 150 mL/min) não foi uma covariável significativa nas análises farmacocinéticas da população que incluíram pacientes com comprometimento renal normal ou leve, moderado ou grave, ou insuficiência renal. Não se espera que BLENREP® seja removido por diálise devido ao seu tamanho molecular. Embora o cys-mcMMAF livre possa ser removido por diálise, a exposição sistêmica ao cys-mcMMAF é muito baixa e não demonstrou estar associada a desfechos de eficácia ou segurança com base na análise de resposta à exposição.
Insuficiência hepática
Não foram realizados estudos formais em pacientes com comprometimento hepático. A função hepática, conforme classificação do Grupo de Trabalho de Disfunção de Órgãos National Cancer Institute, não foi uma covariável significativa nas análises farmacocinéticas da população que incluíram pacientes com função hepática normal, leve (bilirrubina total superior ao ULN a ≤1,5 × ULN e qualquer AST ou bilirrubina total ≤ ULN com AST > ULN) ou comprometimento hepático moderado (bilirrubina total superior a 1,5 x ULN a ≤3 × ULN e qualquer AST).
Peso corporal
O peso corporal (37 a 170 kg) foi uma covariável significativa nas análises farmacocinéticas da população, mas esse efeito não foi clinicamente relevante com o regime de administração proporcional ao peso. Para alterações no peso corporal > 10% durante o tratamento, recalcule a dose com base no peso corporal real no momento da dosagem.
4. CONTRAINDICAÇÕES
BLENREP® é contraindicado nos casos de hipersensibilidade à substância ativa ou a qualquer componente da fórmula (consultar COMPOSIÇÃO).
Este medicamento é contraindicado para menores de 18 anos.
5. ADVERTÊNCIAS E PRECAUÇÕES
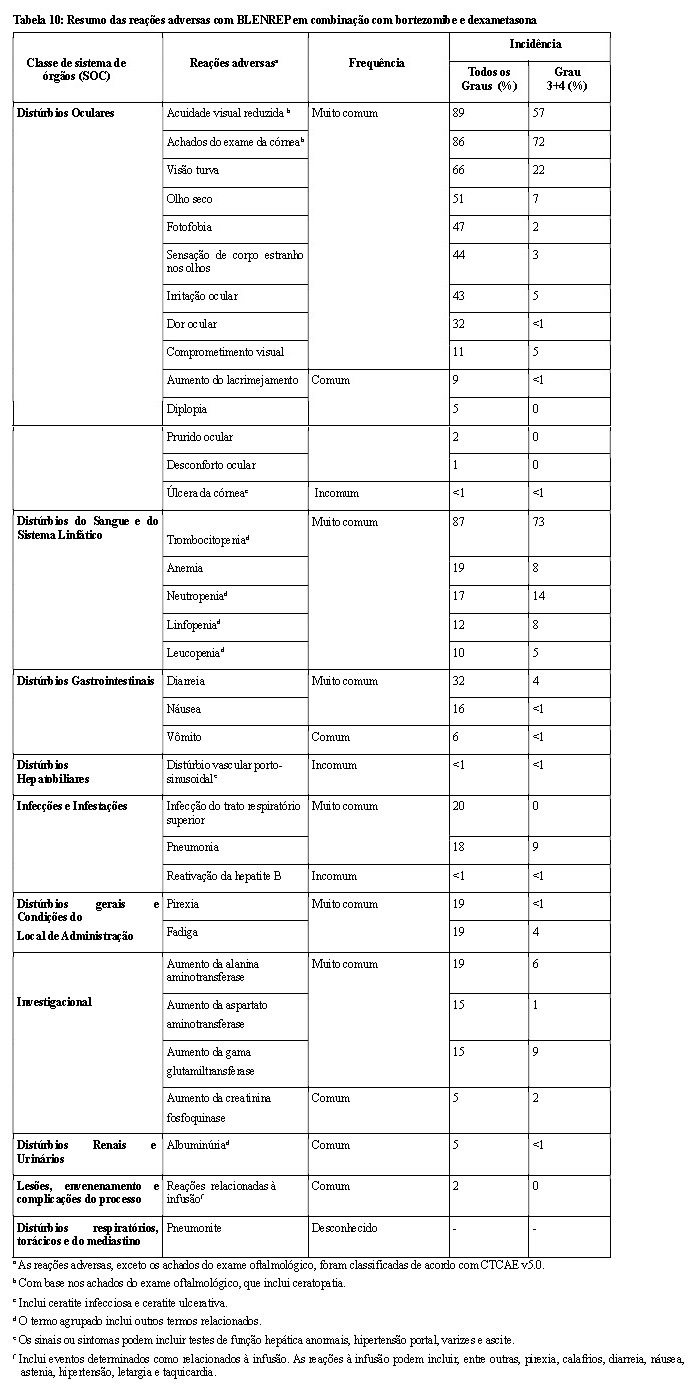
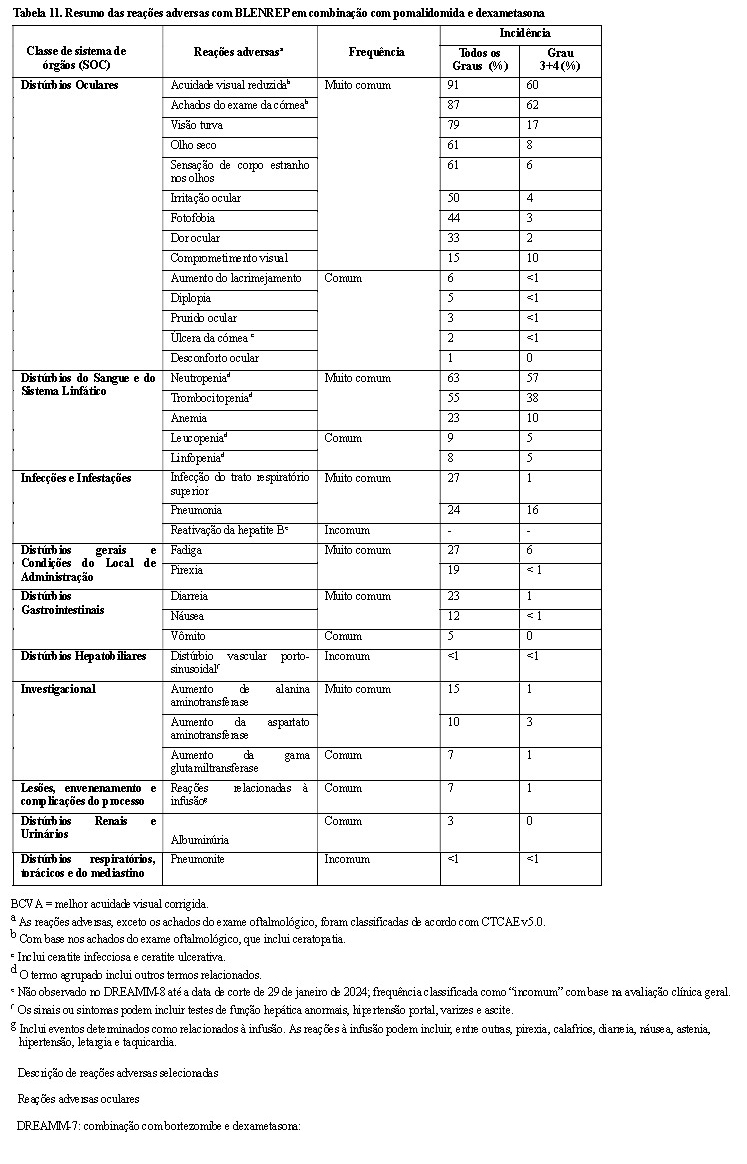
Reações Adversas Oculares
Reações adversas oculares (por ex., visão turva, olho seco, irritação ocular e fotofobia) foram relatadas com o uso de BLENREP® .
Os achados mais comumente relatados no exame da córnea incluíram ceratopatia puntiforme superficial, alterações epiteliais semelhantes a microcistos e opacidade, com ou sem alterações na acuidade visual. Alterações clinicamente relevantes na acuidade visual podem estar associadas à dificuldade de dirigir ou operar máquinas.
Exames oftalmológicos, incluindo avaliação da acuidade visual e exame com lâmpada de fenda, devem ser realizados antes de cada uma das primeiras 6 doses de BLENREP® e durante o tratamento, conforme indicação clínica.
Os pacientes devem ser orientados a administrar lágrimas artificiais sem conservantes pelo menos 4 vezes ao dia durante o tratamento (consulte Posologia e Administração). Os pacientes devem evitar o uso de lentes de contato até o final do tratamento. As lentes de contato de bandagem podem ser usadas sob a orientação de um oftalmologista.
Os pacientes que apresentarem achados no exame da córnea (ceratopatias, como ceratopatia puntiforme superficial ou depósitos semelhantes a microcistos) com ou sem alterações na acuidade visual podem precisar de modificação da dose (atraso e/ou redução) ou descontinuação do tratamento com base na gravidade dos achados (consulte Posologia e Modo de usar).
Foram relatados casos de úlcera de córnea (ceratite ulcerativa e infecciosa) (consulte Reações Adversas). Elas devem ser tratadas prontamente e conforme indicação clínica por um oftalmologista. O tratamento com BLENREP® deve ser interrompido até que a úlcera da córnea tenha cicatrizado (consulte Posologia e Modo de usar).
Trombocitopenia
Eventos trombocitopênicos (trombocitopenia e diminuição da contagem de plaquetas) foram relatados com BLENREP®. A trombocitopenia pode levar a eventos hemorrágicos graves, incluindo sangramento gastrointestinal e intracraniano.
O hemograma completo deve ser obtido no período basal e monitorado durante o tratamento, conforme indicação clínica. Os pacientes com trombocitopenia de Grau 3 ou 4 ou aqueles em tratamento anticoagulante concomitante podem precisar de monitoramento mais frequente e devem ser tratados com atraso ou redução da dose (consulte Posologia e Modo de usar).
A terapia de suporte (por exemplo, transfusões de plaquetas) deve ser fornecida de acordo com a prática médica padrão.
Reações à Infusão
Reações relacionadas à infusão (IRR) foram relatadas com BLENREP®. A maioria das IRRs foi de Grau 1 ou 2 e resolvida no mesmo dia (consulte Reações Adversas). Se ocorrer uma IRR de grau 2 ou superior durante a administração, reduza a taxa de infusão ou interrompa a infusão, dependendo da gravidade dos sintomas. Instituir tratamento médico adequado e reiniciar a infusão em uma taxa mais lenta, se a condição do paciente estiver estável. Se ocorrer IRR de Grau 2 ou superior, administrar pré-medicação para as infusões subsequentes (consulte Posologia e Modo de usar).
Pneumonite
Casos de pneumonite, incluindo eventos fatais, foram observados com BLENREP®, embora uma associação causal não tenha sido estabelecida. A avaliação de pacientes com sintomas pulmonares inexplicáveis novos ou agravados (por ex., tosse, dispneia) deve ser realizada para excluir uma possível pneumonite. Em caso de suspeita de pneumonite de Grau 3 ou superior, o BLENREP® deve ser permanentemente descontinuado e o tratamento adequado deve ser iniciado.
Reativação do vírus da hepatite B
A reativação do vírus da hepatite B (HBV) pode ocorrer em pacientes tratados com produtos medicinais direcionados a células B, incluindo BLENREP®. Pacientes com evidência de sorologia positiva para HBV devem ser monitorados quanto a sinais clínicos e laboratoriais de HBV reativação. Se a reativação do HBV ocorrer enquanto estiver em BLENREP®, os pacientes devem ser tratados de acordo com as diretrizes clínicas.
Gestação e Lactação
Gestação
Fertilidade
Com base nos achados em animais e no mecanismo de ação, BLENREP® pode prejudicar a fertilidade em mulheres e homens férteis (consulte Características Farmacológicas).
Mulheres férteis/Contracepção em homens e mulheres
O estado de gravidez de mulheres férteis deve ser verificado antes do início da terapia com BLENREP®. Mulheres férteis devem usar métodos contraceptivos eficazes durante o tratamento com BLENREP® e por 4 meses após a última dose.
Homens com parceiras férteis devem usar métodos contraceptivos eficazes durante o tratamento com BLENREP® e por 6 meses após a última dose.
Gestação
Não existem dados quanto ao uso de BLENREP® em mulheres grávidas. Com base no mecanismo de ação do componente citotóxico monometil auristatina F (MMAF), BLENREP® pode causar danos ao feto-embrião quando administrado a uma mulher grávida (consulte Características Farmacológicas). Sabe-se que a imunoglobulina G (IgG) humana atravessa a placenta; portanto, BLENREP® pode ser transmitido da mãe para o feto em desenvolvimento.
BLENREP® não deve ser usado durante a gravidez, a menos que o benefício para a mãe supere os potenciais riscos para o feto. Se uma mulher grávida precisar ser tratada, ela deve ser informada claramente sobre o potencial risco para o feto.
Lactação
Não se sabe se BLENREP® é excretado no leite humano. A imunoglobulina G (IgG) está presente no leite humano em pequenas quantidades. Como o BLENREP® é um anticorpo monoclonal IgG humanizado, e com base no mecanismo de ação, ele pode causar reações adversas graves em crianças amamentadas. As mulheres devem ser aconselhadas a interromper a amamentação antes de iniciar o tratamento com BLENREP® e por 3 meses após a última dose.
Categoria C
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgião-dentista.
Uso contraindicado no aleitamento ou na doação de leite humano. Este medicamento é contraindicado durante o aleitamento ou doação de leite, pois pode ser excretado no leite humano e pode causar reações indesejáveis no bebê. Seu médico ou cirurgião-dentista deve apresentar alternativas para o seu tratamento ou para a alimentação do bebê.
Efeitos sobre a capacidade de dirigir veículos ou operar máquinas
A piora da acuidade visual foi relatada em alguns pacientes tratados com BLENREP® durante os estudos clínicos (consulte Advertências e Precauções, Reações Adversas). Os pacientes devem ser aconselhados a ter cuidado ao dirigir ou operar máquinas, pois BLENREP® pode afetar a visão.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo de interação medicamentosa foi realizado. Com base nos dados in vitro e clínicos disponíveis, há um baixo risco de interações medicamentosas farmacocinéticas ou farmacodinâmicas para belantamabe mafodotina. Terapias combinadas com bortezomibe, lenalidomida, pomalidomida e/ou dexametasona não afetam as propriedades farmacocinéticas de belantamabe mafodotina (veja a seção Propriedades Farmacocinéticas).
7. CUIDADOS DE ARMAZENAMENTO DO MEDICAMENTO
Cuidados de Armazenamento
Armazenar no recipiente original. Conservar na geladeira (2 °C a 8 °C). Não congelar.
O prazo de validade do medicamento é de 48 meses a contar da data de fabricação.
Solução reconstituída:
A solução reconstituída pode ser armazenada em temperatura ambiente (20 °C a 25 °C) ou em um refrigerador (2 °C a 8 °C) por até 4 horas. Não congelar.
Solução diluída:
Se não for usada imediatamente, a solução diluída pode ser armazenada em geladeira (2 °C a 8 °C) antes da administração por até 24 horas. Não congelar. Se for refrigerada, deixe a solução diluída atingir a temperatura ambiente antes da administração. A solução de infusão diluída pode ser mantida em temperatura ambiente (20 °C a 25 °C) por um período máximo de 6 horas (incluindo o tempo de infusão).
Número de lote e datas de fabricação e validade: vide embalagem
Não use medicamento com o prazo de validade vencido. Guarde-o em sua embalagem original.
Aspectos físicos / Características organolépticas
BLENREP® é apresentado como um pó liofilizado branco a amarelado.
A solução reconstituída deve ser um líquido claro a opalescente, incolor a amarelo a marrom.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
8. POSOLOGIA E MODO DE USAR
Pelo menos uma agulha é necessária para a reconstituição de cada frasco, e uma agulha adicional para a retirada e transferência da solução reconstituída para a bolsa de infusão. O número total de agulhas necessárias dependerá do número de frascos utilizados, que é determinado com base no peso do paciente. Não há requisitos especiais para administração; considere a prática clínica local.
Uso e Manuseio
BLENREP® é um medicamento citotóxico anticâncer. Os procedimentos adequados de manuseio devem ser seguidos. Use uma técnica asséptica para a reconstituição e diluição da solução de administração.
Calcule a dose (mg), o volume (mL) total de solução necessário e o número de frascos necessários com base no peso (kg) corporal real do paciente.
Reconstituição
1 Retire o(s) frasco(s) de BLENREP® do refrigerador e deixe o(s) em repouso por aproximadamente 10 minutos para atingir a temperatura ambiente.
2 Reconstitua cada frasco de 70 mg com 1,4 mL de Água Estéril para Injeção para obter uma concentração final de 50 mg/mL. Reconstitua cada frasco de 100 mg com 2 mL de Água Estéril para Injeção para obter uma concentração final de 50 mg/mL. Gire suavemente o frasco para ajudar na dissolução. Não agite.
3 Inspecione visualmente a solução reconstituída quanto a partículas e descoloração. A solução reconstituída deve ser um líquido claro a opalescente, incolor a amarelo a marrom. Descarte o frasco reconstituído se forem observadas partículas estranhas que não sejam partículas proteicas translúcidas a brancas.
Instruções de Diluição para Uso Intravenoso
1 Retire de cada frasco o volume necessário para a dose calculada.
2 Adicione a quantidade necessária de BLENREP® à bolsa de infusão contendo 250 mL de solução injetável de cloreto de sódio 9 mg/mL (0,9%). Misture a solução diluída invertendo-a suavemente. A concentração final da solução diluída deve estar entre 0,2 mg/mL e 2 mg/mL. NÃO AGITE.
3 Descarte qualquer solução reconstituída de BLENREP® não utilizada que tenha ficado no frasco.
Se a solução diluída não for usada imediatamente, ela pode ser armazenada em um refrigerador (2 °C a 8 °C) por até 24 horas antes da administração. Se for refrigerada, deixe a solução diluída atingir a temperatura ambiente antes da administração. A solução diluída pode ser mantida em temperatura ambiente (20 °C a 25 °C) por um período máximo de 6 horas (incluindo o tempo de infusão).
Instruções de Administração
1 Administre a solução diluída por infusão intravenosa durante aproximadamente 30 minutos usando um conjunto de infusão feito de cloreto de polivinila ou poliolefina.
2 Não é necessário filtrar a solução diluída. No entanto, se a solução diluída for filtrada, recomenda-se o uso de um filtro à base de polietersulfona (PES).
Descarte
Qualquer medicamento não utilizado ou resíduos devem ser descartados de acordo com as exigências locais.
Posologia e administração
O tratamento com BLENREP® deve ser iniciado e monitorado por médicos com experiência no tratamento de mieloma múltiplo.
Método de Administração
BLENREP® é um medicamento citotóxico anticâncer. Os procedimentos adequados de manuseio devem ser seguidos. As instruções sobre reconstituição e diluição adicional são fornecidas na seção Uso e Manuseio.
BLENREP® é administrado como uma infusão intravenosa durante aproximadamente 30 minutos.
Tratamento de Suporte Recomendado
Os pacientes devem ser submetidos a um exame oftalmológico (incluindo exames de acuidade visual e com lâmpada de fenda) realizado por um oftalmologista antes de cada uma das primeiras 6 doses de BLENREP® e, posteriormente, conforme indicação clínica (consulte Advertências e Precauções).
Os médicos devem incentivar os pacientes a informá-los sobre quaisquer sintomas oculares. Além disso, eles devem orientar os pacientes a administrarem lágrimas artificiais sem conservantes pelo menos 4 vezes ao dia, começando no primeiro dia de infusão e continuando até a conclusão do tratamento, pois isso pode reduzir os sintomas oculares (consulte Advertências e Precauções).
Para pacientes com sintomas de olho seco, terapias adicionais podem ser consideradas, conforme recomendado pelo oftalmologista.
Adultos
A administração de BLENREP® deve ser continuada até a progressão da doença ou toxicidade inaceitável.
Combinação com outras terapias
O esquema posológico de dose inicial recomendado de BLENREP® em combinação com outras terapias está apresentada na Tabela 7.
Quando BLENREP® for administrado em combinação com outras terapias, consulte a seção Resultados de Eficácia e as respectivas informações de prescrição, conforme aplicável.
Modificações de Dose
A dose de BLENREP® deve ser individualizada para cada paciente. As modificações de dose recomendadas são fornecidas nas Tabelas 8 e 9 para reações adversas.
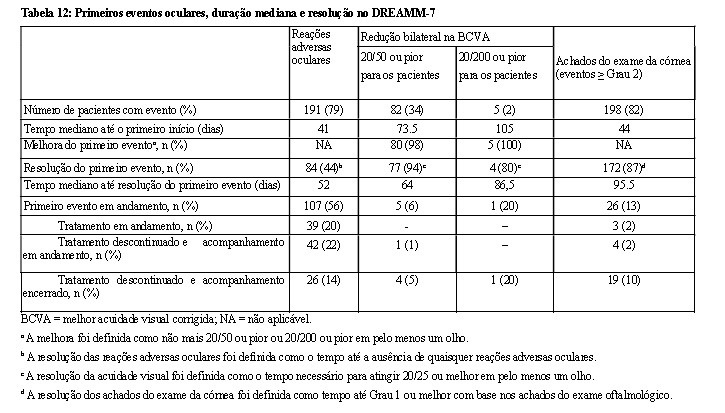
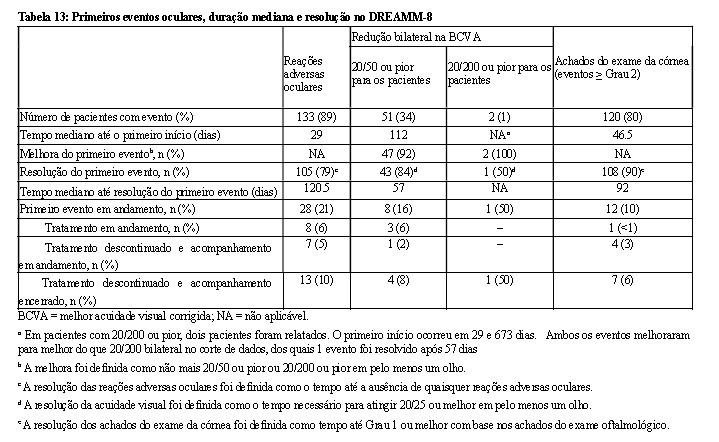
Os eventos oculares foram classificados com base nos achados do exame oftalmológico, que incluem a combinação dos achados do exame da córnea e a melhor acuidade visual corrigida (BCVA).
O médico responsável pelo tratamento deve analisar os achados do exame oftalmológico do paciente antes da administração e determinar a dose de BLENREP® com base nos resultados (Tabela 9). Durante o exame oftalmológico, o oftalmologista deve avaliar o seguinte:
• O(s) achado(s) do exame da córnea e o declínio da BCVA. ·
• Se houver um declínio na BCVA, a relação com o BLENREP® deve ser determinada. ·
• A classificação da categoria desses achados de exame e da BCVA deve ser comunicada ao médico responsável pelo tratamento.
Os achados do exame da córnea podem ou não ser acompanhados de alterações na BCVA. Observação: Um olho pode ser afetado mais gravemente do que o outro. É importante que os médicos considerem não apenas os achados do exame da córnea, mas também as alterações da acuidade visual e os sintomas relatados ao avaliarem os atrasos e as reduções de dose.
Não reescalone a dose de BLENREP® após a redução da dose devido a reações adversas oculares.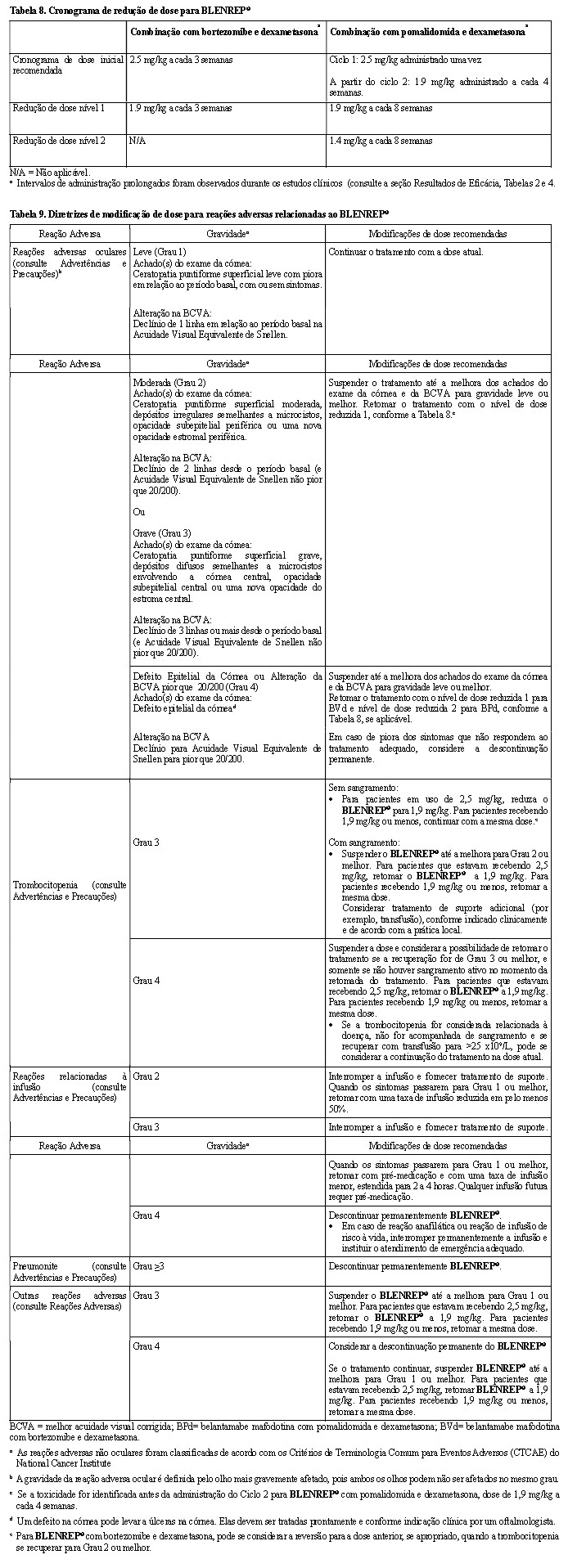
Crianças A segurança e a eficácia do BLENREP® não foram estabelecidas em crianças com menos de 18 anos de idade.
Idosos
Não é necessário ajuste de dose em pacientes com mais de 65 anos de idade (consulte Características Farmacológicas).
Insuficiência renal
Não é necessário ajuste de dose em pacientes com comprometimento renal leve, moderado ou grave ou insuficiência renal (eGFR < 30 mL/min) (Características Farmacológicas).
Insuficiência hepática
Não é necessário ajuste de dose em pacientes com comprometimento hepático leve (bilirrubina total maior que ULN a ≤1,5 × ULN e qualquer aspartato transaminase [AST] ou bilirrubina total ≤ ULN com AST > ULN). Há dados limitados em pacientes com insuficiência hepática moderada e não há dados suficientes em pacientes com comprometimento hepático grave para apoiar uma recomendação de dose (Características Farmacológicas).
Incompatibilidades
Na ausência de estudos de compatibilidade, o conc