BIKTARVY
GILEAD
tenofovir alafenamida, hemifumarato de + entricitabina + bictegravir sodico
Antiviral.
Apresentações.
Biktarvy é apresentado em frascos contendo 30 comprimidos revestidos.
USO ORAL
USO ADULTO E PEDIÁTRICO ACIMA DE 6 ANOS, COM PESO CORPÓREO DE PELO MENOS 25 KG
Composição.
Cada comprimido revestido contém bictegravir sódico equivalente a 50 mg de bictegravir (52,5 mg de bictegravir sódico equivalem a 50 mg de bictegravir), 200 mg de entricitabina e hemifumarato de tenofovir alafenamida equivalente a 25 mg de tenofovir alafenamida (28,04 mg de hemifumarato de tenofovir alafenamida equivalem a 25 mg de tenofovir alafenamida).
Excipientes: celulose microcristalina, croscarmelose sódica, estearato de magnésio, álcool polivinílico, dióxido de titânio, macrogol, talco, óxido de ferro vermelho e óxido de ferro preto.
Informações técnicas.
1. INDICAÇÕES
Biktarvy é indicado para o tratamento de adultos e pacientes pediátricos, 6 anos ou mais e com peso corporal de pelo menos 25 kg, com infeção pelo vírus da imunodeficiência humana do tipo 1 (HIV-1), sem evidências, presentes ou passadas, de resistência à classe dos inibidores da integrase, entricitabina ou tenofovir.
2. RESULTADOS DE EFICÁCIA
A eficácia e segurança de Biktarvy em adultos infectados pelo HIV-1, virgens de tratamento, são baseadas em dados de 48 semanas e 96 semanas de dois estudos, randomizados, duplo-cego, controlados, GS-US-380-1489 (n=629) and GS-US-380-1490 (n=645).
A eficácia e segurança de Biktarvy em adultos infectados pelo HIV-1 e virologicamente suprimidos são baseadas em dados de 48 semanas de um estudo de randomizados, duplo cego, controlado, GS-US-380-1844 (n=563) e um estudo randomizado, aberto, controlado GS-US-380-1878 (n=577).
A eficácia e segurança de Biktarvy em pacientes pediátricos infectados pelo HIV são baseadas em dados de um estudo aberto, GS-US-380-1474, em pacientes pediátricos virologicamente suprimidos com idades entre 12 a < 18 anos (≥ 35 kg) (N=50) e com idades de 6 a < 12 anos (≥ 25 kg) (N=50).
- Pacientes infectados pelo HIV-1 virgens de tratamento
No estudo GS-US-380-1489, os pacientes foram randomizados na proporção de 1:1 para receberem ou B/F/TAF (n=314) ou abacavir/dolutegravir/lamivudina (600/50/300 mg) (n=315) uma vez ao dia. No estudo GS-US-380-1490, os pacientes foram randomizados para proporção de 1:1 para receber ou Biktarvy (n=320) ou dolutegravir + entricitabina/tenofovir alafenamida (50+200/25 mg) (n=325) uma vez ao dia.
Nos estudos GS-US-380-1489 e GS-US-380-1490, a idade média era 35 anos (intervalo 18-77), 89% eram homens, 58% eram caucasianos, 33% eram negros and 3% eram asiáticos. Vinte e quatro por cento dos pacientes foram identificados como hispânicos/latinos. A prevalência de diferentes subtipos foi comparável entre todos os três grupos, com o subtipo B predominante em ambos os grupos de tratamento; 11% eram subtipos não-B. A média de RNA HIV-1 no início do estudo foi 4.4 log10 cópias/mL (intervalo 1.3-6.6). A média da contagem de células CD4+ no início do estudo foi 460 células/mm3 (intervalo 0-1636) e 11% tinha contagem de células CD4+ menor do que 200 células /mm3. Dezoito por cento dos pacientes tinham cargas virais no início do estudo maiores que 100.000 cópias/mL. Em ambos os estudos, os pacientes foram estratificados pelo RNA HIV-1 no início do estudo (menor ou igual 100.000 cópias/mL, maior que 100.000 cópias /mL para menor que ou igual 400.000 cópias/mL, ou maior que 400.000 cópias/mL), pela contagem de CD4 (menor que 50 células /mL, 50-199 células /mL, ou maior ou igual 200 células/mL), e por região (EUA ou não-EUA).
Os desfechos de tratamento dos Estudos GS-US-380-1489 e GS-US-380-1490 após as Semanas 48 e 96 são apresentados na Tabela 1.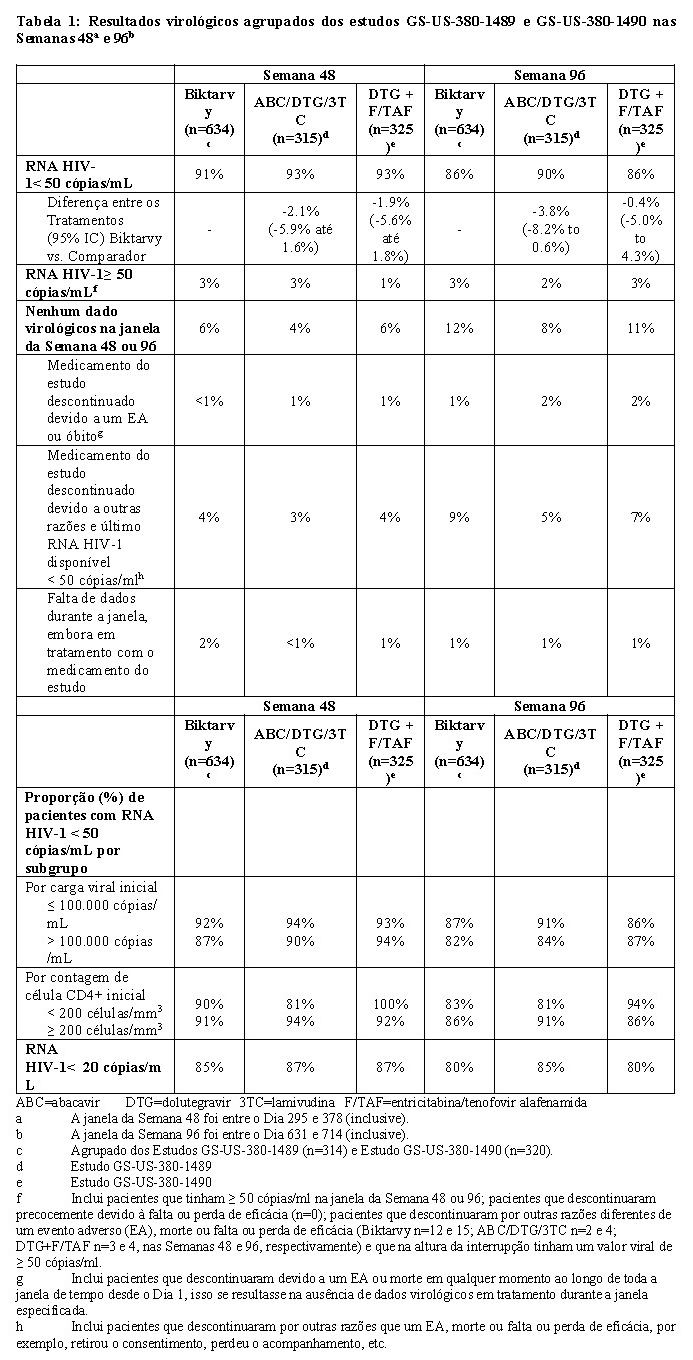
Biktarvy foi não-inferior em atingir o RNA HIV-1 RNA < 50 cópias/mL em ambas as Semanas 48 e 96 quando comparado ao abacavir/dolutegravir/lamivudina e dolutegravir+entricitabina/tenofovir alafenamida, respectivamente. O estudo atingiu o critério de não-inferioridade de -12% como desfecho primário na Semana 48. Os desfechos de tratamento foram similares entre os subgrupos por idade, sexo, raça, carga viral inicial e contagem de células CD4+ inicial.
Nos Estudos GS-US-380-1489 e GS-US-380-1490, o aumento médio na contagem de CD4+ desde o início do estudo até à Semana 96 foi de 262, 288 e 281 células/mm3 nos grupos agrupados de Biktarvy, abacavir/dolutegravir/lamivudina e dolutegravir+entricitabina/tenofovir alafenamida, respectivamente.
- Pacientes infectados pelo HIV-1 e suprimidos virologicamente
No estudo GS-US-380-1844, a eficácia e segurança na troca de um regime de dolutegravir+abacavir/lamivudina ou abacavir/dolutegravir/lamivudina para Biktarvy foram avaliadas em um estudo randomizado e duplo-cego com adultos (n=563) infectados pelo HIV-1 e virologicamente suprimidos (RNA HIV-1 < 50 cópias/mL). Os pacientes tinham que estar estavelmente suprimidos (RNA HIV-1 < 50 cópias/mL) em seu regime inicial por pelo menos 3 meses antes de entrar no estudo. Os pacientes foram randomizados numa proporção de 1:1 para ou trocar para Biktarvy no início do estudo (n=282) ou continuar com seu tratamento antirretroviral inicial (n=281). Os pacientes tinham uma idade média de 45 anos (intervalo 20-71), 89% eram homens, 73% eram Brancos e 22% eram Negros. Dezessete por cento dos pacientes identificados como Hispânicos/Latinos. A prevalência dos diferentes subtipos do HIV-1 foi comparável entre os grupos de tratamento, com o subtipo B predominante em ambos os grupos; 5% era subtipo não-B. A contagem média de células CD4+ no início do estudo era 723 células/mm3 (intervalo 124-2444).
No estudo GS-US-380-1878, a eficácia e segurança na troca de abacavir/lamivudina ou entricitabina/fumarato de tenofovir desoproxila (200/300 mg) mais atazanavir ou darunavir (potencializado por ou cobicistate ou ritonavir) para Biktarvy foram avaliadas em um estudo randomizado e aberto, com adultos infectados pelo HIV-1 e virologicamente suprimidos (n=577). Os pacientes tinham que estar estavelmente suprimidos em seu regime inicial por pelo menos 6 meses e não podiam ter sidos tratados anteriormente com qualquer inibidor da transferência de cadeia da integrase (INI - Inibidores da integrase). Os pacientes foram randomizados em uma proporção de 1:1 para ou trocar para Biktarvy (n=290) ou continuarem com seu tratamento antirretroviral inicial (n=287). Os pacientes tinham uma idade média de 46 anos (intervalo 20-79), 83% eram homens, 66% eram Brancos e 26% eram Negros. Dezenove por cento dos pacientes identificados como Hispânico/Latino. A contagem média das células CD4+ no início do tratamento era 663 células/mm3 (intervalo 62-2582). A prevalência dos diferentes subtipos foi comparável entre os grupos de tratamento, com o subtipo B predominante em ambos os grupos; 11% eram subtipos não-B. Os pacientes foram estratificados por regime de tratamento prévio. Na fase de seleção, 15% dos pacientes estavam recebendo abacavir/lamivudina mais atazanavir ou darunavir (potencializado por ou cobicistate ou ritonavir) e 85% dos pacientes estavam recebendo entricitabina/fumarato de tenofovir desoproxila mais atazanavir ou darunavir (potencializado por ou cobicistate ou ritonavir).
Os desfechos de tratamento dos Estudos GS-US-380-1844 e GS-US-380-1878 até a Semana 48 são apresentadas na Tabela 2.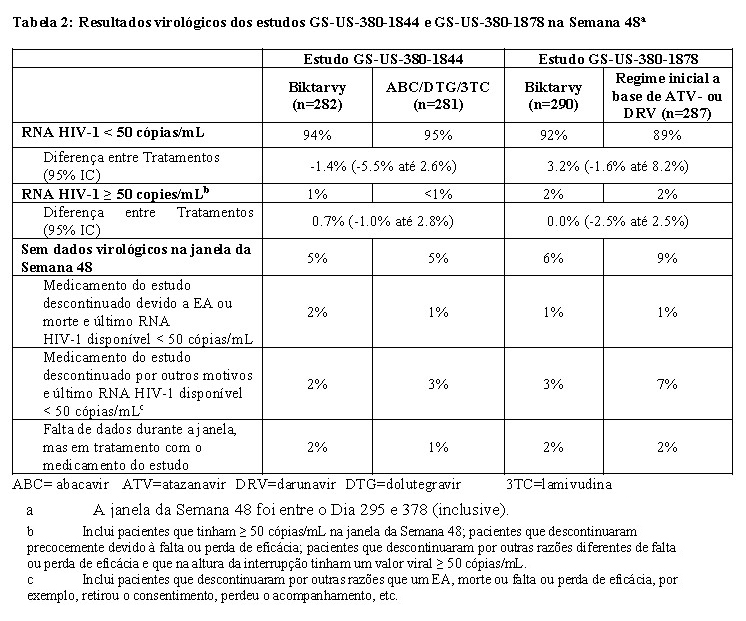
Biktarvy foi não-inferior ao tratamento controle em ambos os estudos de acordo com a margem de não- inferioridade pré-especificada de 4%. Os desfechos de tratamentos entre os grupos foram similares entre os subgrupos por idade, sexo, raça e região.
No GS-US-380-1844, a alteração média na contagem de CD4+ desde o começo do estudo até a Semana 48 foi -31 células/mm3 nos pacientes que trocaram para Biktarvy e 4 células/mm3 nos pacientes que continuaram no tratamento com abacavir/dolutegravir/lamivudina. No GS-US-380-1878, a alteração média na contagem de CD4+ desde o início do estudo até a Semana 48 foi 25 células/mm3 nos pacientes que trocaram para B/F/TAF e 0 célula/mm3 nos pacientes que permaneceram em seus tratamentos iniciais.
- Pacientes coinfectados pelo HIV e VHB
O número de pacientes coinfectados com HIV e HBV tratados com B/F/TAF é limitado. No Estudo GS-US-380-1490, 8 pacientes coinfectados com HIV/VHB no início do estudo foram randomizados para receber Biktarvy. Na semana 48, 7 pacientes apresentaram supressão do VHB (DNA VHB < 29 IU/mL) e tinham RNA HIV-1 < 50 copies/mL. Um paciente tinha dados faltando quanto ao DNA VHB na Semana 48. Na semana 96, 4 pacientes apresentavam supressão do VHB e tinham RNA HIV-1 < 50 cópias / mL. Quatro pacientes apresentavam dados em falta quanto ao DNA-VHB na semana 96 (um perdido para o acompanhamento desde a semana 48, 1 perdido para o acompanhamento depois da semana 72, 1 com dados em falta quanto ao VHB, mas com RNA HIV-1 < 50 cópias/mL e 1 com dados em falta na janela da semana 96).
No Estudo GS-US-380-1878, na Semana 48, 100% (8/8) dos pacientes coinfectados com HIV/VHB no início do estudo no braço com B/F/TAF mantiveram o DNA VHB < 29 IU/mL (faltando = análise excluída) e RNA HIV < 50 cópias/mL.
- Pacientes pediátricos
No Estudo GS-US-380-1474, a eficácia, a segurança e a farmacocinética de Biktarvy em pacientes pediátricos com supressão virológica infectados por HIV-1 entre as idades de 12 a < 18 anos (≥ 35 kg) (N = 50) e as idades de 6 a < 12 anos (≥ 25 kg) (N = 50) foram avaliadas.
Coorte 1: Adolescentes com supressão virológica (12 a < 18 anos; ≥ 35 kg)
Os pacientes da coorte 1 tinham uma idade média de 14 anos (12 a 17 anos) e um peso inicial médio de 51,7 kg (35 a 123), 64% eram mulheres, 27% eram asiáticos e 65% eram negros. No início do estudo, a contagem mediana de células CD4 + foi de 750 células / mm3 (intervalo: 337 a 1207) e a mediana de CD4 +% foi de 33% (intervalo: 19% a 45%).
Após a mudança para o Biktarvy, 98% (49/50) dos pacientes na coorte 1 permaneceram suprimidos (RNA do HIV-1 < 50 cópias / mL) na Semana 48. A alteração média desde o início na contagem de células CD4 + na Semana 48 foi de -22 células/mm3 Dois de 50 indivíduos preencheram os critérios para inclusão na população de análise de resistência até a Semana 48. Nenhuma resistência emergente ao Biktarvy foi detectada até a Semana 48.
Coorte 2: Crianças virologicamente suprimidas (6 a < 12 anos; ≥ 25 kg)
Os pacientes da coorte 2 tinham uma idade média de 10 anos (variação de 6 a 11) e um peso inicial médio de 31,9 kg (variação de 25 a 69), 54% eram mulheres, 22% eram asiáticos e 72% eram negros. No início do estudo, a contagem mediana de células CD4 + foi de 898 células/mm3 (intervalo de 390 a 1991) e a mediana de CD4 +% foi de 37% (intervalo: 19% a 53%).
Após a mudança para o Biktarvy, 100% (50/50) dos pacientes na coorte 2 permaneceram suprimidos (RNA do HIV-1 < 50 cópias / mL) na Semana 24. A alteração média desde o início na contagem de células CD4 + na Semana 24 foi de -24 células / mm3. Nenhum paciente se qualificou para análise de resistência até a Semana 24.
3. CARACTERÍSTICAS FARMACOLÓGICAS
- Propriedades farmacodinâmicas
Grupo farmacoterapêutico: Antiviral para uso sistêmico; antivirais para o tratamento de infecções pelo HIV, combinações, código ATC: J05AR20
Mecanismo de ação e efeitos farmacodinâmicos
O bictegravir é um inibidor da transferência de cadeia da integrase (INI - Inibidores da integrase) que se liga ao local ativo da integrase e bloqueia a etapa de transferência da cadeia da integração do ácido desoxirribonucleico (DNA) retroviral, o qual é essencial para o ciclo de replicação do HIV. O bictegravir possui atividade contra o HIV-1 e o HIV-2.
A entricitabina é um inibidor nucleosídeo da transcriptase reversa (ITRN) e um análogo nucleosídeo da 2'-deoxicitidina. A entricitabina é fosforilada por enzimas celulares para formar a entricitabina trifosfato. A entricitabina trifosfato inibe a replicação do HIV por incorporação no ácido desoxirribonucleico (DNA) viral através da transcriptase reversa (TR) do HIV, resultando na terminação da cadeia de DNA. A entricitabina possui atividade contra o HIV-1, o HIV-2 e o VHB.
O tenofovir alafenamida é um inibidor nucleotídeo da transcriptase reversa (INRN) e um pró-fármaco fosfonamidato do tenofovir (análogo 2'-deoxiadenosina monofosfato). O tenofovir alafenamida é permeável nas células e, devido a um aumento da estabilidade plasmática e ativação intracelular através de hidrólise pela catepsina A, o tenofovir alafenamida é mais eficaz do que o TDF em concentrar o tenofovir nas células mononucleares do sangue periférico (CsMSP) (incluindo linfócitos e outras células alvo do HIV) e macrófagos. O tenofovir intracelular é subsequentemente fosforilado ao metabólito farmacologicamente ativo tenofovir difosfato. O tenofovir difosfato inibe a replicação do HIV por incorporação no DNA viral através da TR do HIV, o que resulta na terminação da cadeia de DNA. O tenofovir possui atividade contra o HIB-1, o HIV-2 e o VHB.
Atividade antiviral in vitro
A atividade antiviral do bictegravir contra isolados laboratoriais e clínicos do HIV-1 foi analisada em linhagens celulares linfoblastoides, CsMSP, monócitos/macrófagos primários e linfócitos T CD4+. Os valores da concentração efetiva 50% (CE50) para o bictegravir encontravam-se no intervalo de < 0,05 a 6,6 nM. A CE95 proteínas-ajustada do bictegravir foi de 361 nM (0,162 microgramas/mL) para o HIV-1 do tipo selvagem. O bictegravir apresentou atividade antiviral em culturas celulares contra o grupo do HIV-1 (M, N, O), incluindo os subtipos A, B, C, D, E, F e G (os valores da CE50 variaram de < 0,05 a 1,71 nM) e demonstrou atividade contra o HIV-2 (CE50 = 1,1 nM).
A atividade antiviral de entricitabina contra isolados laboratoriais e clínicos do HIV-1 foi analisada em linhagens celulares linfoblastoides, na linhagem celular MAGI CCR5 e nas CsMSP. Os valores da CE50 para a entricitabina encontravam-se no intervalo de 0,0013 a 0,64 mM. A entricitabina apresentou atividade antiviral em culturas celulares contra os subtipos A, B, C, D, E, F e G do HIV-1 (os valores da CE50 variaram de 0,007 a 0,075 mM) e demonstrou atividade específica contra as cepas de HIV-2 (os valores da CE50 variaram de 0,007 a 1,5 mM).
A atividade antiviral de tenofovir alafenamida contra isolados laboratoriais e clínicos do HIV-1 subtipo B foi analisada em linhagens celulares linfoblastoides, CsMSP, monócitos/macrófagos primários e linfócitos T CD4+. Os valores da CE50 para o tenofovir alafenamida encontravam-se no intervalo de 2,0 a 14,7 nM. O tenofovir alafenamida apresentou atividade antiviral em culturas de células contra todos os grupos do HIV-1 (M, N e O), incluindo os subtipos A, B, C, D, E, F e G (os valores da CE50 variaram entre 0,10 e 12,0 nM) e atividade contra o HIV-2 (os valores da CE50 variaram de 0,91 a 2,63 nM).
Resistência
In vitro
Os isolados do HIV-1 com sensibilidade diminuída ao bictegravir foram selecionados em cultura celular. Em uma seleção emergiram as substituições de aminoácidos M50I e R263K e a suscetibilidade fenotípica ao bictegravir foi reduzida 1,3; 2,2- e 2,9- vezes para a M50I, R263K e M50I+R263K, respectivamente. Em uma segunda seleção, emergiram substituições de aminoácidos T66I and S153F e a suscetibilidade fenotípica ao bictegravir alterou-se 0,4-; 1,9- e 0,5- vezes para T66I, S153F e T66I+S153F, respectivamente.
Isolados de HIV-1 com sensibilidade diminuída à entricitabina foram selecionados em culturas celulares e tinham mutações M184V/I na TR do HIV-1.
Os isolados do HIV-1 com sensibilidade diminuída ao tenofovir alafenamida foram selecionados em cultura celular e tinham a mutação K65R na TR do HIV-1; além disso, observou-se transitoriamente uma mutação K70E na TR do HIV-1. Os isolados do HIV-1 com a mutação K65R apresentam sensibilidade diminuída de baixo nível ao abacavir, entricitabina, tenofovir e lamivudina. Estudos in vitro de seleção de resistência a fármacos com o tenofovir alafenamida não mostraram nenhum desenvolvimento de resistência de alto-nível após cultura prolongada.
Em pacientes sem tratamento prévio (estudos GS-US-380-1489 e GS-US-380-1490) e virologicamente suprimidos (estudos GS-US-380-1844 e GS-US-380-1878), nenhum paciente que recebeu Biktarvy apresentou HIV-1 com resistência genotípica ou fenotípica emergente do tratamento com bictegravir, entricitabina ou ao tenofovir alafenamida na população de análise da resistência final (n=10) com RNA HIV-1 ≥ 200 cópias/mL quando da confirmação da falha virológica, na Semana 48, Semana 96 (somente estudos com pacientes sem tratamento prévio) ou no momento da descontinuação precoce do medicamento do estudo. No momento da entrada no estudo, um paciente sem experiência terapêutica prévia tinha mutações pré-existentes associadas à resistência a INI (Q148H + G140S) e tinha RNA HIV-1 < 50 cópias/mL da semana 4 à semana 96. Além disso, 6 pacientes tinham a mutação pré-existente associada à resistência a INI T97A; todos os pacientes tinham RNA HIV-1 < 50 cópias/mL na semana 96 ou na última visita.
Resistência-cruzada
A suscetibilidade ao bictegravir foi testada contra 64 isolados clínicos resistentes a INI (20 com uma única substituição e 44 com 2 ou mais substituições). Destes, todos os isolados com mutações únicas ou duplas sem a Q148H/K/R e 10 dos 24 isolados com a Q148H/K/R com substituições adicionais associadas à resistência a INI tinham uma suscetibilidade ≤ 2,5 vezes diminuída ao bictegravir; foi encontrada uma suscetibilidade > 2,5 vezes diminuída ao bictegravir em 14 dos 24 isolados com substituições G140A/C/S e Q148H/R/K na integrase. Destes, nove dos 14 isolados tinham mutações adicionais em L74M, T97A ou E138A/K. Em um estudo separado, a mutagênese sitio-dirigida com a G118R e T97A+G118R apresentaram uma suscetibilidade 3,4 e 2,8 vezes diminuída ao bictegravir, respectivamente. A relevância destes dados de resistência cruzada in vitro para a prática clínica ainda não foi estabelecida.
O bictegravir demonstrou atividade antiviral equivalente contra 5 clones mutantes de HIV-1 resistentes a inibidores não nucleosídeos da transcriptase reversa (ITRNNs), 3 clones mutantes de HIV-1 resistentes a ITRN e 4 clones mutantes de HIV-1 resistentes a inibidores da protease (IP), comparativamente com a estirpe selvagem.
Os vírus resistentes à entricitabina com a substituição M184V/I apresentaram resistência cruzada à lamivudina, mas mantiveram a sensibilidade à didanosina, estavudina, tenofovir e à zidovudina.
As mutações K65R e K70E resultam numa sensibilidade diminuída ao abacavir, didanosina, lamivudina, entricitabina e tenofovir, mas mantêm a sensibilidade à zidovudina. O HIV-1 resistente a multinucleosidos com uma mutação T69S com inserção dupla ou com um complexo de mutações Q151M, incluindo a K65R, apresentou suscetibilidade diminuída ao tenofovir alafenamida.
- Propriedades farmacocinéticas
Absorção
O bictegravir é absorvido após administração oral com as concentrações plasmáticas máximas ocorrendo 2,0-4,0 horas após a administração de B/F/TAF. Em relação ao estado de jejum, a administração de B/F/TAF com uma refeição com teor de gorduras moderado (~600 kcal, 27% de gordura) ou elevado (~800 kcal, 50% de gordura) resultou num aumento da AUC (24%) do bictegravir. Esta alteração modesta não é considerada como sendo clinicamente significativa e B/F/TAF pode ser administrado com ou sem alimentos.
Após a administração oral de B/F/TAF com ou sem alimentos em adultos infectados pelo HIV-1, os parâmetros farmacocinéticos médios (CV%) de doses múltiplas de bictegravir foram Cmáximo = 6,15 mg/mL (22,9%), AUCtau = 102 mg•h/mL (26,9%) e Cmínino = 2,61 mg/mL (35,2%).
A entricitabina é rápida e extensamente absorvida após administração oral, com as concentrações plasmáticas máximas ocorrendo 1,5-2,0 horas após a administração de B/F/TAF. A biodisponibilidade absoluta média da entricitabina sob a forma de cápsulas duras de 200 mg foi de 93%. A exposição sistêmica à entricitabina não foi afetada quando administrada com alimentos e B/F/TAF pode ser administrado com ou sem alimentos.
Após a administração oral de B/F/TAF com ou sem alimentos em adultos infectados pelo HIV-1, os parâmetros farmacocinéticos médios (CV%) de doses múltiplas de entricitabina foram Cmax = 2,13 mg /mL (34,7%), AUCtau = 12,3 mg•h/mL (29,2%) e Cmínimo = 0,096 mg/mL (37,4%).
O tenofovir alafenamida é rapidamente absorvido após administração oral, com as concentrações plasmáticas máximas ocorrendo 0,5-2,0 horas após a administração de B/F/TAF. Em relação ao estado de jejum, a administração de tenofovir alafenamida com uma refeição com teor de gorduras moderado (~600 kcal, 27% de gordura) e teor de gorduras elevado (~800 kcal, 50% de gordura) resultou num aumento da AUCfinal de 48% e 63%, respectivamente. Estas alterações modestas não são consideradas como sendo clinicamente significativas e B/F/TAF pode ser administrado com ou sem alimentos.
Após a administração oral de B/F/TAF com ou sem alimentos em adultos infectados pelo HIV-1, os parâmetros farmacocinéticos médios (CV%) de doses múltiplas de tenofovir alafenamida foram: Cmax = 0,121 mg/ml (15,4%) e AUCtau = 0,142 mg•h/ml (17,3%).
Distribuição
A ligação in vitro do bictegravir às proteínas plasmáticas humanas foi > 99% (fração livre ~0,25%). A razão in vitro da concentração de bictegravir entre sangue e plasma humanos foi de 0,64.
A ligação in vitro da entricitabina às proteínas plasmáticas humanas foi < 4% e independente de concentração dentro do intervalo 0,02 -200 mg/mL. Na concentração plasmática máxima, a razão das concentrações médias do fármaco entre plasma e sangue foi de ~1,0 e a razão das concentrações médias do fármaco entre sêmen e plasma foi de ~4,0.
A ligação in vitro do tenofovir às proteínas plasmáticas humanas é inferior a 0,7% e é independente de concentração no intervalo de 0,01-25 mg/ml. A ligação ex-vivo de tenofovir alafenamida às proteínas plasmáticas humanas, em amostras recolhidas durante os estudos clínicos, foi de aproximadamente 80%.
Biotransformação
O metabolismo é a principal via de depuração do bictegravir em humanos. Estudos de fenotipagem in vitro demonstraram que o bictegravir é metabolizado principalmente pelo CYP3A e a UGT1A1. Após a administração de uma dose oral única de [14C]-bictegravir, ~60% da dose nas fezes incluía o fármaco inalterado, o conjugado desfluoro-hidroxi-BIC-císteina e outros metabólitos oxidativos menores. Trinta e cinco por centro da dose foi recuperada na urina e consistia principalmente do glicuronido do bictegravir e de outros metabólitos oxidativos menores e dos seus conjugados de fase II. A depuração renal do fármaco inalterado foi mínima.
Após a administração de [14C]-entricitabina, a recuperação completa da dose de entricitabina foi efetuada na urina (~ 86%) e nas fezes (~ 14%). Treze por cento da dose foi recuperada na urina sob a forma de três metabólitos putativos. O metabolismo da entricitabina inclui a oxidação da fração tiol para formar os diastereoisomeros 3´-sulfóxido (~9% da dose) e a conjugação com o ácido glicurônico para formar o 2´-O-glicoronido (~ 4% da dose). Não foram identificados outros metabólitos.
O metabolismo é uma importante via de eliminação para o tenofovir alafenamida no ser humano, sendo responsável por > 80% de uma dose oral. Estudos in vitro demonstraram que o tenofovir alafenamida é metabolizado dando origem ao tenofovir (metabólito principal) pela catepsina A nas CsMSP (incluindo os linfócitos e outras células alvo do HIV) e macrófagos; e pela carboxilesterase-1 nos hepatócitos. In vivo, o tenofovir alafenamida é hidrolisado nas células de modo a formar tenofovir (metabólito principal), o qual é fosforilado dando origem ao metabólito ativo tenofovir difosfato. Em estudos clínicos com humanos, uma dose oral de 25 mg de tenofovir alafenamida resultou em concentrações de tenofovir difosfato > 4 vezes superiores nas CsMSP e concentrações > 90% inferiores de tenofovir no plasma em comparação com uma dose oral de 300 mg de fumarato tenofovir desoproxila.
Eliminação
O bictegravir é primariamente eliminado por metabolismo hepático. Excreção renal de bictegravir intacto é uma via menor (~1% da dose). O tempo de meia-vida plasmático de bictegravir foi 17,3 horas.
A entricitabina é primariamente excretada pelos rins tanto por filtração glomerular como por secreção tubular ativa. O tempo de meia-vida plasmática de entricitabina foi de aproximadamente de 10 horas.
O tenofovir alafenamida é eliminado após o metabolismo para tenofovir. O tenofovir alafenamida e o tenofovir têm a meia-vida plasmática de 0,51 e 32,37 horas, respectivamente. O tenofovir é eliminado pelos rins por filtração glomerular e por secreção tubular ativa. A excreção renal do tenofovir alafenamida intacto é uma via menor em que menos de 1% da dose é eliminada na urina.
Linearidade
A farmacocinética de doses múltiplas de bictegravir é proporcional ao longo do intervalo de doses de 25 a 100 mg. A farmacocinética de doses múltiplas de entricitabina é proporcional ao longo de doses de 25 a 200 mg. As exposições de tenofovir alafenamida são dose-proporcional ao longo do intervalo de doses 8 a 125 mg.
Outras populações especiais
- Comprometimento renal
Não se observaram diferenças clinicamente relevantes na farmacocinética do bictegravir, tenofovir alafenamida ou tenofovir entre indivíduos sadios e pacientes com comprometimento renal grave (CrCl estimada < 30 ml/min). Não há dados farmacocinéticos de bictegravir ou tenofovir alafenamida em pacientes com clearance de creatinina menor que 15 mL/min. A exposição sistêmica média da entricitabina foi mais elevada em pacientes com comprometimento renal grave (CrCl < 30 ml/min) (33,7 mg•h/ml) do que em indivíduos com função renal normal (11,8 mg•h/ml).
- Comprometimento hepático
Não se observaram alterações clinicamente relevantes na farmacocinética do bictegravir em indivíduos com comprometimento hepático moderado. A farmacocinética da entricitabina não foi estudada em indivíduos com compromisso hepático; entretanto, a entricitabina não é significativamente metabolizada pelas enzimas hepáticas, então o impacto do comprometimento hepático deve ser limitado. Não se observaram alterações clinicamente relevantes na farmacocinética do tenofovir alafenamida ou do seu metabólito tenofovir em pacientes com comprometimento hepático leve, moderado ou grave.
- Idade, sexo e raça
A farmacocinética do bictegravir, da entricitabina e do tenofovir não foi totalmente avaliada em idosos (≥ 65 anos de idade). As análises populacionais utilizando dados de farmacocinética agrupados dos estudos em adultos não identificaram quaisquer diferenças clinicamente relevantes devido à idade, sexo ou raça nas exposições do bictegravir, entricitabina ou do tenofovir alafenamida.
A média do Cmínimo de bictegravir foi inferior em 50 pacientes pediátricos com idade entre 12 e < 18 anos (≥35 kg) que receberam Biktarvy no Estudo GS-US-380-1474 em relação aos adultos após administração de Biktarvy, mas não foi considerada clinicamente significativa com base na relação exposição-resposta; As exposições de entricitabina e hemifumarato de tenofovir alafenamida nestes pacientes pediátricos foram semelhantes às dos adultos. A média da Cmax do bictegravir e as exposições de entricitabina e tenofovir alafenamida (AUC e Cmax), obtidas em 50 pacientes pediátricos com idades entre 6 e < 12 anos (≥ 25 kg) que receberam Biktarvy no Estudo GS-US-380-1474 foram superiores do que exposições em adultos; no entanto, o aumento não foi considerado clinicamente significativo, pois os perfis de segurança foram semelhantes em pacientes adultos e pediátricos.
- Dados de segurança pré-clínicos
O bictegravir não foi mutagênico nem clastogênico em estudos convencionais de genotoxicidade.
O bictegravir não foi carcinogênico num estudo de 6 meses em camundongos transgênicos rasH2 (com doses até 100 mg/kg/dia nos machos e de 300 mg/kg/dia nas fêmeas, as quais resultaram em exposições de aproximadamente 15 e 23 vezes, nos machos e fêmeas, respectivamente, a exposição em humanos com a dose humana recomendada) e nem no estudo de 2 anos em ratos (com doses até 300 mg/kg/dia), as quais resultaram em exposições de aproximadamente 31 vezes a exposição humana.
Os estudos de bictegravir em macacos revelaram que o fígado é o principal órgão alvo da toxicidade. Foi descrita toxicidade hepatobiliar num estudo de 39 semanas com uma dose de 1.000 mg/kg/dia, a qual resultou em exposições de aproximadamente 16 vezes a exposição em humanos com a dose humana recomendada e que era parcialmente reversível após um período de recuperação de 4 semanas.
Estudos em animais com bictegravir não demonstraram evidência de teratogenicidade ou um efeito na função reprodutora. Na descendência de fêmeas de ratos e coelhos tratadas com bictegravir durante a gravidez, não houve efeitos toxicologicamente significativos nos parâmetros de avaliação do desenvolvimento.
Os dados não-clínicos da entricitabina não revelam riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança, toxicidade de dose repetida, genotoxicidade, potencial carcinogênico, toxicidade reprodutiva e no desenvolvimento. A entricitabina tem demonstrado um baixo potencial carcinogênico em camundongos e em ratos.
Os estudos não-clínicos de tenofovir alafenamida no rato e no cão revelaram que os ossos e os rins são os órgãos alvos primários de toxicidade. A toxicidade óssea foi observada como redução da densidade mineral óssea no rato e no cão, com exposições de tenofovir, pelo menos, 43 vezes superiores às que são esperadas após a administração de B/F/TAF. Observou-se a presença de uma infiltração mínima de histiócitos no olho em cães com exposições de tenofovir alafenamida e de tenofovir aproximadamente 14- e 43- vezes superiores, respectivamente, às que são esperadas após a administração de B/F/TAF.
O tenofovir alafenamida não foi mutagênico nem clastogênico em estudos convencionais de genotoxicidade. Uma vez que a exposição de tenofovir é menor em ratos e camundongos após a administração de tenofovir alafenamida em comparação ao fumarato de tenofovir desoproxila, apenas foram realizados estudos de carcinogenicidade e um estudo peri/pós-natal no rato com o fumarato de tenofovir desoproxila. Não se demonstraram riscos especiais para o ser humano segundo estudos convencionais de potencial carcinogênico e toxicidade reprodutiva e desenvolvimento. Os estudos de toxicidade reprodutiva em ratos e coelhos não demonstraram alterações nos parâmetros de acasalamento, fertilidade, gravidez ou nos parâmetros fetais. No entanto, o fumarato de tenofovir desoproxila reduziu o índice de viabilidade e o peso das crias num estudo de toxicidade peri/pós-natal com doses tóxicas maternas.
4. CONTRAINDICAÇÕES
Este medicamento é contraindicado em pacientes com histórico de hipersensibilidade às substâncias ativas ou a qualquer um dos excipientes (ver item Composição).
Coadministração com rifampicina, dofetilida e Erva-de-São-João (Hypericum perforatum) (ver seção 6. INTERAÇÕES MEDICAMENTOSAS).
5. ADVERTÊNCIAS E PRECAUÇÕES
Embora uma supressão virológica efetiva com terapia antirretroviral tenha provado reduzir substancialmente o risco de transmissão sexual, a existência de um risco residual não pode ser excluída. Devem ser tomadas precauções para prevenir a transmissão, de acordo com as Guias de Recomendação Nacionais.
- Pacientes coinfectados pelo HIV e vírus da hepatite B ou C
Os pacientes com hepatite crônica B ou C em tratamento com terapia antirretroviral têm um risco maior de sofrerem reações adversas hepáticas graves e potencialmente fatais.
Existem dados limitados sobre a segurança e eficácia de Biktarvy em pacientes coinfectados pelo HIV-1 e o vírus hepatite C (VHC).
Biktarvy contém tenofovir alafenamida, o qual é ativo contra o vírus da hepatite B (VHB).
A descontinuação do tratamento com Biktarvy em pacientes coinfectados pelo HIV e pelo VHB pode estar associada a exacerbações agudas graves de hepatite. Os pacientes coinfectados pelo HIV e VHB que descontinuarem o tratamento com Biktarvy devem ser cuidadosamente monitorados, com acompanhamento tanto clínico como laboratorial durante vários meses após a interrupção do tratamento.
- Doença hepática
A segurança e a eficácia de Biktarvy em pacientes com doenças hepáticas significativas subjacentes não foram estabelecidas.
Os pacientes com disfunção hepática pré-existente, incluindo hepatite crônica ativa, têm uma frequência aumentada de alterações da função hepática durante a terapia antirretroviral combinada e devem ser monitorados de acordo com a prática clínica. Se nestes pacientes existir evidência de agravamento da doença hepática, deve ser considerada a interrupção ou descontinuação do tratamento.
- Peso e parâmetros metabólicos
Durante a terapia antirretroviral pode ocorrer um aumento do peso e dos níveis de lipídios e glicose no sangue. Estas alterações podem estar em parte associadas ao controle da doença e ao estilo de vida. Para os lipídios, existe em alguns casos evidência de um efeito do tratamento, enquanto para o aumento do peso não existe uma evidência forte que o relacione com um tratamento em particular. Para o monitoramento dos lipídeos e glicose no sangue deve-se seguir as orientações estabelecidas para o tratamento do HIV. As alterações lipídicas devem ser tratadas de modo clinicamente apropriado.
- Disfunção mitocondrial após exposição in utero
Os análogos dos nucleosídeos e nucleotídeos podem, num grau variável, ter um impacto na função mitocondrial, o que é mais pronunciado com a estavudina, didanosina e zidovudina. Há relatos de disfunção mitocondrial em bebês HIV negativos, expostos in utero e/ou após o nascimento, a análogos dos nucleosídeos; estes estavam relacionados predominantemente com regimes contendo zidovudina. As principais reações adversas notificadas são alterações hematológicas (anemia, neutropenia) e alterações metabólicas (hiperlactatemia, hiperlipasemia). Estes eventos são geralmente transitórios. Foram relatadas raramente alterações neurológicas de início tardio (hipertonia, convulsões, comportamento anormal). Desconhece-se atualmente se estas alterações neurológicas são transitórias ou permanentes. Estes achados devem ser levados em consideração para qualquer criança exposta in utero a análogos dos nucleosídeos e nucleotídeos que apresentar sinais clínicos graves de etiologia desconhecida, especialmente sinais neurológicos. Estes resultados não alteram as recomendações nacionais atuais sobre a utilização de terapia antirretroviral em mulheres grávidas, para prevenir a transmissão vertical do HIV.
- Síndrome inflamatória de reconstituição imune
Em pacientes infectados pelo HIV com deficiência imunológica grave à data da instituição da terapia antirretroviral combinada, pode ocorrer uma reação inflamatória a infeções oportunistas assintomáticas ou residuais e causar várias situações clínicas graves ou o agravamento dos sintomas. Tipicamente, estas reações foram observadas durante as primeiras semanas ou meses após início da terapia antirretroviral combinada. Os exemplos relevantes incluem a retinite por citomegalovírus, as infeções micobacterianas generalizadas e/ou focais e a pneumonia por Pneumocystis jirovecii. Qualquer sintoma de inflamação deve ser avaliado e, quando necessário, instituído o tratamento.
A ocorrência de doenças autoimunes (como a doença de Graves) também foi notificada no enquadramento de reativação imunológica; contudo, o tempo notificado para o início da ocorrência destas doenças é mais variável e estes acontecimentos podem ocorrer muitos meses após o início do tratamento.
- Infecções oportunistas
Os pacientes devem ser avisados que Biktarvy ou qualquer outra terapia antirretroviral não cura a infecção pelo HIV e que eles podem continuar desenvolvendo infecções oportunistas e outras complicações da infecção pelo HIV. Assim, os pacientes devem permanecer sob observação clínica cuidadosa de médicos com experiência no tratamento de pacientes com doenças associadas ao HIV.
- Osteonecrose
Apesar da etiologia ser considerada multifatorial (incluindo a utilização de corticosteroides, o consumo de álcool, a imunossupressão grave, um índice de massa corporal aumentado), foram notificados casos de osteonecrose, particularmente em pacientes com doença por HIV avançada e/ou exposição prolongada a terapia antirretroviral combinada. Os pacientes devem ser instruídos a procurar aconselhamento médico caso sintam mal-estar e dor articular, rigidez articular ou dificuldade de movimentos.
- Nefrotoxicidade
Não se pode excluir um risco potencial de nefrotoxicidade resultante da exposição crônica a níveis baixos de tenofovir devido à administração de tenofovir alafenamida (ver subitem Dados de Segurança Pré-Clínica em "3. CARACTERÍSTICAS FARMACOLÓGICAS").
Pacientes utilizando pró-fármacos de tenofovir que tenham comprometimento da função renal e aqueles que tomam medicamentos nefrotóxicos, incluindo medicamentos anti-inflamatórios não esteroidais, apresentam um risco aumentado de desenvolverem reações adversas renais.
- Comprometimento renal
Biktarvy não deve ser iniciado em pacientes com uma CrCl estimada < 30 ml/min já que os dados disponíveis sobre a utilização de Biktarvy nesta população são limitados (ver subitem "Propriedades Farmacocinéticas" em "3. PROPRIEDADES FARMACOLÓGICAS").
- População pediátrica
Não há dados disponíveis para se fazer uma recomendação de dose para pacientes pediátricos pesando menos que 25 kgs e com menos de 6 anos.
- Coadministração com outros medicamentos
Biktarvy não deve ser coadministrado simultaneamente com antiácidos contendo magnésio/alumínio ou suplementos de ferro em jejum. Biktarvy deve ser administrado, pelo menos, 2 horas antes ou com alimentos 2 horas depois de antiácidos contendo magnésio e/ou alumínio. Biktarvy deve ser administrado, pelo menos, 2 horas antes de suplementos de ferro ou tomado juntamente com alimentos (ver seção 6. INTERAÇÕES MEDICAMENTOSAS).
Alguns medicamentos não devem ser coadministrados com Biktarvy: atazanavir, boceprevir, carbamazepina, ciclosporina (uso IV ou oral), oxcarbazepina, fenobarbital, fenitoína, rifabutina, rifapentina ou sucralfato.
Biktarvy não deve ser coadministrado com outros medicamentos antirretrovirais.
-Fertilidade, Gravidez e Lactação
Gravidez
Os dados sobre o uso de bictegravir ou tenofovir alafenamida em mulheres grávidas são inexistentes ou são limitados (menos que 300 resultados de gravidezes). Uma grande quantidade de dados em mulheres grávidas (mais de 1.000 gravidezes expostas) indica ausência de toxicidade malformativa ou fetal/neonatal associada à entricitabina.
Os estudos em animais não indicam efeitos nocivos diretos ou indiretos de entricitabin