BESPONSA
PFIZER
inotuzumabe ozogamicina
Anticorpo monoclonal. Antineoplásico.
Apresentações.
Besponsa® 1 mg de inotuzumabe ozogamicina na forma de pó liofilizado para solução injetável em embalagem contendo 1 frasco-ampola.
VIA DE ADMINISTRAÇÃO: VIA INTRAVENOSA
USO ADULTO
Composição.
Cada frasco-ampola de dose única contém o equivalente a 1 mg de inotuzumabe ozogamicina. Após reconstituição com 4 mL de água estéril, a concentração final é de 0,25mg/mL de inotuzumabe ozogamicina. O volume extraível de cada frasco-ampola é de no mínimo 3,6mL, correspondente à 0,9 mg de inotuzumabe ozogamicina. Excipientes: sacarose, polissorbato 80, cloreto de sódio, trometamina (trometamol)
Informações técnicas.
1. INDICAÇÕES
Besponsa® (inotuzumabe ozogamicina) está indicado como monoterapia para o tratamento de adultos com leucemia linfoblástica aguda (LLA) de células B precursoras, recidivada ou refratária, CD22 positivo. O tratamento de pacientes adultos com LLA de células B precursoras, recidivada ou refratária, positivo para cromossomo Filadélfia (Ph+) só é indicado após falha do tratamento com pelo menos um inibidor de tirosina quinase.
2. RESULTADOS DE EFICÁCIA
Eficácia e segurança clínica
Pacientes com LLA recidivada ou refratária que receberam um ou dois regimes de tratamento prévios para LLA - estudo 1
A segurança e a eficácia do inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária foram avaliadas em um estudo randomizado, aberto, internacional, multicêntrico de fase 3 (estudo 1). Os pacientes elegíveis tinham ≥18 anos de idade e apresentavam LLA precursora de células B recidivada ou refratária cromossomo Filadélfia negativo ou cromossomo Filadélfia positivo. Todos os pacientes tinham que ter ≥5% de blastos na medula e ter recebido um ou dois regimes prévios de quimioterapia de indução para LLA. Os pacientes com LLA precursora de células B cromossomo Filadélfia positivo tinham que ter uma doença cujo tratamento tivesse falhado, com, pelo menos, um inibidor de tirosina quinase e quimioterapia padrão. A tabela 2 mostra o regime de administração utilizado para tratar os pacientes.
No total, 326 pacientes foram randomizados para o estudo.
Entre os 326 pacientes que foram randomizados para receber inotuzumabe ozogamicina (N = 164) ou a quimioterapia de escolha do investigador (N = 162), 215 (66%) pacientes tinham recebido um regime prévio de tratamento para LLA e 108 (33%) pacientes tinham recebido dois regimes prévios de tratamento para LLA. A idade mediana foi de 47 anos (faixa de variação: 18-79 anos), 276 (85%) pacientes apresentavam LLA cromossomo Filadélfia negativo, 206 (63%) pacientes tiveram uma duração da primeira remissão < 12 meses e 55 (18%) pacientes foram submetidos a um TCTH (transplante de células tronco hematopoiéticas) antes de receber inotuzumabe ozogamicina ou a quimioterapia de escolha do investigador. Os dois grupos de tratamento eram geralmente equilibrados em relação aos dados demográficos da avaliação inicial e às características da doença.
Todos os pacientes avaliáveis apresentavam LLA precursora de células B com expressão de CD22, com > 90% dos pacientes avaliáveis apresentando ≥70% de blastos leucêmicos positivos para CD22 antes do tratamento, conforme avaliado por citometria de fluxo em um laboratório central.
Os endpoints primários foram sobrevida global (SG) e RC/RCi, avaliados por um EAC (comitê de avaliação de endpoint) cego e independente. Os endpoints secundários incluíram negatividade da DRM, duração da remissão (DoR), taxa de TCTH e sobrevida livre de progressão (PFS). RC/RCi, DRM e DoR foram analisados nos 218 pacientes inicialmente randomizados e SG, PFS e TCTH foram analisados em todos os 326 pacientes randomizados.
A tabela 1 mostra os resultados de eficácia desse estudo.
Entre os 218 pacientes inicialmente randomizados, segundo avaliação do EAC, 64/88 (73%) e 21/88 (24%) dos pacientes respondedores obtiveram RC/RCi nos ciclos 1 e 2, respectivamente, no braço de inotuzumabe ozogamicina, e 29/32 (91%) e 1/32 (3%) dos pacientes respondedores obtiveram RC/RCi nos ciclos 1 e 2, respectivamente, no braço da quimioterapia de escolha do investigador.
Os resultados de RC/RCi, DRM e DoR nos 218 pacientes iniciais randomizados foram consistentes com aqueles observados nos 326 pacientes randomizados.
Entre os 326 pacientes randomizados, a probabilidade de sobrevida aos 24 meses foi de 22,8% no braço de inotuzumabe ozogamicina e de 10,0% no braço da quimioterapia de escolha do investigador. Apesar da maior frequência de mortes prematuras após o TCTH no braço de inotuzumabe ozogamicina, foi observada uma melhora na sobrevida para inotuzumabe ozogamicina em comparação com a quimioterapia de escolha do investigador em pacientes submetidos à TCTH.
Em geral, 79/164 (48,2%) dos pacientes do braço de inotuzumabe ozogamicina e 35/162 (21,6%) dos pacientes do braço da quimioterapia de escolha do investigador foram submetidos à TCTH de acompanhamento. Isso incluiu 71 e 18 pacientes do braço de inotuzumabe ozogamicina e do braço da quimioterapia de escolha do investigador, respectivamente, que foram diretamente submetidos à TCTH. Nestes pacientes diretamente submetidos à TCTH, houve um intervalo mediano de 4,9 semanas (faixa de variação: 1-19 semanas) entre a última dose de inotuzumabe ozogamicina e o TCTH. No braço de inotuzumabe ozogamicina, as causas mais comuns de mortalidade não relacionada à recidiva após o TCTH incluem VOD e infecções (vide item 5. Advertências e Precauções). Cinco dos 18 eventos de VOD/SOS ocorridos após o TCTH foram fatais (vide item 5. Advertências e Precauções). Seis pacientes apresentaram VOD persistente no momento da morte e morreram devido à falência de múltiplos órgãos (MOF) ou infecção (três pacientes morreram devido a MOF, dois pacientes morreram por infecção e um paciente morreu devido a MOF e infecção). Em pacientes submetidos a um TCTH de acompanhamento, a SG mediana foi de 11,9 meses (IC de 95%: 9,2, 20,6) versus 18,6 meses (IC de 95%: 14,6, 27,8) e a probabilidade de sobrevida no mês 24 foi de 37,8% (IC de 95%: 27,2, 48,4) versus 36,2% (IC de 95%: 20,3, 52,3) para inotuzumabe ozogamicina versus quimioterapia de escolha do investigador, respectivamente.
Os resultados relatados pelos pacientes foram medidos com o uso do Questionário de Qualidade de Vida da Organização Europeia para Pesquisa e Tratamento do Câncer (EORTC QLQ-C30). O inotuzumabe ozogamicina resultou em escores médios significativamente melhores após a avaliação inicial (inotuzumabe ozogamicina e quimioterapia de escolha do investigador, respectivamente) no funcionamento físico (75,0 versus 68,1; p = 0,0139), capacidade funcional (64,7 versus 53,4; p = 0,0065), funcionamento social (68,1 versus 59,8; p = 0,0336) e perda de apetite (17,6 versus 26,3; p = 0,0193) em comparação com a quimioterapia de escolha do investigador.
Pacientes com LLA recidivada ou refratária que receberam dois ou mais regimes de tratamento prévios para LLA - estudo 2
A segurança e a eficácia do inotuzumabe ozogamicina foram avaliadas em um estudo multicêntrico, aberto, de braço único e de fase 1/2 (estudo 2). Os pacientes elegíveis tinham ≥18 anos de idade e apresentavam LLA precursora de células B recidivada ou refratária.
Na parte da fase 1 do estudo, 37 pacientes receberam Besponsa® com uma dose total de 1,2 mg/m2 (n = 3), 1,6 mg/m2 (n = 12) ou 1,8 mg/m2 (n = 22). A dose recomendada de inotuzumabe ozogamicina foi de 1,8 mg/m2/ciclo administrada a uma dose de 0,8 mg/m2 no dia 1 e 0,5 mg/m2 nos dias 8 e 15 de um ciclo de 28 dias com redução da dose quando da obtenção de RC/RCi.
Entre os 35 pacientes na parte da fase 2 que receberam inotuzumabe ozogamicina na dose recomendada, 15 (43%) e 17 (49%) pacientes receberam dois ou > 2 regimes de tratamento prévios para LLA, respectivamente. A idade mediana foi de 34 anos (faixa de variação: 20-79 anos) e seis (17%) pacientes tiveram duração da primeira remissão < 12 meses. Desses 35 pacientes, 24/35 (69% e 95% de intervalo de confiança [IC]: 50,7-83,2) pacientes obtiveram RC/RCi. Dos 24 pacientes que obtiveram RC/RCi, 18/24 (75%) dos pacientes também obtiveram negatividade da DRM. A DoR mediana foi de 3,8 meses (IC de 95%: 2,2-5,8), a PFS mediana foi de 3,7 meses (IC de 95%: 2,6-4,7) e a SG mediana foi de 6,4 meses (IC de 95%: 4,5-7,9).
Referências
1. Kantarjian HM, DeAngelo DJ, Stelljes M, et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N Engl J Med 2016; 375:740-53
2. Kantarjian, H. M., Su, Y., Jabbour, E. J., Bhattacharyya, H., Yan, E., Cappelleri, J. C. and Marks, D. I., Patient-reported outcomes from a phase 3 randomized controlled trial of inotuzumab ozogamicin versus standard therapy for relapsed/refractory acute lymphoblastic leukemia. Cancer 2018, 124: 2151-2160.
3. DeAngelo DJ, Stock W, Stein AS, et al. Inotuzumab ozogamicin in adults with relapsed or refractory CD22-positive acute lymphoblastic leukemia: a phase 1/2 study. Blood Advances. 2017;1(15):1167-1180.
3. CARACTERÍSTICAS FARMACOLÓGICAS
Propriedades farmacodinâmicas
• Mecanismo de ação
O inotuzumabe ozogamicina é um conjugado de anticorpo-medicamento (CAM) cujo alvo é o CD22. O inotuzumabe é um anticorpo IgG4 humanizado que reconhece especificamente o CD22 humano. A molécula pequena, N-acetil-gama-caliqueamicina, é um produto natural citotóxico semissintético. A N-acetil-gama-caliqueamicina é covalentemente ligada ao anticorpo por meio de um ligante. Dados não clínicos sugerem que a atividade anticancerígena do inotuzumabe ozogamicina se deve à ligação do CAM a células tumorais com expressão de CD22, seguida da internalização do complexo CAM-CD22 e da liberação intracelular de N-acetil-gama-caliqueamicina dimetilidrazida por meio da clivagem hidrolítica do ligante. A ativação de N-acetil-gama-caliqueamicina dimetilidrazida induz quebras de DNA de fita dupla, subsequentemente induzindo uma parada do ciclo celular e morte celular programada (apoptose).
• Efeitos farmacodinâmicos
Durante o período de tratamento, a resposta farmacodinâmica ao inotuzumabe ozogamicina foi caracterizada pela depleção de blastos leucêmicos positivos para CD22.
Propriedades Farmacocinéticas
Em pacientes com LLA recidivada ou refratária, a exposição no estado de equilíbrio foi alcançada no ciclo 4. A concentração máxima média observada (Cmáx) do inotuzumabe ozogamicina foi de 308 ng/mL. A área total média simulada sob a curva de concentração versus tempo (ASC) por ciclo foi de 100.000 ng•h/mL.
• Distribuição
In vitro, a ligação de N-acetil-gama-caliqueamicina dimetilidrazida a proteínas plasmáticas humanas é de, aproximadamente, 97%. In vitro, N-acetil-gama-caliqueamicina dimetilidrazida é um substrato da P-glicoproteína (Pgp). Em humanos, o volume total de distribuição do inotuzumabe ozogamicina foi de, aproximadamente, 12 L.
• Metabolismo
In vitro, N-acetil-gama-caliqueamicina dimetilidrazida foi principalmente metabolizada por redução não enzimática. Em humanos, os níveis séricos de N-acetil-gama-caliqueamicina dimetilidrazida estavam normalmente abaixo do limite de quantificação.
• Eliminação
A farmacocinética do inotuzumabe ozogamicina foi bem caracterizada por um modelo de dois compartimentos com componentes de depuração lineares e dependentes do tempo. Em 234 pacientes com LLA recidivada ou refratária, a depuração do inotuzumabe ozogamicina no estado de equilíbrio foi de 0,0333 L/h, e a meia vida terminal (t½) foi de 12,3 dias. Após a administração de doses múltiplas, foi previsto um acúmulo de 5,3 vezes de inotuzumabe ozogamicina até o ciclo 4.
Com base em uma análise farmacocinética populacional em 765 pacientes, a área da superfície corporal demonstrou afetar significativamente a disposição do inotuzumabe ozogamicina. A dose de inotuzumabe ozogamicina é administrada com base na área de superfície corporal (vide item 8. Posologia e Modo de Usar).
Farmacocinética em grupos específicos de participantes ou pacientes
• Idade, raça e sexo
Com base em uma análise farmacocinética populacional, idade, raça e sexo não afetaram significativamente a disposição do inotuzumabe ozogamicina.
• Insuficiência hepática
Não foi conduzido nenhum estudo farmacocinético formal de inotuzumabe ozogamicina em pacientes com insuficiência hepática.
Com base em uma análise farmacocinética populacional em 765 pacientes, a depuração do inotuzumabe ozogamicina em pacientes com insuficiência hepática definida pelo Grupo de Trabalho de Disfunção de Órgãos do Instituto Nacional do Cancer (NCI ODWG) categoria B1 (bilirrubina total ≤ LSN e AST > LSN; n = 133) ou B2 (bilirrubina total > 1,0-1,5 × LSN e AST em qualquer nível; n = 17) foi semelhante aos pacientes com função hepática normal (bilirrubina total/AST ≤ LSN; n = 611) (vide item 8. Posologia e Modo de Usar). Em três pacientes com insuficiência hepática definida pelo NCI ODWG categoria C (bilirrubina total > 1,5-3 × LSN e AST em qualquer nível) e um paciente com NCI ODWG categoria D (bilirrubina total > 3 × LSN e AST em qualquer nível), não pareceu que a depuração do inotuzumabe ozogamicina foi reduzida.
• Insuficiência renal
Nenhum estudo farmacocinético formal de inotuzumabe ozogamicina foi conduzido em pacientes com insuficiência renal.
Com base na análise farmacocinética populacional em 765 pacientes, a depuração do inotuzumabe ozogamicina em pacientes com insuficiência renal leve (CLcr 60-89 mL/min; n = 237), insuficiência renal moderada (CLcr 30-59 mL/min; n = 122) ou insuficiência renal grave (CLcr 15-29 mL/min; n = 4) foi similar para pacientes com função renal normal (CLcr ≥90 mL/min; n = 402) (vide item 8. Posologia e Modo de Usar). A segurança e a eficácia do inotuzumabe ozogamicina não foram estudadas em pacientes com insuficiência renal terminal (vide item 8. Posologia e Modo de Usar).
• Eletrofisiologia cardíaca
Com base em uma análise farmacocinética de exposição-resposta em 250 pacientes com LLA recidivada ou refratária ou outras malignidades hematológicas que receberam 1,8 mg/m2/ciclo de inotuzumabe ozogamicina, administrado em três doses divididas nos dias 1 (0,8 mg/m2), 8 (0,5 mg/m2) e 15 (0,5 mg/m2) de um ciclo de 21 a 28 dias ou 1,8 mg/m2/ciclo, administrado uma vez a cada quatro semanas, respectivamente, o intervalo de QTcF mediano aumentou em 2,53 milissegundos (ms) em relação à avaliação inicial (97,5° percentil: 4,92 ms) na Cmáx média estimada para pacientes com LLA recidivada ou refratária (371 ng/mL) e em 3,87 ms em relação à avaliação inicial (97,5° percentil: 7,54 ms) na Cmáx média 1,5 vez maior (569 ng/mL).
Em um estudo clínico randomizado em pacientes com LLA recidivada ou refratária (Estudo 1), os aumentos máximos do intervalo QTcF de ≥ 30 mseg e ≥ 60 msec do valor basal foram medidos em 30/162 (19%) e 4/162 (3%) pacientes no braço de ozogamicina inotuzumabe, respectivamente, versus 18/124 (15%) e 3/124 (2%) no braço de quimioterapia de escolha do investigador, respectivamente. Aumentos no intervalo QTcF > 450 mseg e > 500 mseg foram observados em 26/162 (16%) e nenhum dos pacientes no braço de inotuzumabe ozogamicina versus 12/124 (10%) e 1/124 (1%) pacientes no braço de quimioterapia de escolha do investigador, respectivamente (ver seção 9. REAÇÕES ADVERSAS). As alterações máximas médias (IC de 90%) em QTcF a partir da avaliação inicial foram de 16,5 ms (14,3-18,7) no braço de inotuzumabe ozogamicina e de 10,8 ms (8,0-13,6) no braço da quimioterapia de escolha do investigador. A análise da tendência central das alterações no intervalo de QTcF em relação à avaliação inicial demonstrou que o limite máximo superior do IC de 90% bilateral de QTcF foi de 21,1 ms (observado no ciclo 4/Dia 1/uma hora) no braço de inotuzumabe ozogamicina e de 21,2 ms (observado no ciclo 2/Dia 1/uma hora) no braço da quimioterapia de escolha do investigador (vide item 5. Advertências e Precauções).
Dados de Segurança Pré-clínicos
• Toxicidade de dose repetida
Nos animais, os principais órgãos-alvo incluíram fígado, medula óssea e os órgãos linfoides com alterações hematológicas associadas e alterações no sistema nervoso e renal. Outras alterações observadas incluíram efeitos de órgãos reprodutivos masculinos e femininos (consulte abaixo). A maioria dos efeitos foi reversível a parcialmente reversível, exceto os efeitos no fígado e no sistema nervoso. A relevância dos achados irreversíveis de animais para humanos é incerta.
• Genotoxicidade
O inotuzumabe ozogamicina foi clastogênico in vivo na medula óssea de camundongos machos. Isso é consistente com a indução conhecida de quebras de DNA por caliqueamicina. N-acetil-gama-caliqueamicina dimetilidrazida (o agente citotóxico liberado do inotuzumabe ozogamicina) foi mutagênico em um ensaio de mutação reversa bacteriana in vitro (Ames).
• Carcinogenicidade
Não foram conduzidos estudos formais de carcinogenicidade com inotuzumabe ozogamicina. Em estudos de toxicidade, os ratos desenvolveram hiperplasia de células ovais, focos hepatocelulares alterados e adenomas hepatocelulares no fígado em, aproximadamente, 0,3 vezes a exposição clínica humana com base na ASC. Em um macaco, um foco de alteração hepatocelular foi detectado em, aproximadamente, 3,1 vezes a exposição clínica humana com base na ASC no final do período de administração de 26 semanas. A relevância desses achados em animais para humanos é incerta.
• Toxicidade reprodutiva
A administração de inotuzumabe ozogamicina a ratas na dose maternalmente tóxica (aproximadamente 2,3 vezes a exposição clínica humana com base na ASC) antes do acasalamento e durante a primeira semana de gestação resultou em toxicidade embrio fetal, incluindo aumento de reabsorção e diminuição de embriões viáveis. A dose maternalmente tóxica (aproximadamente 2,3 vezes a exposição clínica humana com base na ASC) também resultou em retardo do crescimento fetal, incluindo diminuição do peso fetal e atraso da ossificação esquelética. Também ocorreu um leve retardo do crescimento fetal em ratos em, aproximadamente, 0,4 vezes a exposição clínica humana com base na ASC (vide item 5. Advertências e Precauções - Fertilidade, gravidez e lactação).
Considera-se que o inotuzumabe ozogamicina tem potencial para prejudicar a função reprodutiva e a fertilidade em homens e mulheres com base em achados não clínicos. Em estudos de toxicidade de doses repetidas em ratos e macacos, os achados reprodutivos em fêmeas incluíram atrofia dos ovários, útero, vagina e glândula mamária. O nível de efeito adverso não observado (NOAEL) para os efeitos sobre os órgãos reprodutivos de fêmeas foi de, aproximadamente, 2,2 e 3,1 vezes a exposição clínica humana com base na ASC, respectivamente. Em estudos de toxicidade de doses repetidas em ratos, os achados reprodutivos em machos incluíram degeneração testicular associada à hipospermia e atrofia da vesícula prostática e seminal. O NOAEL não foi identificado para os efeitos nos órgãos reprodutores de machos, que foram observados em, aproximadamente, 0,3 vezes a exposição clínica humana com base na ASC. (vide item 5. Advertências e Precauções - Fertilidade, gravidez e lactação).
4. CONTRAINDICAÇÕES
Besponsa® é contraindicado nas seguintes situações: pacientes com hipersensibilidade conhecida a inotuzumabe ozogamicina e a outros componentes da fórmula.
5. ADVERTÊNCIAS E PRECAUÇÕES
Hepatotoxicidade, incluindo doença veno-oclusiva hepática/síndrome da obstrução sinusoidal (VOD/SOS)
Em um estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), foi relatada hepatotoxicidade, incluindo VOD/SOS hepática grave, com risco de vida e, às vezes, fatal e aumentos nos testes hepáticos (vide item 9. Reações Adversas).
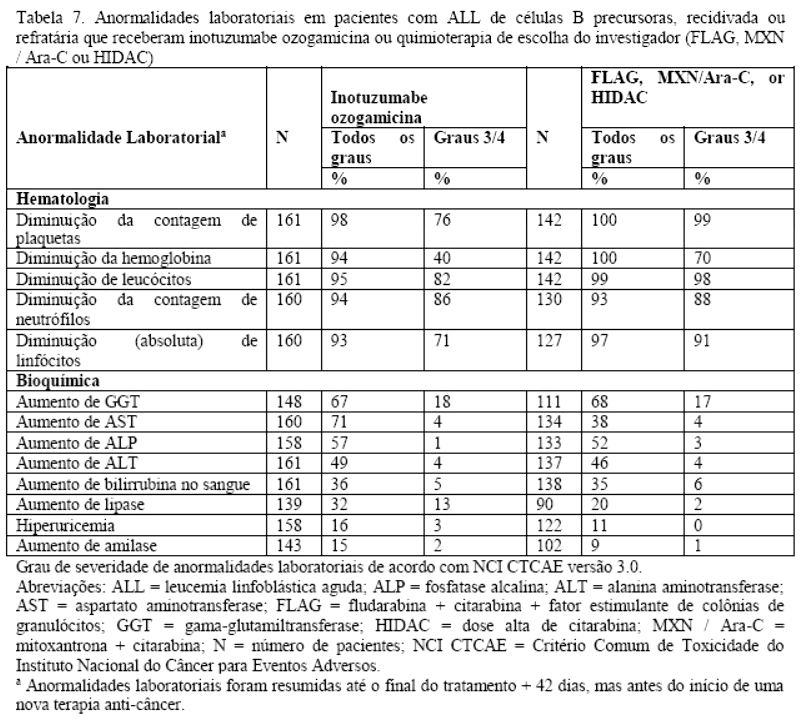
VOD/SOS foi relatada em 23/164 (14%) dos pacientes durante ou após o tratamento, ou depois de um TCTH após a conclusão do tratamento. Testes hepáticos com resultados anormais de AST, ALT e de bilirrubina total de grau 3/4 ocorreram em 7/160 (4%), 7/161 (4%) e 8/161 (5%) dos pacientes, respectivamente.
Dos 164 pacientes tratados, foi relatada VOD/SOS em 5/164 (3%) dos pacientes durante a terapia do estudo ou no acompanhamento sem um TCTH interveniente. Dos 79 pacientes que receberam TCTH subsequente (8 dos quais receberam terapia adicional de resgate após o tratamento com inotuzumabe ozogamicina antes de receberem TCTH), foi relatada VOD/SOS em 18/79 (23%) dos pacientes. Cinco dos 18 eventos de VOD/SOS ocorridos após o TCTH foram fatais (vide item 3. Propriedades Farmacológicas - Propriedades Farmacodinâmicas).
VOD/SOS foi relatada até 56 dias após a última dose durante o tratamento ou durante o acompanhamento sem TCTH interveniente. O tempo médio para o TCTH a partir início da VOD/SOS foi de 15 dias (faixa de variação: 3-57 dias).
Alguns pacientes podem ter um maior risco de desenvolvimento de VOD/SOS.
Os pacientes que tiveram VOD/SOS prévia ou que têm doença hepática grave e persistente (por exemplo, cirrose, hiperplasia regenerativa nodular, hepatite ativa) podem ter um maior risco de piora da doença hepática, incluindo o desenvolvimento de VOD/SOS, após o tratamento com inotuzumabe ozogamicina.
O TCTH prévio pode estar associado a um maior risco de VOD/SOS. Dos 5 pacientes que apresentaram VOD/SOS durante o tratamento com inotuzumabe ozogamicina, mas sem TCTH interveniente, dois pacientes também receberam TCTH antes do tratamento com inotuzumabe ozogamicina. Entre os pacientes que receberam TCTH, foi relatada VOD/SOS após o TCTH subsequente ao tratamento com inotuzumabe ozogamicina em 5/11 (46%) dos pacientes que receberam TCTH antes e após o tratamento com inotuzumabe ozogamicina e em 13/68 (19%) dos pacientes que receberam TCTH somente após o tratamento com inotuzumabe ozogamicina.
Entre os pacientes que recebem TCTH, o uso de regimes de condicionamento do TCTH contendo dois agentes alquilantes e o último nível de bilirrubina total ≥LSN antes do TCTH de acompanhamento estão significativamente associados a um maior risco de VOD/SOS após o TCTH. Outros fatores que também podem estar associados a um maior risco de VOD/SOS após o TCTH incluem idade avançada, histórico de doença hepática e/ou hepatite antes do tratamento, linhas de resgate mais tardias e maior número de ciclos de tratamento.
Devido ao risco de VOD/SOS, especialmente após o TCTH, deve-se monitorar de perto os sinais e os sintomas de VOD/SOS, os quais podem incluir elevações na bilirrubina total, hepatomegalia (que pode ser dolorosa), aumento rápido de peso e ascite. Monitorar apenas a bilirrubina total pode não identificar todos os pacientes com risco de VOD/SOS. Em todos os pacientes, deve-se monitorar os exames laboratoriais hepáticos, incluindo ALT, AST, bilirrubina total e fosfatase alcalina, antes e após cada dose de inotuzumabe ozogamicina. Para os pacientes que apresentarem exames hepáticos alterados, recomenda-se monitoramento mais frequente de testes laboratoriais, sinais clínicos e sintomas de hepatotoxicidade. Para pacientes que recebem TCTH, deve-se monitorar cuidadosamente os exames laboratoriais hepáticos durante o primeiro mês após o TCTH, com menos frequência a partir de então, de acordo com a prática clínica padrão.
A elevação dos testes hepáticos pode exigir a interrupção do tratamento, redução da dose ou interrupção permanente do inotuzumabe ozogamicina (vide item 8. Posologia e Modo de Usar).
Considere cuidadosamente a relação benefício/risco antes de administrar inotuzumabe ozogamicina em pacientes com experiência prévia de VOD/SOS ou em pacientes com doença hepática grave e persistente (por exemplo, cirrose, hiperplasia nodular regenerativa, hepatite ativa). Se esses pacientes forem tratados com inotuzumabe ozogamicina, monitore de perto os sinais e os sintomas de VOD/SOS e interrompa permanentemente o tratamento em caso de VOD/SOS (vide item 8. Posologia e Modo de Usar).
Deve-se ter especial atenção quando administrar inotuzumabe ozogamicina em pacientes mais velhos, que tiveram TCTH anterior e estão em linhas de resgate posteriores ou com histórico de doença hepática e/ou hepatite. Devido ao risco de VOD, para os pacientes que receberão TCTH, a duração recomendada do tratamento com inotuzumabe ozogamicina é de dois ciclos; um terceiro ciclo deve ser considerado para pacientes que não atingem RC ou RCi e negatividade da DRM após dois ciclos (vide item 8. Posologia e Modo de Usar). Evite o uso de regimes de condicionamento do TCTH contendo dois agentes alquilantes.
Interrompa permanentemente o tratamento em caso de VOD/SOS (vide item 8. Posologia e Modo de Usar). Em caso de VOD/SOS grave, os pacientes deverão ser tratados de acordo com a prática clínica padrão.
Risco aumentado de mortalidade pós-transplante não relacionada à recidiva
Num estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), observou-se uma taxa de mortalidade pós-TCTH não relacionada à recidiva mais elevada em pacientes que receberam Besponsa® comparativamente ao braço da quimioterapia escolhida pelo investigador, resultando em uma taxa de mortalidade pós-TCTH mais elevada no Dia 100.
No geral 79/164 pacientes (48%) no braço de Besponsa® e 35/162 pacientes (22%) no braço da quimioterapia escolhida pelo investigador foram submetidos a um TCTH no seguimento. A taxa de mortalidade pós-TCTH não relacionada à recidiva foi de 31/79 (39%) e 8/35 (23%) no braço de Besponsa®, em comparação com o braço da quimioterapia escolhida pelo investigador, respetivamente.
No braço Besponsa® as causas mais comuns de mortalidade pós-TCTH não relacionada à recidiva incluíram VOD e infecções. Cinco dos 18 eventos VOD que ocorreram após o TCTH foram fatais. No braço do Besponsa®, entre pacientes com VOD em curso no momento do óbito, 6 pacientes morreram devido à falência de múltiplos órgãos (MOF) ou infecção (3 pacientes morreram devido a MOF, 2 pacientes morreram devido a infecção e 1 paciente morreu devido a MOF e infecção).
Monitorar atentamente as toxicidades pós-TCTH, incluindo sinais e sintomas de infecção e VOD [ver item 5. Advertências e Precauções - Hepatotoxicidade, incluindo doença veno-oclusiva hepática/síndrome da obstrução sinusoidal (VOD/SOS) e Mielossupressão/citopenia].
Mielossupressão/citopenia
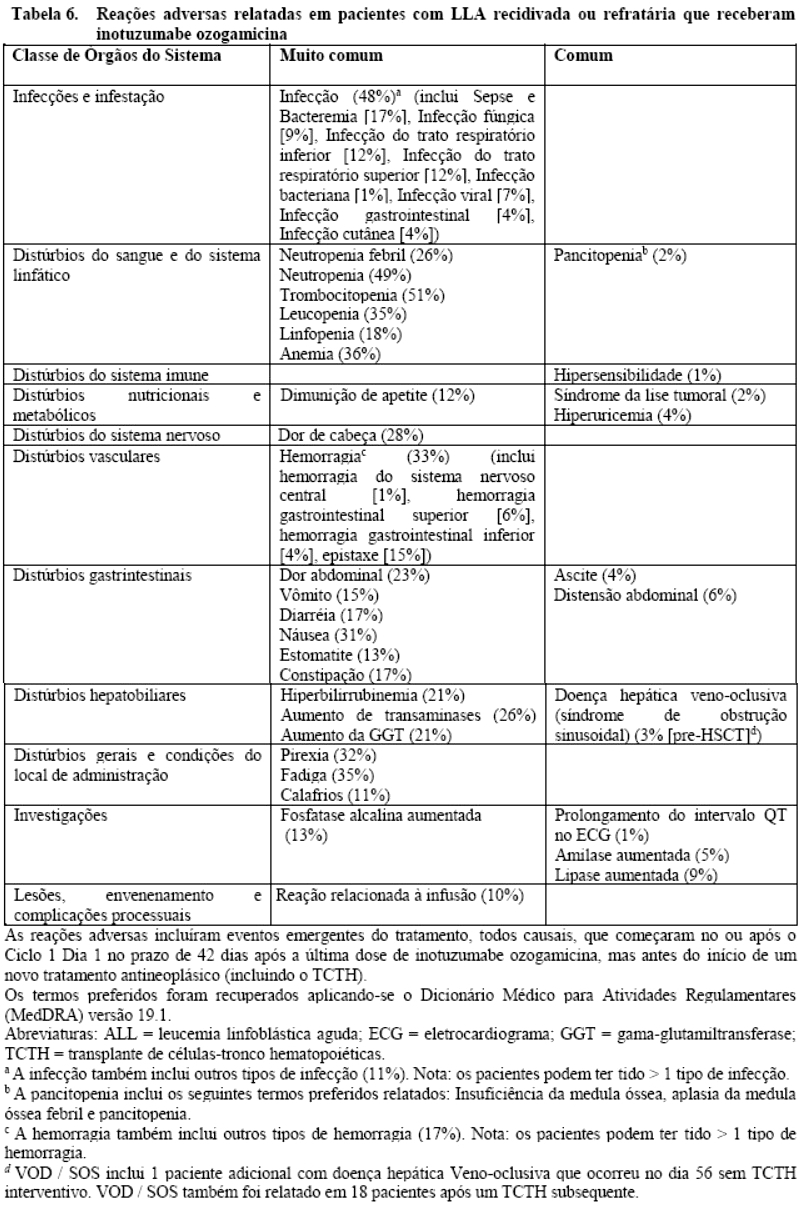
Em um estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), foram relatadas neutropenia, trombocitopenia, anemia, leucopenia, neutropenia febril, linfopenia e pancitopenia, algumas das quais com risco vida (vide item 9. Reações Adversas).
Trombocitopenia e neutropenia foram relatadas em 83/164 (51%) e 81/164 (49%) dos pacientes, respectivamente. Trombocitopenia e neutropenia de grau 3 foram relatadas em 23/164 pacientes (14%) e 33/164 pacientes (20%), respectivamente. Trombocitopenia e neutropenia de grau 4 foram relatadas em 46/164 pacientes (28%) e 45/164 pacientes (27%), respectivamente. Neutropenia febril, que pode ser fatal, foi relatada em 43/164 (26%) dos pacientes.
Foram relatadas complicações associadas à neutropenia e à trombocitopenia (incluindo infecções e eventos de sangramento/hemorrágicos, respectivamente) em alguns pacientes (vide item 9. Reações Adversas).
Infecções, incluindo infecções graves, algumas das quais com risco de vida ou fatais, foram relatadas em 79/164 (48%) dos pacientes. Infecções fatais, incluindo pneumonia, sepse neutropênica, sepse, choque séptico e sepse por pseudômonas foram relatadas em 8/164 (5%) dos pacientes. Foram relatadas infecções bacterianas, virais e fúngicas.
Eventos de sangramento/hemorrágicos, com gravidade leve na maior parte, foram relatados em 54/164 (33%) dos pacientes. Eventos de sangramento/hemorrágicos de grau 3/4 foram relatados em 8/164 (5%) dos pacientes. Um evento de sangramento/hemorrágico de grau 5 (hemorragia intra-abdominal) foi relatado em 1/164 (1%) pacientes. O evento hemorrágico mais comum foi epistaxe, a qual foi relatada em 24/164 (15%) dos pacientes.
Monitore a contagem de sangue total antes de cada dose de inotuzumabe ozogamicina e controle sinais e sintomas de infecção durante o tratamento e após o TCTH (vide item 3. Propriedades Farmacológicas - Propriedades Farmacodinâmicas), sangramento/hemorragia ou outros efeitos da mielossupressão durante o tratamento com inotuzumabe ozogamicina. Administre anti-infecciosos profiláticos de maneira adequada e realize testes de vigilância durante e após o tratamento com inotuzumabe ozogamicina. O tratamento de pacientes com infecção grave, sangramento/hemorragia ou outros efeitos da mielossupressão, incluindo neutropenia grave ou trombocitopenia, pode exigir interrupção da administração, redução da dose ou interrupção permanente de inotuzumabe ozogamicina (vide item 8. Posologia e Modo de Usar).
Monitore hemograma completo antes de cada dose de inotuzumabe ozogamicina, bem como os sinais e sintomas de infecção, sangramento/hemorragia ou outros efeitos de mielossupressão durante o tratamento com inotuzumabe ozogamicina. Conforme apropriado, administrar agentes anti-infecciosos profiláticos e empregar testes de monitoramento durante e após o tratamento com inotuzumabe ozogamicina. O tratamento de infecção grave, sangramento/hemorragia ou outros efeitos de mielossupressão, incluindo neutropenia grave ou trombocitopenia, podem requerer a interrupção da dose, a redução da dose ou a descontinuação permanente do tratamento com inotuzumabe ozogamicina (Ver seção 8. Posologia e Modo de Uso).
Reações relacionadas à infusão
Em um estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), reações relacionadas à infusão, todas com gravidade de grau 2, foram relatadas em 4/164 (2%) dos pacientes (vide item 9. Reações Adversas). As reações relacionadas à infusão geralmente ocorreram no ciclo 1 logo após o final da infusão de inotuzumabe ozogamicina, sendo resolvidas de forma espontânea ou com tratamento médico.
É recomendada a pré-medicação com um corticosteroide, antipirético e anti-histamínico antes da administração (vide item 8. Posologia e Modo de Usar).
Monitore os pacientes de perto durante a infusão e durante, pelo menos, uma hora após o término da infusão para um possível início de reações relacionadas à infusão, incluindo sintomas como hipotensão, ondas de calor, erupções cutâneas ou problemas respiratórios. Caso ocorra uma reação relacionada à infusão, interrompa a infusão e institua o tratamento médico adequado. Dependendo da gravidade da reação relacionada à infusão, considere a interrupção da infusão ou a administração de esteroides e anti-histamínicos. Para reações à infusão graves ou com risco de vida, interrompa permanentemente o inotuzumabe ozogamicina (vide item 8. Posologia e Modo de Usar).
Síndrome de lise tumoral
Em um estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), foi relatada síndrome de lise tumoral (SLT), que pode ter risco de vida ou ser fatal, em 4/164 (2%) dos pacientes (vide item 9. Reações Adversas). SLT de grau 3/4 foi relatada em 3/164 (2%) dos pacientes. A SLT ocorreu logo após o final da infusão de inotuzumabe ozogamicina, sendo resolvida com tratamento médico.
Monitore os sinais e os sintomas de SLT, tratando-a de acordo com a prática clínica padrão.
Medicação prévia para reduzir os níveis de ácido úrico e hidratação é recomendada antes do início do tratamento para pacientes com uma alta carga de doença (Veja seção 8. Posologia e Modo de Usar).
Prolongamento do intervalo de QT
Em um estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), aumentos no intervalo de QT corrigido para a frequência cardíaca utilizando a fórmula de Fridericia (QTcF) ≥60 ms na avaliação inicial foram medidos em 4/162 (3%) dos pacientes. Nenhum paciente tinha valores de QTcF > 500 ms. Foi relatado um prolongamento no intervalo de QT de grau 2 em 2/164 (1%) dos pacientes. Não foi relatado nenhum prolongamento de QT de grau ≥3 ou eventos de Torsade de Pointes (vide itens 9. Reações Adversas e 3. Propriedades Farmacológicas - Propriedades Farmacodinâmicas).
O inotuzumabe ozogamicina deve ser administrado com precaução em pacientes com histórico ou predisposição para prolongamento do intervalo de QT, recebendo medicamentos que sabidamente prolongam o intervalo de QT e em pacientes com transtornos eletrolíticos (vide item 6. Interações Medicamentosas). Eletrocardiogramas (ECGs) e eletrólitos devem ser obtidos antes do início do tratamento, bem como periodicamente monitorados durante o tratamento.
Aumento da amilase e lipase
Nos pacientes que receberam inotuzumabe ozogamicina, foram relatados aumentos de amilase e lipase (ver item 9. Reações Adversas).
Os pacientes devem ser monitorados quanto ao aumento de amilase e lipase. A doença hepatobiliar potencial deve ser avaliada e tratada de acordo com a prática médica padrão.
Imunizações
A segurança da imunização com vacinas virais vivas durante ou após o tratamento com Besponsa® não foi estudada. A vacinação com vacinas virais vivas não é recomendada durante pelo menos 2 semanas antes do início do tratamento com Besponsa®, durante o tratamento e até a recuperação de linfócitos B após o último ciclo de tratamento.
Fertilidade, gravidez e lactação
• Mulheres em idade fértil/contracepção em homens e mulheres
Mulheres em idade fértil devem ser orientadas a evitar engravidar durante o uso de inotuzumabe ozogamicina.
As mulheres devem ser orientadas a usar contracepção eficaz durante o tratamento com inotuzumabe ozogamicina e durante, pelo menos, oito meses após a última dose. Homens com parceiras do sexo feminino em idade fértil devem usar anticoncepção eficaz durante o tratamento com inotuzumabe ozogamicina e durante, pelo menos, cinco meses após a última dose.
• Gravidez
Não há dados sobre mulheres grávidas usando inotuzumabe ozogamicina. Com base em achados de segurança não clínica, o inotuzumabe ozogamicina pode causar danos embrionários quando administrado a uma mulher grávida. Estudos em animais mostraram toxicidade reprodutiva (vide item 3. Propriedades Farmacológicas - Dados de segurança pré-clínicos).
O inotuzumabe ozogamicina não deve ser usado durante a gravidez, a menos que o possível benefício para a mãe supere os riscos potenciais para o feto. Mulheres grávidas ou pacientes que engravidam enquanto recebem inotuzumabe ozogamicina ou pacientes do sexo masculino tratados como parceiros de mulheres grávidas devem ser informados do perigo potencial para o feto.
• Amamentação
Não há informações sobre a presença de inotuzumabe ozogamicina ou de seus metabólitos no leite humano, os efeitos sobre bebês amamentados ou os efeitos sobre a produção de leite. O risco a recém-nascidos/bebês não pode ser excluído. Devido à possibilidade de reações adversas em bebês amamentados, as mulheres não devem amamentar durante o tratamento com inotuzumabe ozogamicina e durante, pelo menos, dois meses após a dose final (vide item 3. Propriedades Farmacológicas - Dados de segurança pré-clínicos).
• Fertilidade
Com base em achados não clínicos, a fertilidade masculina e feminina pode ser comprometida pelo tratamento com inotuzumabe ozogamicina (vide item 3. Propriedades Farmacológicas - Dados de segurança pré-clínicos). Homens e mulheres devem procurar orientação para a preservação da fertilidade antes do tratamento.
Besponsa® é um medicamento classificado na categoria D de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. A paciente deve informar imediatamente seu médico em caso de suspeita de gravidez.
Efeitos na capacidade de dirigir e operar máquinas
O inotuzumabe ozogamicina pode terinfluência na capacidade de dirigir e operar máquinas. Os pacientes podem apresentar fadiga durante o tratamento com inotuzumabe ozogamicina (vide item 9. Reações Adversas). Portanto, deve ser recomendado cuidado ao dirigir ou operar máquinas.
Incompatibilidades
Na ausência de estudos de compatibilidade, este medicamento não deve ser administrado com outros medicamentos exceto aqueles mencionados no item 8. Posologia e Modo de Usar.
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
6. INTERAÇÕES MEDICAMENTOSAS
Nenhum estudo clínico de interações medicamentosas foi realizado com o inotuzumabe ozogamicina (vide item 3. Propriedades Farmacológicas - Propriedades Farmacocinéticas).
Em um estudo clínico randomizado de inotuzumabe ozogamicina em pacientes com LLA recidivada ou refratária (estudo 1), foi observado um intervalo de QT prolongado com inotuzumabe ozogamicina (vide itens 3. Propriedades Farmacológicas - Propriedades Farmacodinâmicas e vide item 5. Advertências e Precauções). Portanto, o uso concomitante de inotuzumabe ozogamicina com medicamentos que sabidamente prolongam o intervalo de QT ou que sejam capazes de induzir Torsades de Pointes deve ser cuidadosamente considerado. Monitore o intervalo de QT no caso de combinações desses medicamentos (vide item 5. Advertências e Precauções).
Efeito de outros medicamentos sobre o inotuzumabe ozogamicina
In vitro, N-acetil-gama-caliqueamicina dimetilidrazida é principalmente metabolizada por redução não enzimática. Portanto, é improvável que a coadministração de inotuzumabe ozogamicina com inibidores ou indutores das enzimas metabolizadoras de fármacos do citocromo P450 (CYP) ou da uridina difosfato glucuronosiltransferase (UGT) altere a exposição a N-acetil-gama-caliqueamicina dimetilidrazida.
Com base na análise farmacocinética populacional em 736 pacientes, a administração concomitante de medicamentos citorredutores, incluindo hidroxiureia, fatores estimulantes de colônias de granulócitos, incluindo filgrastim, lenograstim e inibidores da P-gp, não tiveram efeito aparente sobre a depuração do inotuzumabe ozogamicina.
Efeito de inotuzumabe ozogamicina sobre outros medicamentos
Efeito sobre os substratos de CYP:
In vitro, a N-acetil-gama-caliqueamicina dimetilidrazida e o inotuzumabe ozogamicina têm baixo potencial para inibir as atividades de CYP1A2, CYP2A6 (testado usando somente inotuzumabe ozogamicina), CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 e CYP3A4/5 ou para induzir as atividades de CYP1A2, CYP2B6 e CYP3A4 em concentrações clinicamente relevantes.
Efeito sobre os substratos de UGT:
In vitro, a N-acetil-gama-caliqueamicina dimetilidrazida teve baixo potencial para inibir as atividades de UGT1A1, UGT1A4, UGT1A6, UGT1A9 e UGT2B7 em concentrações clinicamente relevantes.
Efeitos sobre os substratos do transportador de medicamento:
In vitro, a N-acetil-gama-caliqueamicina dimetilidrazida teve baixo potencia