BENEFIX
PFIZER
fator ix
Anti-hemofílico.
Apresentações.
BeneFixTM 250 UI, 500 UI, 1000 UI ou 2000 UI, pó liófilo injetável, em embalagens contendo 1 frascoampola + 1 seringa preenchida com 5 mL de diluente + 1 adaptador do frasco-ampola (para reconstituição) + 1 conjunto de infusão + 2 lenços umedecidos com álcool + 1 curativo + 1 gaze.
VIA DE ADMINISTRAÇÃO: INTRAVENOSA
USO ADULTO E PEDIÁTRICO
Composição.
Cada frasco-ampola de BeneFixTM contém 250 UI, 500 UI, 1000 UI ou 2000 UI de alfanonacogue. Excipientes: L-histidina, glicina, sacarose, polissorbato 80 e ácido clorídrico. Diluente: água para injeção (solução de cloreto de sódio 0,234%).
Indicações.
BeneFixTM (alfanonacogue) está indicado para o controle e prevenção de episódios hemorrágicos e para profilaxia de atividades rotineiras e cirúrgicas de pacientes com hemofilia B (deficiência congênita de fator IX ou doença de Christmas), incluindo controle e prevenção de sangramento em procedimentos cirúrgicos.
Resultados de eficácia.
Em 4 estudos clínicos de alfanonacogue, um total de 128 indivíduos (56 pacientes previamente tratados, 9 indivíduos participantes apenas no estudo cirúrgico, e 63 pacientes não tratados previamente) receberam mais de 28 milhões de UI administrados durante um período de até 64 meses. Os estudos incluíram 121 indivíduos HIV-negativos e 7 indivíduos HIV-positivos.
Cinquenta e seis pacientes previamente tratados receberam aproximadamente 20,9 milhões de UI de alfanonacogue em dois estudos clínicos. O número médio de dias de exposição foi 83,5. Estes pacientes previamente tratados foram tratados sob demanda ou profilaticamente durante episódios hemorrágicos e foram acompanhados durante um intervalo médio de 24 meses (intervalo de 1 a 29 meses, média de 23,4 ± 5,34 meses). Cinquenta e cinco destes pacientes previamente tratados receberam uma média de 42,8 UI/kg (variação de 6,5 a 224,6 UI/kg, média de 46,6 ± 23,5 UI/kg) por infusão; para os episódios hemorrágicos. Todos os indivíduos foram avaliados quanto à eficácia. Um indivíduo interrompeu o estudo após um mês de tratamento devido a episódios de sangramento difíceis de controlar; ele não tinha um inibidor detectável. A dose desse indivíduo não tinha sido adequadamente titulada. Os outros 55 indivíduos restantes foram tratados com sucesso. Episódios hemorrágicos (sem relatos quanto a sua gravidade) como hemartroses, sangramento em partes moles e músculo foram tratados com sucesso. Oitenta e oito por cento das infusões totais administradas para episódios de sangramento foram classificadas como geradoras de uma resposta "excelente" ou "boa". Oitenta e um por cento de todos os episódios de sangramento foram tratados com uma única infusão de alfanonacogue. Um indivíduo desenvolveu um inibidor transitório de baixa titulação (titulação máxima de 1,5 BU). Este indivíduo já havia recebido produtos derivados do plasma sem um histórico de desenvolvimento de inibidores. Ele foi capaz de continuar o tratamento com alfanonacogue sem aumento do inibidor ou anafilaxia, no entanto, foi necessário o aumento da frequência de administração de alfanonacogue; posteriormente, o inibidor do fator IX do indivíduo e seu efeito sobre a meia-vida de alfanonacogue foram resolvidos. Quarenta e um pacientes tiveram medidas de fibrinopeptídeo A e fragmento de protrombina 1 + 2 antes da infusão, 4 a 8 horas e 24 horas após a infusão. Vinte e nove dos indivíduos tiveram elevações em fibrinopeptídeo A com um valor máximo de 35,3 nmol/L (22 dos 29 indivíduos tiveram valores basais elevados). Dez dos indivíduos tinham fragmentos de protrombina 1 + 2 elevados com um valor máximo de 1,82 nmol/L (3 dos 10 indivíduos tiveram valores basais elevados).
Um total de 20 pacientes previamente tratados receberam alfanonacogue para profilaxia secundária (administração regular de terapia de reposição de fator IX para prevenir hemorragias em pacientes que já demonstraram evidência clínica de artropatia hemofílica ou doença articular) com alguma periodicidade; durante o estudo a média foi de 2,0 infusões por semana. Foi administrado alfanonacogue em 19 pacientes para profilaxia secundária de rotina (pelo menos duas vezes por semana) para um total de 345 pacientes-meses com um período médio de acompanhamento de 24 meses por indivíduo. A dose média utilizada por estes 19 indivíduos foi de 40,3 UI/kg, variando de 13 a 78 UI/kg. Um indivíduo adicional foi tratado semanalmente, utilizando uma dose média de 33,3 UI/kg, durante um período de 21 meses. Noventa e três por cento das respostas foram classificadas como "excelente" ou "eficaz". Estes 20 pacientes previamente tratados receberam um total de 2.985 infusões de alfanonacogue para profilaxia de rotina. Sete destes pacientes previamente tratados experimentaram um total de 26 episódios de sangramento espontâneos dentro de 48 horas após a infusão.
O controle de hemostasia foi avaliado no contexto cirúrgico. Trinta e seis procedimentos cirúrgicos foram realizados em 28 pacientes. Treze procedimentos cirúrgicos de pequeno porte foram realizados em 12 indivíduos, incluindo sete procedimentos odontológicos, uma biópsia da pele, uma remoção do cisto, uma vasectomia, uma remoção de nevus, e 2 remoções de unha encravada do pé. Vinte e três procedimentos cirúrgicos maiores foram realizados em 19 indivíduos, incluindo um transplante de fígado, uma esplenectomia, 3 herniorrafias inguinais, 11 procedimentos ortopédicos, um desbridamento em panturrilha e 6 extrações dentárias complicadas.
Vinte e três indivíduos foram submetidos a 27 procedimentos cirúrgicos com um regime de reposição de pulso. A dose média perioperatória (pré e intra-operatória) para esses procedimentos foi de 85 ± 32,8 UI/kg (variação de 25 a 154,9 UI/kg). A dose média total pós-operatória (hospitalar e ambulatorial) foi de 63,1 ± 22,0 UI/kg (variação de 28,6 a 129,0).
A cobertura total de alfanonacogue durante o período cirúrgico para os principais procedimentos variou de 4.230 a 385.800 UI. A dose no pré-operatório para os procedimentos principais variou de 75 a 155 UI/kg. Nove dos principais procedimentos cirúrgicos foram realizados em 8 indivíduos usando um regime de infusão contínua.
Após doses em bolus no pré-operatório (94,1 a 144,5 UI/kg), infusão contínua de alfanonacogue foi administrada a uma taxa média de 6,7 UI/kg/hr (variação das taxas médias: 4,3 a 8,6 UI/kg/hr, média de 6,4 ± 1,5 UI/kg/hr) para uma duração média de 5 dias (intervalo de 1 a 11 dias, média de 4,9 ± 3,1). Seis dos oito indivíduos que receberam infusão contínua de alfanonacogue para cirurgias de grande porte foram transferidos para os regimes de pulso intermitente na dose média de 56,3 UI/kg (intervalo 33,6 a 89,1 UI/kg, média de 57,8 ± 18,1 UI/ kg Desvio Padrão) por uma média de 3,5 dias de exposição (intervalo de 1 a 5 dias, com média de 3,3 ± 1,4 Desvio Padrão) durante o período pós-operatório. Embora os níveis de fator IX circulantes direcionados para restaurar e manter a hemostasia foram alcançados tanto com o regime de reposição de pulso quanto com o regime de infusão contínua, experiência em ensaios clínicos com infusão contínua de alfanonacogue para a profilaxia cirúrgica na hemofilia B tem sido muito limitada para estabelecer a sua eficácia clínica e segurança. Indivíduos que receberam administração por infusão contínua de alfanonacogue para a profilaxia cirúrgica também receberam infusões em bolus intermitentes do produto.
Entre os pacientes cirúrgicos, o aumento médio da atividade do fator IX circulante foi de 0,7 UI/dL por UI/kg infundida (intervalo de 0,3 a 1,2 UI/dL, média 0,8 ± 0,2 UI/dL por UI/kg). A eliminação média da meia-vida para os pacientes cirúrgicos foi de 19,4 horas (intervalo de 10 a 37 horas, média 21,3 ± 8,1 horas).
A hemostasia foi mantida durante todo o período cirúrgico, no entanto, um paciente necessitou de uma drenagem de hematoma em ferida cirúrgica local, e um outro paciente que recebeu alfanonacogue após uma extração de dente necessitou de intervenção cirúrgica adicional devido a uma coleção no local da extração. Não houve evidência clínica de complicações trombóticas em qualquer um dos sujeitos. Em sete indivíduos nos quais fibrinopeptídeo A e fragmento de protrombina 1 + 2 foram medidos antes da infusão, em 4 a 8 horas, e diariamente até 96 horas, não houve evidência de aumento significativo na ativação da coagulação. Dados de dois outros indivíduos foram considerados não avaliáveis.
Sessenta e três pacientes não tratados previamente receberam aproximadamente 6,2 milhões de UI de alfanonacogue em estudo aberto de eficácia e segurança acima de uma média de 89 dias de exposição. Estes pacientes não tratados previamente foram acompanhados durante um intervalo médio de 37 meses (intervalo de 4 a 64 meses, média de 38,1 ± 16,4 meses). Cinquenta e quatro destes pacientes não tratados previamente receberam uma dose média de 62,7 UI/kg (variação de 8,2 a 292,0 UI/kg, média de 75,6 ± 42,5 UI/kg) por infusão para episódios de sangramento. Dados relativos à gravidade dos episódios de sangramento não foram relatados. Noventa e quatro por cento das infusões administradas para iniciar o tratamento do sangramento foram classificadas como resposta "excelente" ou "boa".
Setenta e cinco por cento de todos os episódios de sangramento foram tratados com uma única infusão de alfanonacogue. Três destes 54 indivíduos não foram tratados com sucesso, incluindo um episódio em um indivíduo devido ao tempo de atraso da infusão e dose insuficiente, e em 2 indivíduos devido à formação de inibidor. Um indivíduo desenvolveu um inibidor de alta titulação (titulação máxima de 42 BU) no dia 7 da exposição. Um segundo indivíduo desenvolveu um inibidor de alta titulação (titulação máxima de 18 BU) após 15 dias de exposição. Ambos os indivíduos experimentaram manifestações alérgicas em associação temporal com o desenvolvimento de inibidores.
Trinta e dois pacientes previamente não tratados receberam alfanonacogue para a profilaxia de rotina. Vinte e quatro pacientes previamente não tratados receberam alfanonacogue pelo menos duas vezes por semana para um total de 2.587 infusões. A dose média por infusão foi de 72,5 ± 37,1 UI/kg, e a duração média da profilaxia foi de 13,4 ± 8,2 meses. Oito pacientes previamente não tratados receberam alfanonacogue uma vez por semana para um total de 571 infusões. A dose média por infusão foi 75,9 ± 17,9 UI/kg, e a duração média da profilaxia foi de 17,6 ± 7,4 meses. Cinco pacientes previamente não tratados experimentaram um total de 6 episódios de sangramento espontâneos dentro de 48 horas após a infusão.
Vinte e três pacientes previamente não tratados receberam alfanonacogue para profilaxia cirúrgica em 30 procedimentos cirúrgicos. Todos os procedimentos cirúrgicos foram de pequeno porte, exceto duas herniorrafias. A dose em bolus pré-operatória variou de 32,3 UI/kg para 247,2 UI/kg. A dose total perioperatória variou de 385 a 23.280 UI. Cinco dos procedimentos cirúrgicos foram realizados utilizando um esquema de infusão contínua de 3 a 5 dias. Experiência em ensaios clínicos de alfanonacogue para a profilaxia cirúrgica na hemofilia B é muito limitada para estabelecer a eficácia clínica e segurança da administração do produto por infusão contínua.
População pediátrica
A segurança e eficácia foram demonstradas em pacientes pediátricos previamente tratados ou não tratados. Consulte o item 8. Posologia e modo de usar.
Para informações de dose relacionadas ao uso em crianças, consulte o item 8. Posologia e modo de usar.
Caract. farmacológicas.
Mecanismo de Ação
BeneFixTM contém fator IX de coagulação recombinante (alfanonacogue). Esse fator recombinante é uma glicoproteína de cadeia única com massa molecular aproximada de 55.000 Daltons que pertence à família das serinoproteases dos fatores de coagulação dependentes da vitamina K. O fator IX de coagulação recombinante é uma glicoproteína à base de DNA recombinante com características estruturais e funcionais equivalentes às do fator IX endógeno. O fator IX é ativado tanto pelo complexo fator VII/fator tecidual na via extrínseca, quanto pelo fator XIa na via intrínseca. O fator IX ativado, em combinação com o fator VIII ativado, ativa o fator X. Isso resulta na conversão da protrombina em trombina. A trombina, então, converte o fibrinogênio em fibrina e ocorre a formação de um coágulo. A atividade do fator IX está ausente ou bastante reduzida em pacientes com hemofilia B, podendo ser necessário instituir terapia de reposição.
A hemofilia B é um distúrbio de coagulação sanguínea hereditária ligada ao sexo e devido à diminuição dos níveis de fator IX, resulta em hemorragia profusa nas articulações, músculos ou órgãos internos, tanto espontaneamente como em decorrência de trauma acidental ou cirúrgico. Com a terapia de reposição, os níveis plasmáticos do fator IX aumentam, possibilitando a correção temporária da deficiência do fator e a correção da tendência a episódios hemorrágicos.
Farmacocinética
Uma infusão única de BeneFixTM em pacientes (dados de base) com hemofilia B mostra valores médios de recuperação de ± desvio padrão, determinados pela idade, 0,78 ± 0,23 UI/dL/UI/kg (variação de 0,4 a 1,4
UI/dL por UI/kg) para indivíduos ≥15 anos de idade (n=37) e 0,68 ± 0,30 UI/dL por UI/kg (variação de 0,18 a
2,08 UI/dL por UI/kg) para indivíduos < 15 anos de idade (n=56).
Em um estudo farmacocinético cruzado e randomizado, BeneFixTM reconstituído em 0,234% de diluente de cloreto de sódio mostrou-se ser farmacocineticamente equivalente ao BeneFixTM previamente comercializado (reconstituído com água estéril para injeção) em 24 pacientes previamente tratados (≥12 anos) a uma dose de 75 UI/kg. Além disso, os parâmetros farmacocinéticos foram acompanhados em 23 dos mesmos pacientes previamente tratados após a administração repetida de BeneFixTM por seis meses e se mostraram inalterados em comparação àquelas obtidos no início da avaliação. Um resumo dos dados farmacocinéticos pode ser encontrado na Tabela 1:
Contraindicações.
BeneFixTM é contraindicado em pacientes com história conhecida de hipersensibilidade a qualquer constituinte da fórmula ou em pacientes com história conhecida de hipersensibilidade a proteínas de hamster.
Advertências e precauções.
Hipersensibilidade
Foram relatadas reações de hipersensibilidade do tipo alérgica, incluindo anafilaxia, com todos os produtos com fator IX, inclusive BeneFixTM. Frequentemente, esses eventos ocorreram em relação temporal estreita com o desenvolvimento de inibidores do fator IX. Os pacientes devem ser informados sobre os sinais e sintomas iniciais das reações de hipersensibilidade, incluindo urticária, urticária generalizada, calafrios, rubor, angioedema, aperto no peito, laringoespasmo, broncoespasmo, dispneia, chiados, desmaio, hipotensão, taquicardia, visão turva e anafilaxia. Se ocorrerem reações alérgicas ou anafiláticas, a administração de BeneFixTM deve ser imediatamente suspensa e devem ser adotadas condutas clínicas adequadas, que pode incluir tratamento para choque. Caso ocorra algum desses sintomas, os pacientes devem ser aconselhados a descontinuar o uso do produto e entrar em contato com o médico e/ou procurar atendimento de emergência imediato, dependendo do tipo/gravidade da reação.
Foi relatada síndrome nefrótica após a indução de tolerância imunológica com produtos com fator IX em pacientes com hemofilia B com inibidores do fator IX e história de reações alérgicas ao fator IX. A segurança e a eficácia do uso de BeneFixTM para indução de tolerância imunológica ainda não foram estabelecidas. No caso de reações alérgicas graves, medidas hemostáticas alternativas devem ser consideradas.
Anticorpos neutralizadores de atividade
Foram detectados inibidores em pacientes tratados com produtos com fator IX. Assim como ocorre com todos os produtos com fator IX, os pacientes em uso de BeneFixTM devem ser monitorizados quanto ao desenvolvimento de inibidores de fator IX. Os pacientes com inibidores do fator IX podem apresentar maior risco de anafilaxia com uma exposição futura ao fator IX. Os pacientes que apresentarem reações alérgicas devem ser avaliados quanto à presença de inibidor. Informações preliminares sugerem que pode existir uma relação entre a presença de mutações (deleções) importantes no gene do fator IX e risco aumentado de formação de inibidor e de reações de hipersensibilidade aguda. Os pacientes que sabidamente apresentam mutações como deleções importantes do gene do fator IX devem ser observados atentamente quanto a sinais e sintomas de reações de hipersensibilidade aguda, particularmente durante as fases iniciais da primeira exposição ao produto. Como existe a possibilidade de reações alérgicas com os concentrados de fator IX, as administrações iniciais (aproximadamente 10-20) de fator IX devem ser realizadas sob supervisão médica em locais que dispõem de recursos médicos adequados para tratamento de reações alérgicas.
Trombose
Historicamente, a administração dos concentrados complexos do fator IX derivados do plasma humano, contendo os fatores II, VII, IX e X, estava associada com o desenvolvimento de complicações tromboembólicas. Embora BeneFixTM não contenha fatores coagulantes, com exceção do fator IX, o risco potencial de trombose e de coagulação intravascular disseminada observado em outros produtos contendo o fator IX deve ser reconhecido. Em razão do risco potencial de complicações tromboembólicas, deve-se ter cautela ao se administrar este produto em pacientes com doença hepática, pacientes no pós-operatório, recémnascidos ou pacientes com risco de fenômenos tromboembólicos ou coagulação intravascular disseminada.
A segurança e eficácia da administração de BeneFixTM através da infusão contínua não foi definida. Consulte os itens 8. Posologia e Modo de Usar e 9. Reações Adversas. Houve relatos pós-comercialização de eventos trombóticos, incluindo a síndrome fatal de veia cava superior com risco de morte em recém-nascidos criticamente doentes, ao receber infusão contínua de BeneFixTM através de um cateter venoso central.
TM
Gravidez: Estudos de reprodução e lactação em animais não foram conduzidos com o BeneFix. Não existe experiência suficiente com o uso dos produtos com fator IX em mulheres grávidas. Portanto, o fator IX deve ser administrado a mulheres grávidas apenas se a indicação for clara.
BeneFixTM é um medicamento classificado na categoria C de risco de gravidez. Portanto, este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica ou do cirurgiãodentista.
Lactação: Não existe experiência suficiente com o uso dos produtos com fator IX em mulheres lactantes. Portanto, o fator IX deve ser administrado a mulheres em fase de amamentação apenas se a indicação for clara.
Efeitos na capacidade de dirigir e usar máquinas
Com base no perfil farmacodinâmico e farmacocinético e nas reações adversas reportadas, o BeneFixTM tem pouca ou nenhuma influência na capacidade de dirigir ou usar máquinas.
Uso Pediátrico: Foram avaliadas a segurança e eficácia em pacientes pediátricos previamente tratados e previamente não tratados. Para informações posológicas em crianças veja também o item 8. Posologia e Modo de Usar - Posologia para Tratamento Profilático.
Uso Geriátrico: Os estudos clínicos de BeneFixTM não incluíram números suficientes de indivíduos com idade maior ou igual a 65 anos para determinar se eles respondem de modo diferente dos indivíduos mais jovens. Assim como ocorre com qualquer paciente tratado com BeneFixTM, a escolha da dose em pacientes idosos deve ser individualizada.
Dados de Segurança Pré-clínicos
BeneFixTM mostrou ser não mutagênico no teste de Ames e não clastogênico em ensaio de aberrações cromossômicas. Não foram realizados estudos sobre carcinogênese ou prejuízos da fertilidade.
Compatibilidades, Incompatibilidades
Como não existem estudos de incompatibilidade, BeneFixTM reconstituído não deve ser administrado pelo equipo ou recipiente com outros medicamentos. Utilizar apenas o conjunto de infusão fornecido. Pode ocorrer falha de tratamento como consequência da adsorção do fator IX de coagulação humano na superfície interna de certos equipos de infusão.
Atenção: Este medicamento contém Açúcar, portanto, deve ser usado com cautela em portadores de Diabetes.
Interações medicamentosas.
Não são conhecidas interações entre o fator IX de coagulação recombinante com outros medicamentos.
Interferências com Exames Laboratoriais e Testes Diagnósticos
Foi observada correção temporária de tempo de tromboplastina parcial (TTPa) anormal; nenhum efeito sobre TTPa normal foi observado.
Cuidados de armazenamento.
BeneFixTM pó liófilo injetável deve ser conservado sob refrigeração (temperatura entre 2 e 8°C) e pode ser utilizado por 36 meses a partir da data de fabricação.
Deve-se evitar o congelamento para prevenir danos à seringa preenchida de diluente.
BeneFixTM, quando reconstituído, contém polissorbato-80, que é conhecido por aumentar a taxa de extração de di-(2-etilheil)ftalato (DEHP) a partir do cloreto de polivinilo (PVC). Isso deve ser considerado durante a preparação e administração do BeneFixTM, incluindo o tempo de armazenamento em um recipiente de PVC após a reconstituição. É importante que as recomendações no item 8. Posologia e modo de usar sejam seguidas rigorosamente.
Após reconstituição, manter em temperatura ambiente por no máximo 3 horas.O medicamento reconstituído deve ser administrado dentro de três horas após a reconstituição.
Número de lote e datas de fabricação e validade: vide embalagem.
Não use medicamento com o prazo de validade vencido.
Guarde-o em sua embalagem original.
Antes de usar, observe o aspecto do medicamento.
Todo medicamento deve ser mantido fora do alcance das crianças.
Características físicas e organolépticas: massa branca essencialmente livre de material particulado claramente visível. Após a reconstituição: solução límpida, incolor e essencialmente livre de material particulado claramente visível.
Posologia e modo de usar.
Posologia
O tratamento deve ser iniciado sob supervisão de um médico com experiência no tratamento da hemofilia B.
O tratamento com produtos que contenham fator IX, inclusive BeneFixTM, requer ajuste de dose individualizado. A dose e a duração do tratamento com todos os produtos com fator IX dependem da gravidade da deficiência desse fator, da localização e extensão da hemorragia e da condição clínica do paciente.
A dose de BeneFixTM pode diferir da utilizada para produtos com fator IX derivado do plasma.
Para garantir que o nível de atividade pretendido do fator IX seja atingido, aconselha-se um monitoramento detalhado utilizando o ensaio da atividade do fator IX, particularmente em intervenções cirúrgicas. Para ajuste posológico adequado, as doses devem ser tituladas levando-se em consideração a atividade do fator IX, os parâmetros farmacocinéticos (tais como meia-vida e recuperação), bem como a condição clínica.
O número de unidades de fator IX administrado é expresso em Unidades Internacionais (UI), em conformidade com o padrão atual da OMS para produtos com fator IX. A atividade plasmática do fator IX é expressa em porcentagem (referente ao plasma humano normal) ou em UI (referente a um padrão internacional para fator IX no plasma). Uma UI de atividade do fator IX equivale à quantidade de fator IX em um mL de plasma humano normal.
A farmacocinética deve ser avaliada regularmente em todos os pacientes e a dose deve ser devidamente ajustada.
O texto para o cálculo da dose encontra-se abaixo:
Pacientes > 15 anos
Em pacientes > 15 anos, em média, uma unidade internacional de BeneFixTM por quilograma de peso corporal aumentou a atividade de fator IX circulante em 0,8 ± 0,2 (intervalo de 0,4 a 1,4) UI/dL. O método de estimativa da dose é ilustrado no exemplo a seguir. Se você estima um aumento médio de 0,8 UI/dL de fator IX por UI/kg administrado por peso corporal, então:
Número de fator IX = Peso x Aumento pretendido de fator x 1,2 (UI/kg por UI/dL)* necessário em UI corpóreo IX (% ou UI/dL) (kg) *Recíproco da recuperação observada (UI/kg por UI/dL)
Pacientes < 15 anos
Em pacientes < 15 anos, em média, uma unidade internacional de BeneFixTM por quilograma de peso corporal aumentou a atividade de fator IX circulante em 0,7 ± 0,3 (intervalo 0,2 a 2,1 UI/dL e mediana de 0,6 UI/dL por UI/kg). O método de estimativa da dose é ilustrado no exemplo a seguir. Se você estima um aumento médio de 0,7 UI/dL de fator IX por UI/kg administrado por peso corporal, então:
Número de fator IX = Peso x Aumento pretendido de fator x 1,4 (UI/kg por UI/dL)* necessário em UI corpóreo IX (% ou UI/dL) (kg) *Recíproco da recuperação observada (UI/kg por UI/dL)
Posologia para Episódios de Sangramento e Cirurgia
No caso dos eventos hemorrágicos mencionados na tabela 2 a seguir, a atividade do fator IX não deve ficar abaixo do nível de atividade plasmática fornecido (em % do valor normal ou em UI/dL) no período correspondente.
Tabela 2: Guia de dose para o controle e prevenção de episódios de sangramento e cirurgia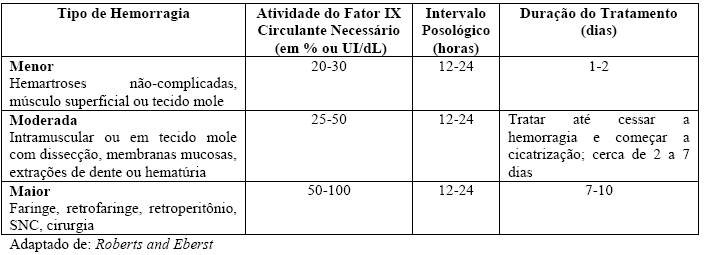
Adaptado de: Roberts and Eberst
Posologia para Tratamento Profilático
Em um estudo clínico de profilaxia secundária de rotina, a dose média para pacientes adultos previamente tratados foi de 40 UI/kg (intervalo de 13 a 78 UI/kg) em intervalos de 3 ou 4 dias. Em pacientes mais jovens, pode ser necessário utilizar intervalos posológicos mais curtos ou doses mais altas.
População idosa
Os estudos clínicos do BeneFixTM não incluíram números suficientes de indivíduos acima de 65 anos ou mais para determinar se eles respondem de maneira diferente dos indivíduos mais jovens. Assim como com todos os pacientes recebendo o BeneFixTM, a seleção de dose para um paciente idoso deve ser individualizada.
Reconstituição
Lave sempre as mãos antes de realizar os seguintes procedimentos. Uma técnica asséptica (ou seja, limpa e livre de germes) deve ser utilizada durante o procedimento de reconstituição. Todos os componentes utilizados na reconstituição e administração deste medicamento devem ser utilizados o mais rapidamente possível após a abertura de seus recipientes esterilizados para minimizar a exposição desnecessária à atmosfera.
BeneFixTM é administrado por infusão intravenosa (IV) após reconstituição com o diluente fornecido (cloreto de sódio 0,234%) na seringa preenchida.
1. Permita que o frasco-ampola de BeneFixTM liofilizado e a seringa preenchida de diluente atinjam a temperatura ambiente.
2. Remova a tampa plástica tipo flip-top do frasco-ampola BeneFixpara expor a porção central da tampa de borracha.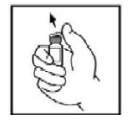
3. Limpe a parte superior do frasco com o lenço umedecido com álcool fornecido, ou utilize outra solução antiséptica e deixe secar. Após a limpeza, não toque na tampa de borracha com a mão ou permita que ela toque em qualquer superfície.
4. Retire a tampa da embalagem plástica transparente do adaptador de frasco-ampola. Não retire o adaptador da embalagem.
5. Coloque o frasco numa superfície plana. Enquanto segura a embalagem com o adaptador, posicione o adaptador do frasco-ampola sobre o frasco-ampola. Pressione firmemente a embalagem até que o adaptador se encaixe na parte superior do frasco, com o grampo do adaptador penetrando na tampa do frasco. Deixe a embalagem do adaptador no lugar.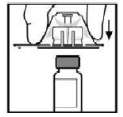
6. Segure a haste do êmbolo como mostrado no diagrama. Evite o contato com o cabo da haste do êmbolo. Conecte a extremidade rosqueada da haste do êmbolo ao êmbolo da seringa preenchida de diluente, empurrando e girando firmemente.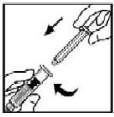
7. Remova a tampa de plástico inviolável da ponta da seringa preenchida de diluente, dobrando a tampa para cima e para baixo para quebrar a perfuração. Não toque no interior da tampa ou na ponta da seringa. Coloque a tampa do seu lado em uma superfície limpa em um local onde seria menos provável de se tornar ambientalmente contaminado.
8. Remova a embalagem para distanciá-la do adaptador e a descarte.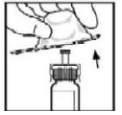
9. Coloque o frasco numa superfície plana. Conecte a seringa preenchida de diluente ao adaptador do frasco, inserindo a ponta na abertura do adaptador enquanto pressiona e gira firmemente a seringa no sentido horário até que a conexão esteja segura.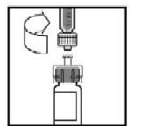
10. Lentamente pressione a haste do êmbolo para injetar todo o diluente no frasco de BeneFixTM.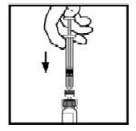
11. Sem retirar a seringa, gire suavemente o conteúdo do frasco até o pó estar dissolvido.
12. Inspecione a solução final para partículas antes da administração. A solução deve aparecer límpida e incolor.
Nota: Se você usar mais de um frasco de BeneFixTM por perfusão, reconstituir cada frasco seguindo as instruções anteriores.
13. Assegure que a haste do êmbolo da seringa ainda esteja totalmente pressionada e inverta o frasco. Tire lentamente a solução para a seringa.
Nota: Se você preparou mais de um frasco de BeneFixTM, retire a seringa preenchida de diluente do adaptador do frasco, deixando o adaptador do frasco ligado ao frasco. Rapidamente anexe uma seringa com bico luerlock grande e aspire de volta o conteúdo reconstituído segundo as instruções acima. Repita este procedimento com cada frasco por vez. Não desconecte as seringas preenchidas com diluente ou seringa com bico luer-lock grande até que esteja pronto para conectar a seringa com bico luer-lock grande ao próximo adaptador do frasco.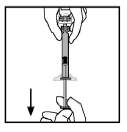
14. Retire a seringa do adaptador do frasco, puxando e girando-a no sentido anti-horário. Descarte o frasco com o adaptador conectado.
Nota: Se a solução não for utilizada de imediato, a tampa da seringa deve ser cuidadosamente recolocada. Não toque na ponta da seringa ou na parte interna da tampa.
Manipulação
Reconstituir o pó liofilizado de BeneFixTM para injeção com o diluente fornecido (cloreto de sódio 0,234%) na seringa preenchida. Uma vez que o diluente foi injetado no frasco, gire gentilmente o frasco até dissolver completamente o pó. Após a reconstituição, a solução é aspirada de volta para a seringa e aplicada via infusão.
A solução deve ser límpida e incolor. A solução deve ser descartada em caso de partículas visíveis ou se for observada descoloração. O produto não contém conservante, e a solução reconstituída deve ser utilizada dentro de 3 horas após a reconstituição.
A solução reconstituída pode ser armazenada em temperatura ambiente até a administração e deve ser administrada em até 3 horas após a reconstituição.
Administração
BeneFixTM é administrado por via intravenosa após a reconstituição do pó liofilizado para solução injetável com o diluente fornecido. A administração do produto deve durar vários minutos (ver item Manipulação). A velocidade de administração deve ser determinada pelo nível de conforto do paciente.
1. Conectar a seringa à extremidade do bico luer do equipo de infusão fornecido.
2. Aplicar um torniquete e preparar o local da injeção limpando bem a pele com o lenço umedecido com álcool fornecido no kit.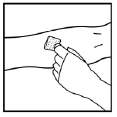
3. Realizar punção venosa. Inserir a agulha no equipo de infusão na veia e retirar o torniquete. O produto BeneFixTM reconstituído deve ser administrado por via intravenosa durante vários minutos. A taxa de administração deve ser determinada pelo nível de conforto do paciente.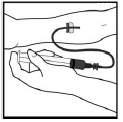
BeneFixTM reconstituído não deve ser administrado por um equipo ou recipiente com outros medicamentos. Após a conclusão do tratamento com BeneFixTM, remova o conjunto de infusão e descarte. Dispor de toda a solução não utilizada, frasco(s) vazio(s), as agulhas e seringas usadas em um recipiente adequado para descarte de resíduos que possam ferir outros se não manipulados e descartados devidamente.
A segurança e eficácia da administração por infusão contínua não foram estabelecidas (ver itens 5.Advertências e Precauções e 9. Reações Adversas).
BeneFixTM deve ser administrado utilizando o conjunto de infusão fornecido no kit, o diluente na seringa preenchida fornecida ou uma seringa de plástico descartável, única e estéril. Além disso, a solução deve ser retirada do frasco utilizando o adaptador de frasco.
Após a reconstituição, o produto deve ser utilizado em um prazo máximo de 3 horas. A solução reconstituída pode ser armazenada em temperatura ambiente até a administração.
Nota: Relatou-se aglutinação de eritrócitos no tubo/seringa com a administração de BeneFixTM. Não foram relatados eventos adversos relacionados a essa observação. Para minimizar o risco de aglutinação, é importante limitar a quantidade de sangue que retorna ao conjunto para infusão. Não deve entrar sangue na seringa. Se ocorrer aglutinação de eritrócitos no equipo ou na seringa, desprezar todo o material (equipo, seringa e solução de BeneFixTM) e reiniciar a administração com uma nova embalagem.
Se alguma reação suspeita de hipersensibilidade for percebida e parecer estar relacionada à administração do BeneFixTM, a taxa de infusão deve ser diminuída ou a infusão deve ser interrompida (consulte itens 5. Advertências e precauções e 9. Reações adversas).
Não há estudos dos efeitos de BeneFixTM administrado por vias não recomendadas. Portanto, para segurança e eficácia desta apresentação, a administração deve ser somente pela via intravenosa.
Dose Omitida:
Caso haja o esquecimento da utilização no horário estabelecido, BeneFixTM pode ser administrado fora do intervalo estabelecido. Entretanto, se já estiver perto do horário de administrar a próxima dose, deve-se desconsiderar a dose esquecida e administrar apenas a próxima dose. Neste caso, o paciente não deve receber dose duplicada. O esquecimento da dose pode comprometer a eficácia do tratamento.
Reações adversas.
A tabela abaixo lista as reações adversas notificadas em estudos clínicos de pacientes previamente tratados e de pacientes previamente não tratados e identificadas na pós-comercialização. As frequências são baseadas em todos os eventos emergentes de tratamento de casualidade em estudos clínicos agrupados com 287 indivíduos.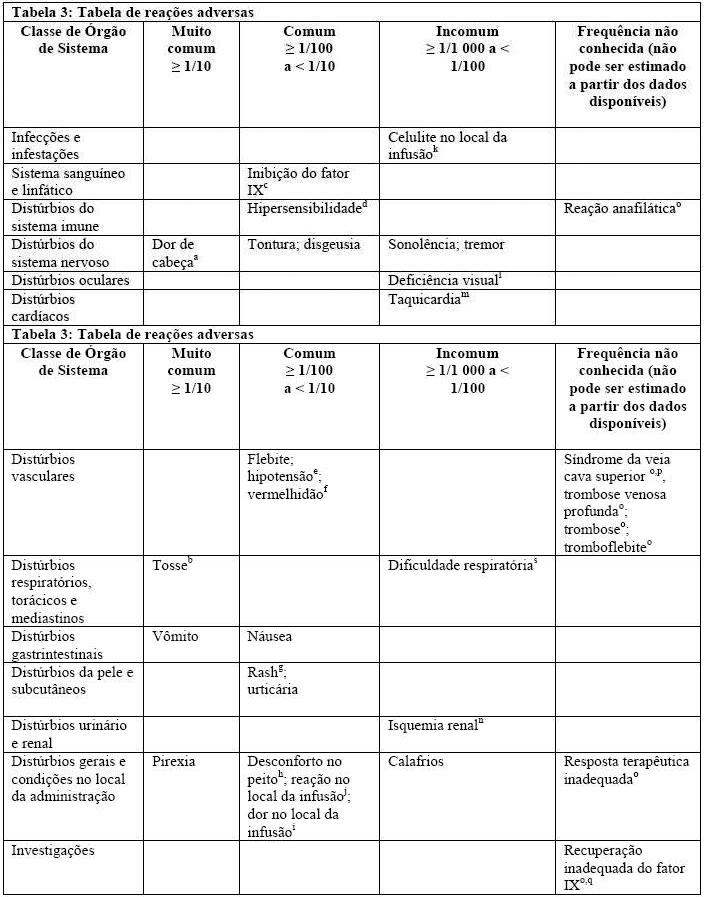
a incluindo enxaqueca, dor de cabeça sinusal.
b incluindo tosse produtiva.
c formação transitória de inibidor de baixa titulação e formação de inibidor de alta titulação.
d incluindo hipersensibilidade a medicamentos, angioedema, broncoespasmo, respiração ofegante, dispneia e espasmo da laringe.
e incluindo diminuição da pressão arterial.
f incluindo rubor, sensação de calor, pele quente.
g incluindo rash macular, papular, maculopapular.
h incluindo dor no peito e aperto no peito.
i incluindo prurido no local da infusão, eritema no local da infusão.
j incluindo dor no local da injeção, desconforto no local da infusão.
k incluindo celulite.
l incluindo escotoma cintilante e visão turva.
m incluindo aumento da frequência cardíaca, taquicardia sinusal.
n desenvolvido em um paciente positivo a anticorpos da hepatite C 12 dias após uma dose de BeneFixTM para um episódio de sangramento. BeneFixTM.
o Reação adversa identificada na pós-comercialização.
p síndrome da veia cava superior em recém-nascidos criticamente doentes, enquanto recebiam infusão contínua do BeneFixTM através de um cateter venoso central.
q Este é um termo verbatim. Nenhum MedDRA 17.1 PT foi recuperado.
Caso ocorra qualquer reação de hipersensibilidade adversa suspeita que se considere relacionada à administração de BeneFixTM, consulte os itens 8. Posologia e modo de usar e 5. Advertências e precauções.
Desenvolvimento de inibidor
Os pacientes com hemofilia B podem desenvolver anticorpos neutralizadores (inibidores).
Detectou-se a presença de inibidor transitório de baixa resposta e clinicamente relevante (título máximo 1,5 BU) em 1 dos 65 pacientes tratados com BeneFixTM (incluindo 9 pacientes participantes somente do estudo cirúrgico) que haviam recebido anteriormente produtos derivados de plasma (pacientes previamente tratados). Esse paciente continuou o tratamento com BeneFixTM sem aumento anamnéstico do inibidor ou anafilaxia.
A partir dos resultados do estudo em pacientes previamente não tratados, 2/63 pacientes desenvolveram inibidores após 7 e 15 dias de exposição. Ambos eram inibidores de alta titulação. Ambos os pacientes apresentaram manifestações alérgicas em associação temporal com o seu desenvolvimento de inibidores.
Atenção: este produto é um medicamento novo e, embora as pesquisas tenham indicado eficácia e segurança aceitáveis, mesmo que indicado e utilizado corretamente, podem ocorrer eventos adversos imprevisíveis ou desconhecidos. Nesse caso, notifique os eventos adversos pelo Sistema de Notificações em Vigilância Sanitária -NOTIVISA, disponível em http://www.anvisa.gov.br/hotsite/notivisa/index.htm, ou para a Vigilância Sanitária Estadual ou Municipal.
Superdose.
Não foram relatados sintomas de superdose com os produtos com fator IX de coagulação recombinante.
Em caso de intoxicação ligue para 08007226001, se você precisar de mais orientações.
Dizeres legais.
MS -1.0216.0233
VENDA SOB PRESCRIÇÃO MÉDICA.
Esta bula foi aprovada pela Anvisa em 02/05/2016.